Recent Advances in Synergistic Antitumor Effects Exploited from the Inhibition of Ataxia Telangiectasia and RAD3-Related Protein Kinase (ATR)


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
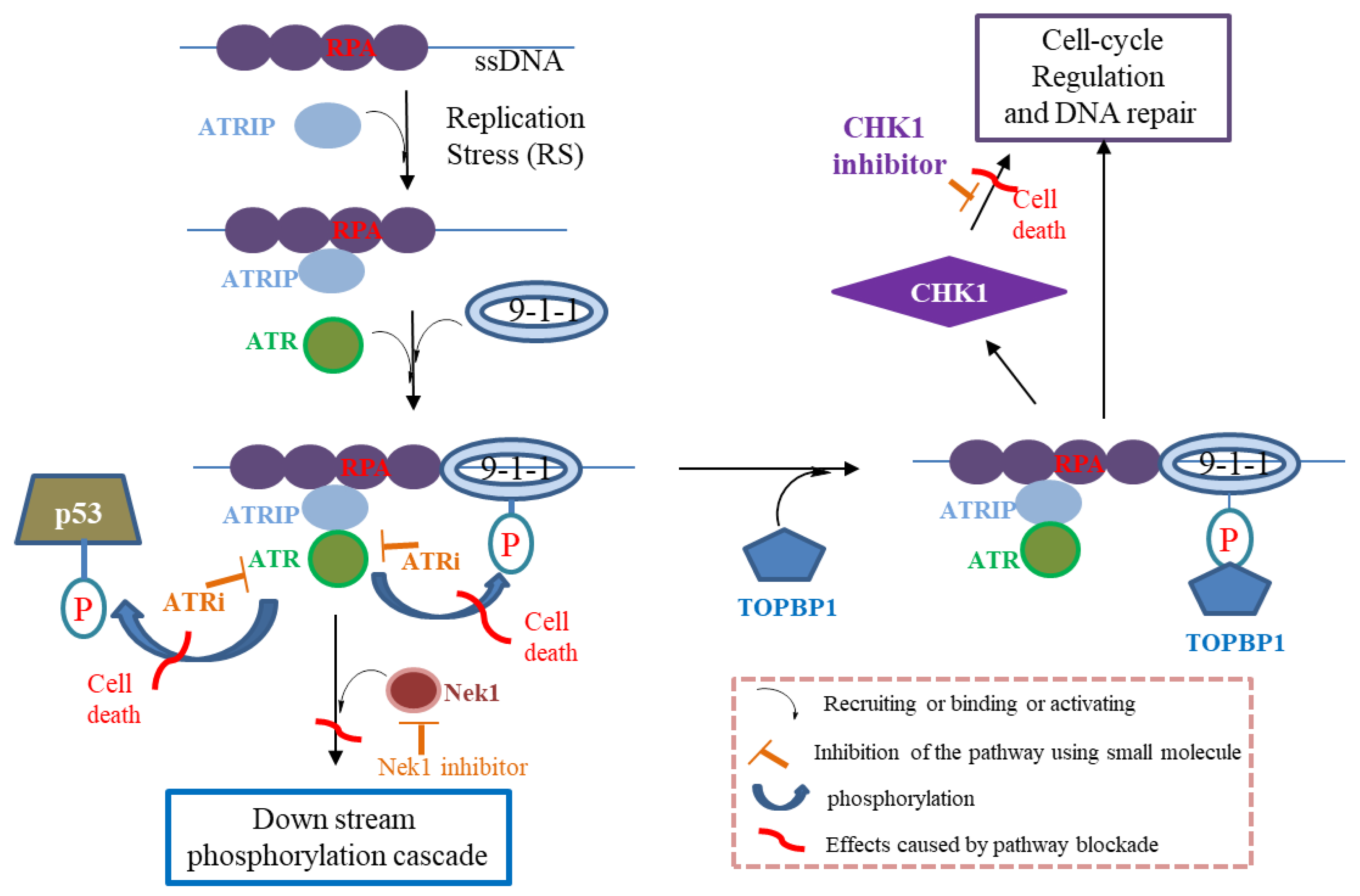
:1. Introduction
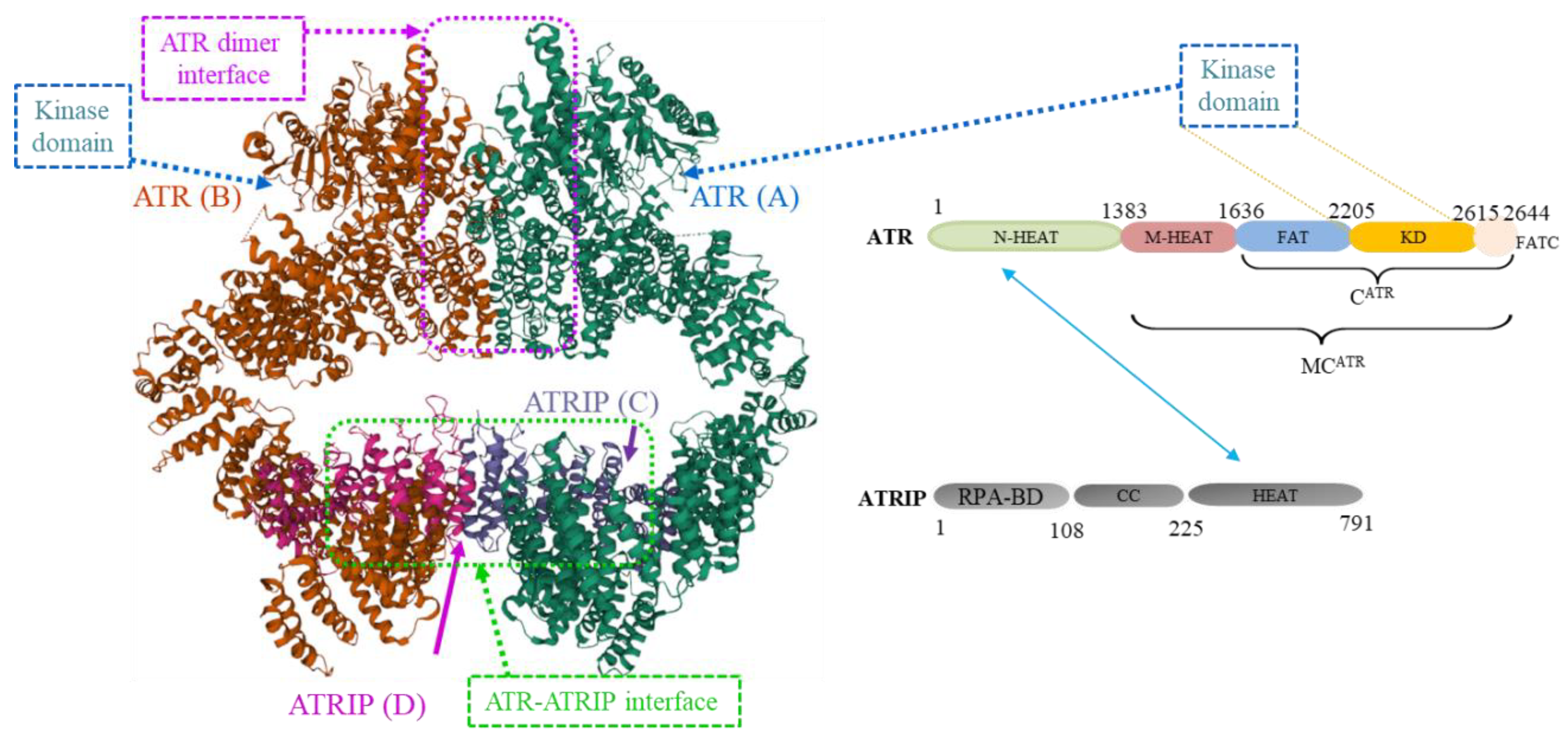
2. ATR Protein Structures
3. ATR Protein and Cancers
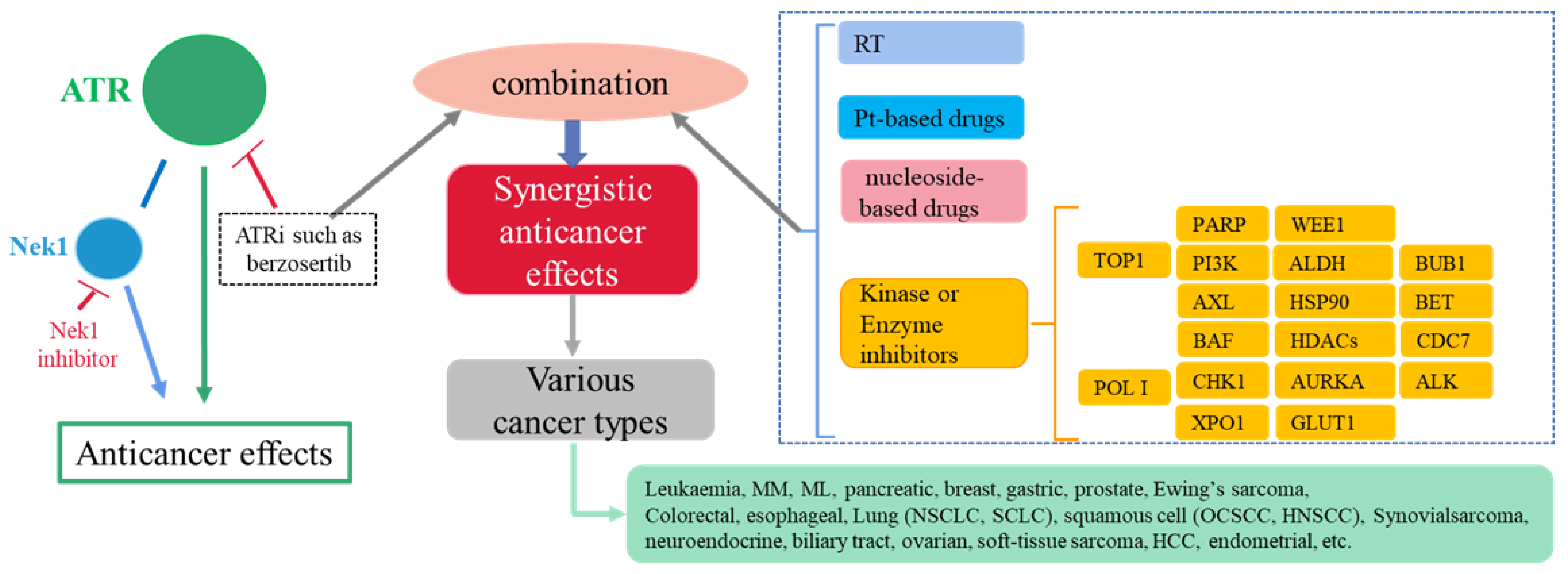
4. Synergistic Anticancer Effects Involving ATR Inhibition
4.1. Radiotherapy (RT)
4.2. Platinum-Based Anticancer Drugs
4.3. Topoisomerase I (TOP1) Inhibitors
4.4. Nucleoside-Based Drugs
4.5. RNA Polymerase I (POL I) Inhibitors
4.6. PARP Inhibitors
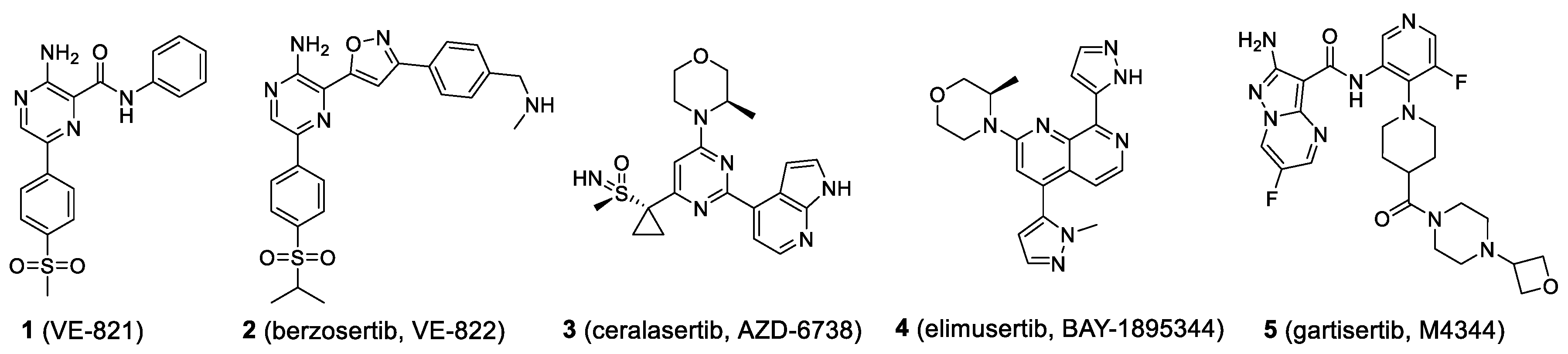
4.6.1. Ceralasertib
4.6.2. Elimusertib
4.6.3. RP-3500
4.6.4. M1774
4.7. PI3K Inhibitors
4.8. AXL Receptor Tyrosine Kinase Inhibitors
4.9. BAF Inhibitors
4.10. CHK1 Inhibitors
4.11. XPO1 Inhibitors
4.12. WEE1 Inhibitor
4.13. ALDH Inhibitors
4.14. HSP90 Inhibitors
4.15. HDACs Inhibitors
4.16. BET Inhibitors
4.17. Aurora Kinase A Inhibitors
4.18. BUB1 Inhibitor
4.19. GLUT1 Inhibitors
4.20. CDC7 Inhibitors
4.21. ALK Inhibitors
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clancy, S. DNA damage & repair: Mechanisms for maintaining DNA integrity. Nat. Educ. 2008, 1, 103. [Google Scholar]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2019, 19, 23–38. [Google Scholar] [CrossRef]
- Li, S.; Topatana, W.; Juengpanich, S.; Cao, J.; Hu, J.; Zhang, B.; Ma, D.; Cai, X.; Chen, M. Development of synthetic lethality in cancer: Molecular and cellular classification. Signal Transduct. Target. Ther. 2020, 5, 241–254. [Google Scholar] [CrossRef]
- Lempiäinen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Hui, Z.; Duan, J.; Garrido, C.; Xie, T.; Ye, X.Y. Discovery of small-molecule ATR inhibitors for potential cancer treatment: A patent review from 2014 to present. Expert Opin. Ther. Pat. 2022, 32, 401–421. [Google Scholar] [CrossRef]
- Hu, S.; Hui, Z.; Lirussi, F.; Garrido, C.; Ye, X.-Y.; Xie, T. Small molecule DNA-PK inhibitors as potential cancer therapy: A patent review (2010–present). Expert Opin. Ther. Patents 2021, 31, 435–452. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Shin, T.B.; Keith, C.T.; Schreiber, S.L. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc. Natl. Acad. Sci. USA 1996, 93, 2850–2855. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ho, C.K.; Ouyang, J.; Zou, L. Nek1 kinase associates with ATR–ATRIP and primes ATR for efficient DNA damage signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 2175–2180. [Google Scholar] [CrossRef] [Green Version]
- Rao, Q.; Liu, M.; Tian, Y.; Wu, Z.; Hao, Y.; Song, L.; Qin, Z.; Ding, C.; Wang, H.-W.; Wang, J.; et al. Cryo-EM structure of human ATR-ATRIP complex. Cell Res. 2018, 28, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Knegtel, R.; Charrier, J.-D.; Durrant, S.; Davis, C.; O’Donnell, M.; Storck, P.; MacCormick, S.; Kay, D.; Pinder, J.; Virani, A.; et al. Rational Design of 5-(4-(Isopropylsulfonyl)phenyl)-3-(3-(4-((methylamino)methyl)phenyl)isoxazol-5-yl)pyrazin-2-amine (VX-970, M6620): Optimization of Intra- and Intermolecular Polar Interactions of a New Ataxia Telangiectasia Mutated and Rad3-Related (ATR) Kinase Inhibitor. J. Med. Chem. 2019, 62, 5547–5561. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ran, T.; Zhang, X.; Xin, J.; Zhang, Z.; Wu, T.; Wang, W.; Cai, G. 3.9 Å structure of the yeast Mec1-Ddc2 complex, a homolog of human ATR-ATRIP. Science 2017, 358, 1206–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannous, E.A.; Yates, L.A.; Zhang, X.; Burgers, P.M. Mechanism of auto-inhibition and activation of Mec1ATR checkpoint kinase. Nat. Struct. Mol. Biol. 2021, 28, 50–61. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000175054-ATR/pathology (accessed on 12 March 2022).
- Hartwell, L.H.; Szankasi, P.; Roberts, C.J.; Murray, A.W.; Friend, S.H. Integrating Genetic Approaches into the Discovery of Anticancer Drugs. Science 1997, 278, 1064–1068. [Google Scholar] [CrossRef]
- Metcalf, K.J.; Alazzeh, A.; Werb, Z.; Weaver, V.M. Leveraging microenvironmental synthetic lethalities to treat cancer. J. Clin. Investig. 2021, 131, e143765. [Google Scholar] [CrossRef]
- Akimov, Y.; Aittokallio, T. Re-defining synthetic lethality by phenotypic profiling for precision oncology. Cell Chemi. Biol. 2021, 28, 246–256. [Google Scholar] [CrossRef]
- Myers, S.H.; Ortega, J.A.; Cavalli, A. Synthetic Lethality through the Lens of Medicinal Chemistry. J. Med. Chem. 2020, 63, 14151–14183. [Google Scholar] [CrossRef]
- Zhang, B.; Tang, C.; Yao, Y.; Chen, X.; Zhou, C.; Wei, Z.; Xing, F.; Chen, L.; Cai, X.; Zhang, Z.; et al. The tumor therapy landscape of synthetic lethality. Nat. Commun. 2021, 12, 1275–1285. [Google Scholar] [CrossRef]
- Rafiei, S.; Fitzpatrick, K.; Liu, D.; Cai, M.-Y.; Elmarakeby, H.A.; Park, J.; Ricker, C.; Kochupurakkal, C.B.S.; Choudhury, A.D.; Hahn, W.C.; et al. ATM Loss Confers Greater Sensitivity to ATR Inhibition Than PARP Inhibition in Prostate Cancer. Cancer Res. 2020, 80, 2094–2100. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, C.R.; Wallez, Y.; Johnson, T.I.; Fernández, S.B.Q.; Durant, S.T.; Cadogan, E.B.; Lau, A.; Richards, F.M.; Jodrell, D.I. Complete loss of ATM function augments replication catastrophe induced by ATR inhibition and gemcitabine in pancreatic cancer models. Br. J. Cancer 2020, 123, 1424–1436. [Google Scholar] [CrossRef]
- Gorski, J.W.; Ueland, F.R.; Kolesar, J.M. CCNE1 Amplification as a Predictive Biomarker of Chemotherapy Resistance in Epithelial Ovarian Cancer. Diagnostics 2020, 10, 279. [Google Scholar] [CrossRef]
- Buisson, R.; Lawrence, M.S.; Benes, C.H.; Zou, L. APOBEC3A and APOBEC3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res. 2017, 77, 4567–4578. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as cancer therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2019, 207, 107450. [Google Scholar] [CrossRef]
- Gamper, A.M.; Rofougaran, R.; Watkins, S.C.; Greenberger, J.S.; Beumer, J.H.; Bakkenist, C.J. ATR kinase activation in G1 phase facilitates the repair of ionizing radiation-induced DNA damage. Nucleic Acids Res. 2013, 41, 10334–10344. [Google Scholar] [CrossRef]
- Sankunny, M.; Parikh, R.A.; Lewis, D.W.; Gooding, W.E.; Saunders, W.S.; Gollin, S.M. Targeted inhibition of ATR or CHEK1 reverses radioresistance in oral squamous cell carcinoma cells with distal chromosome arm 11q loss. Genes Chromosom. Cancer 2013, 53, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, H.; Nakajima, N.I.; Sunada, S.; Lee, Y.; Hirakawa, H.; Yajima, H.; Fujimori, A.; Uesaka, M.; Okayasu, R. VE-821, an ATR inhibitor, causes radiosensitization in human tumor cells irradiated with high LET radiation. Radiat. Oncol. 2015, 10, 175. [Google Scholar] [CrossRef] [Green Version]
- Dunne, V.; Ghita, M.; Small, D.M.; Coffey, C.B.; Weldon, S.; Taggart, C.C.; Osman, S.O.; McGarry, C.K.; Prise, K.M.; Hanna, G.G.; et al. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother. Oncol. 2017, 124, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Baschnagel, A.M.; Elnaggar, J.H.; Van Beek, H.J.; Kromke, A.C.; Skiba, J.H.; Kaushik, S.; Abel, L.; Clark, P.A.; Longhurst, C.A.; Nickelet, K.P.; et al. ATR Inhibitor M6620 (VX-970) Enhances the effect of radiation in non–small cell lung cancer brain me-tastasis patient-derived xenografts. Mol. Cancer Ther. 2021, 20, 2129–2139. [Google Scholar] [CrossRef]
- Tu, X.; Kahila, M.M.; Zhou, Q.; Yu, J.; Kalari, K.R.; Wang, L.; Harmsen, W.S.; Yuan, J.; Boughey, J.C.; Matthew, P.; et al. ATR Inhibition Is a Promising Radiosensitizing Strategy for Triple-Negative Breast Cancer. Mol. Cancer Ther. 2018, 17, 2462–2472. [Google Scholar] [CrossRef] [Green Version]
- Faulhaber, E.-M.; Jost, T.; Symank, J.; Scheper, J.; Bürkel, F.; Fietkau, R.; Hecht, M.; Distel, L. Kinase Inhibitors of DNA-PK, ATM and ATR in Combination with Ionizing Radiation Can Increase Tumor Cell Death in HNSCC Cells While Sparing Normal Tissue Cells. Genes 2021, 12, 925. [Google Scholar] [CrossRef] [PubMed]
- Dok, R.; Glorieux, M.; Bamps, M.; Nuyts, S. Effect of ATR Inhibition in RT Response of HPV-Negative and HPV-Positive Head and Neck Cancers. Int. J. Mol. Sci. 2020, 22, 1504. [Google Scholar] [CrossRef] [PubMed]
- Vitti, E.T.; Kacperek, A.; Parsons, J.L. Targeting DNA Double-Strand Break Repair Enhances Radiosensitivity of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinoma to Photons and Protons. Cancers 2020, 12, 1490. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Huang, Y.; Xiao, Y.; Zhu, Z.; Shen, M.; Zhou, P.; Guo, Z.; Wang, J.; Wang, H.; Dai, W.; et al. ATR inhibitor AZD6738 enhances the anti-tumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune mi-croenvironment in hepatocellular carcinoma. J. Immunother. Cancer 2020, 8, e000340. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.J.; Wijnhoven, P.W.G.; Fok, J.H.L.; Lloyd, R.L.; Cairns, J.; Armenia, J.; Nikkilä, J.; Lau, A.; Bakkenist, C.J.; Galbraith, S.M.; et al. Radiopotentiation Profiling of Multiple Inhibitors of the DNA Damage Response for Early Clinical Development. Mol. Cancer Ther. 2021, 20, 1614–1626. [Google Scholar] [CrossRef]
- Sangster-Guity, N.; Conrad, B.H.; Papadopoulos, N.; Bunz, F. ATR mediates cisplatin resistance in a p53 genotype-specific manner. Oncogene 2011, 30, 2526–2533. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; Luken, M.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with car-boplatin in patients with advanced solid tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; El-Khouiery, A.; Soria, J.-C.; Lopez, J.; Berges, A.; Cheung, S.Y.A.; Irurzun-Arana, I.; Goldwin, A.; et al. Ceralasertib (AZD6738), an Oral ATR Kinase Inhibitor, in Combination with Carboplatin in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2021, 27, 5213–5224. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Wesolowski, R.; Devoe, C.; Lord, S.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R.; Yap, T.A. Phase 1 study of the ATR inhibitor berzosertib in combination with cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 520–527. [Google Scholar] [CrossRef]
- Jossé, R.; Martin, S.E.; Guha, R.; Ormanoglu, P.; Pfister, T.D.; Reaper, P.M.; Barnes, C.S.; Jones, J.; Charlton, P.; Pollard, J.R.; et al. ATR Inhibitors VE-821 and VX-970 Sensitize Cancer Cells to Topoisomerase I Inhibitors by Disabling DNA Replication Initiation and Fork Elongation Responses. Cancer Res. 2014, 74, 6968–6979. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Redon, C.E.; Sciuto, L.; Padiernos, E.; Ji, J.; Lee, M.-J.; Yuno, L.M.A.; Lee, S.; Zhang, Y.; Tran, L.; et al. Phase I Study of ATR Inhibitor M6620 in Combination with Topotecan in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Jo, U.; Senatorov, I.S.; Zimmermann, A.; Saha, L.K.; Murai, Y.; Kim, S.H.; Rajapakse, V.N.; Elloumi, F.; Takahashi, N.; Schultz, C.W.; et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells with Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol. Cancer Ther. 2021, 20, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Ghosh, M.; Kim, T.H.; Park, N.; Pandey, K.; Cho, Y.B.; Hong, S.D.; Katuwal, N.B.; Kang, M.; An, H.J.; et al. Synergism of AZD6738, an ATR Inhibitor, in Combination with Belotecan, a Camptothecin Analogue, in Chemotherapy-Resistant Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 1223. [Google Scholar] [CrossRef] [PubMed]
- Fordham, S.E.; Blair, H.J.; Elstob, C.J.; Plummer, R.; Drew, Y.; Curtin, N.J.; Heidenreich, O.; Pal, D.; Jamieson, D.; Park, C.; et al. Inhibition of ATR acutely sensitizes acute myeloid leukemia cells to nucleoside analogs that target ribonucleotide reductase. Blood Adv. 2018, 2, 1157–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinopoulos, P.A.; da Costa, A.A.B.A.; Gulhan, D.; Lee, E.K.; Cheng, S.-C.; Hendrickson, A.E.W.; Kochupurakkal, B.; Kolin, D.L.; Kohn, E.C.; Liu, J.F.; et al. A Replication stress biomarker is associated with response to gemcitabine versus combined gemcitabine and ATR inhibitor therapy in ovarian cancer. Nat. Commun. 2021, 12, 5574. [Google Scholar] [CrossRef]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef]
- Wang, T.; Shatara, M.; Liu, F.; Knight, T.; Edwards, H.; Wang, G.; Lin, H.; Wang, Y.; Taub, J.W.; Ge, Y. Simultaneous cotar-geting of ATR and RNA Polymerase I transcription demonstrates synergistic antileukemic effects on acute myeloid leukemia. Signal Transduct. Tar. 2019, 4, 44–46. [Google Scholar] [CrossRef] [Green Version]
- Bruno, P.M.; Lu, M.; Dennis, K.A.; Inam, H.; Moore, C.J.; Sheehe, J.; Elledge, S.J.; Hemann, M.T.; Pritchard, J.R. The primary mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is topoisomerase II poisoning. Proc. Natl. Acad. Sci. USA 2020, 117, 4053–4060. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Shah, P.D.; Wethington, S.L.; Pagan, C.; Latif, N.; Tanyi, J.; Martin, L.P.; Morganc, M.; Burgerc, R.A.; Haggertyc, A.; Zarrin, H.; et al. Plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol. Oncol. 2021, 163, 246–253. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, H.; Hafez, N.; Doroshow, D.; Sohal, D.; Keedy, V.; Do, K.T.; LoRusso, P.; Jürgensmeier, J.; Avedissian, M.; Sklar, J.; et al. Ceralasertib-Mediated ATR Inhibition Combined with Olaparib in Advanced Cancers Harboring DNA Damage Response and Repair Alterations (Olaparib Combinations). JCO Precis. Oncol. 2021, 5, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Sule, A.; Van, D.J.; Sundaram, R.K.; Ganesa, S.; Vasquez, J.C.; Bindra, R.S. Targeting IDH1/2 mutant cancers with combinations of ATR and PARP inhibitors. NAR. Cancer 2021, 3, zcab018. [Google Scholar]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, G.B.C.V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, Y.W.A.; Casals, T.; Carmichael, J.; Rodrigues, D.N.; et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, L.P.B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage–Inducing or Repair–Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef] [Green Version]
- Roulston, A.; Zimmermann, M.; Papp, R.; Skeldon, A.; Pellerin, C.; Dumas-Bérube, É.; Dumais, V.; Dorich, S.; Fader, L.D.; Leclaire, L.L.M. RP-3500: A Novel, Potent, and Selective ATR Inhibitor that is Effective in Preclinical Models as a Monotherapy and in Combination with PARP Inhibitors. Mol. Cancer Ther. 2021, 21, 245–256. [Google Scholar] [CrossRef]
- Timothy, A.Y.; Tolcher, A.W.; Elizabeth, R.P.; Andreas, B.; Patricia, F.M.; Locatelli, T.G.G.; Johann, I.G.; Bono, D.S. A first-in-human phase I study of ATR inhibitor M1774 in patients with solid tumors. Presented at ASCO Annual Meeting, Chicago, IL, USA, 4–8 June 2021; p. TPS3153. [Google Scholar]
- Du, W.; Gao, A.; Herman, J.G.; Wang, L.; Zhang, L.; Jiao, S.; Guo, M. Methylation of NRN1 is a novel synthetic lethal marker of PI3K-Akt-mTOR and ATR inhibitors in esophageal cancer. Cancer Sci. 2021, 112, 2870–2883. [Google Scholar] [CrossRef]
- Ramkumar, K.; Stewart, C.A.; Cargill, K.R.; Della, C.C.M.; Wang, Q.; Shen, L.; Diao, L.; Cardnell, R.J.; Peng, D.H.; Rodriguez, B.L.; et al. AXL inhibition induces DNA damage and replication stress in non-small cell lung cancer cells and promotes sensi-tivity to ATR inhibitors. Mol. Cancer Res. 2021, 19, 485–497. [Google Scholar] [CrossRef]
- Chory, E.J.; Kirkland, J.G.; Chang, C.-Y.; D’Andrea, V.D.; Gourisankar, S.; Dykhuizen, E.C.; Crabtree, G.R. Chemical Inhibitors of a Selective SWI/SNF Function Synergize with ATR Inhibition in Cancer Cell Killing. ACS Chem. Biol. 2020, 15, 1685–1696. [Google Scholar] [CrossRef]
- Chaudhuri, L.; Vincelette, N.D.; Koh, B.D.; Naylor, R.M.; Flatten, K.S.; Peterson, K.L.; McNally, A.; Gojo, I.; Karp, J.E.; Mesa, A.R.; et al. CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica 2014, 99, 688–696. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Sanjiv, K.; Hagenkort, A.; Calderón-Montaño, J.M.; Koolmeister, T.; Reaper, P.M.; Jacques, M.O.; Kuiper, R.V.; Schultz, N.; Scobie, M.; Charlton, P.A. Cancer-specific synthetic lethality between ATR and CHK1 kinase activities. Cell Rep. 2016, 14, 298–309. [Google Scholar] [CrossRef]
- Restelli, V.; Lupi, M.; Chilà, R.; Vagni, M.; Tarantelli, C.; Spriano, F.; Gaudio, E.; Bertoni, F.; Damia, G.; Carrassa, L. DNA damage response inhibitor combinations exert synergistic anti-tumor activity in aggressive B-cell lymphomas. Mol. Cancer Ther. 2019, 18, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Robinson, F.S.; Minelli, R.; Tomihara, H.; Rizi, B.S.; Rose, J.L.; Kodama, T.; Srinivasan, S.; Harris, A.L.; Zuniga, A.M.; et al. Sequential administration of XPO1 and ATR inhibitors enhances therapeutic response in TP53-mutated colorectal cancer. Gastroenterology 2021, 161, 196–210. [Google Scholar] [CrossRef]
- Koppenhafer, S.L.; Goss, K.L.; Terry, W.W.; Gordon, D.J. Inhibition of the ATR–CHK1 Pathway in Ewing Sarcoma Cells Causes DNA Damage and Apoptosis via the CDK2-Mediated Degradation of RRM2. Mol. Cancer Ther. 2020, 18, 91–104. [Google Scholar] [CrossRef]
- Wu, X.; Kang, X.; Zhang, X.; Xie, W.; Su, Y.; Liu, X.; Guo, L.; Guo, E.; Li, F.; Hu, D.; et al. WEE1 inhibitor and ataxia telangiectasia and RAD3-related inhibitor trigger stimulator of interferon gene-dependent immune response and enhance tumor treatment efficacy through programmed death-ligand 1 blockade. Cancer Sci. 2021, 112, 4444–4456. [Google Scholar] [CrossRef]
- Rødland, G.E.; Hauge, S.; Hasvold, G.; Bay, L.T.E.; Raabe, T.T.H.; Joel, M.; Syljuåsen, R.G. Differential Effects of Combined ATR/WEE1 Inhibition in Cancer Cells. Cancers 2021, 13, 3790. [Google Scholar] [CrossRef]
- Xu, H.; George, E.; Kinose, Y.; Kim, H.; Shah, J.B.; Peake, J.D.; Ferman, B.; Medvedev, S.; Murtha, T.; Barger, C.J.; et al. CCNE1 copy number is a biomarker for response to combination WEE1-ATR inhibition in ovarian and endometrial cancer models. Cell Rep. Med. 2021, 2, 100394. [Google Scholar] [CrossRef]
- Qi, W.; Xu, X.; Wang, M.; Li, X.; Wang, C.; Sun, L.; Zhao, D.; Sun, L.W. Inhibition of Wee1 sensitizes AML cells to ATR inhibitor VE-822-induced DNA damage and apoptosis. Biochem. Pharmacol. 2019, 164, 273–282. [Google Scholar] [CrossRef]
- Grimley, E.; Cole, A.J.; Luong, T.T.; McGonigal, S.C.; Sinno, S.; Yang, D.; Bernstein, K.A.; Buckanovich, R.J. Aldehyde dehy-drogenase inhibitors promote DNA damage in ovarian cancer and synergize with ATM/ATR inhibitors. Theranostics 2021, 11, 3540–3551. [Google Scholar] [CrossRef]
- Marx, C.; Schaarschmidt, M.U.; Kirkpatrick, J.; Marx-Blümel, L.; Halilovic, M.; Westermann, M.; Hoelzer, D.; Meyer, F.B.; Geng, Y.; Buder, K.; et al. Cooperative treatment effectiveness of ATR and HSP90 inhibition in Ewing’s sarcoma cells. Cell Biosci. 2021, 11, 57–76. [Google Scholar] [CrossRef]
- He, X.R.; Hui, Z.; Xu, L.; Bai, R.R.; Gao, Y.; Wang, Z.C.; Tian, X.; Ye, X.Y. Medicinal chemistry updates of novel HDACs in-hibitors (2020 to present). Eur. J. Med. Chem. 2022, 227, 113946. [Google Scholar] [CrossRef]
- The Designation of the Inventor has Not Yet Been Filed. Combination of a DNA Damage Response Cell Cycle Checkpoint Inhibitors and Belinostat for Treating Cancer. US Patent EP3461480, 3 April 2019.
- Muralidharan, S.V.; Bhadury, J.; Nilsson, L.M.; Green, L.C.; McLure, K.G.; Nilsson, J.A. BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene 2016, 35, 4689–4697. [Google Scholar] [CrossRef]
- Muralidharan, S.V.; Einarsdottir, B.O.; Bhadury, J.; Lindberg, M.F.; Wu, J.; Campeau, E.; Bagge, R.O.; Stierner, U.; Ny, L.; Nilsson, L.M.; et al. BET bromodomain inhibitors synergize with ATR inhibitors in melanoma. Cell Death. Dis. 2017, 8, e2982. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Dulak, A.M.; Hattersley, M.M.; Willis, B.S.; Nikkilä, J.; Wang, A.; Lau, A.; Reimer, C.; Zinda, M.; Fawell, S.E.; et al. BRD4 facilitates replication stress-induced DNA damage response. Oncogene 2018, 37, 3763–3777. [Google Scholar] [CrossRef]
- Tarantelli, C.; Bernasconi, E.; Gaudio, E.; Cascione, L.; Restelli, V.; Arribas, A.J.; Spriano, F.; AndreaRinaldi, A.; Mensah, A.A.; Kwee, I. BET bromodomain inhibitor birabresib in mantle cell lymphoma: In vivo activity and identification of novel combi-nations to overcome adaptive resistance. ESMO Open 2018, 3, e000387. [Google Scholar] [CrossRef] [Green Version]
- Pericole, F.V.; Lazarini, M.; de Paiva, L.B.; Duarte, A.S.S.; Vieira Ferro, K.P.V.; Niemann, F.S.; Roversi, F.M.F.; Olalla Saad, S.T.O. BRD4 Inhibition Enhances Azacitidine Efficacy in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Front. Oncol. 2019, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Roeschert, I.; Poon, E.; Henssen, A.G.; Garcia, H.D.; Gatti, M.; Giansanti, C.; Jamin, Y.; Ade, C.P.; Gallant, P.; Schülein-Völk, C.; et al. Combined inhibition of Aurora-A and ATR kinases results in regression of MYCN-amplified neuroblastoma. Nat. Rev. Cancer 2021, 2, 312–326. [Google Scholar] [CrossRef]
- Scott, A.L.; Paul, T.; Christopher, M.; William, E.W. Patty-Forming Apparatus with Top Feed and Rotary Pump. U.S. Patent 8801427B2, 12 August 2014. [Google Scholar]
- Siemeister, G.; Mengel, A.; Fernández-Montalván, A.E.; Bone, W.; Schröder, J.; Zitzmann-Kolbe, S.; Briem, H.; Prechtl, S.; Holton, S.J.; Mönning, U.; et al. Inhibition of BUB1 Kinase by BAY 1816032 Sensitizes Tumor Cells toward Taxanes, ATR, and PARP Inhibitors In Vitro and In Vivo. Clin. Cancer Res. 2019, 25, 1404–1414. [Google Scholar] [CrossRef] [Green Version]
- Erber, J.; Steiner, J.D.; Isensee, J.; Lobbes, L.A.; Toschka, A.; Beleggia, F.; Schmitt, A.; Kaiser, R.W.J.; Siedek, F.; Persigehl, T.; et al. Dual Inhibition of GLUT1 and the ATR/CHK1 Kinase Axis Displays Synergistic Cytotoxicity in KRAS-Mutant Cancer Cells. Cancer Res. 2019, 79, 4855–4868. [Google Scholar] [CrossRef]
- Rainey, M.D.; Bennett, D.; O’Dea, R.; Zanchetta, M.E.; Voisin, M.; Seoighe, C.; Santocanale, C. ATR Restrains DNA Synthesis and Mitotic Catastrophe in Response to CDC7 Inhibition. Cell Rep. 2020, 32, 108096–108120. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, J.; Benedict, B.; Yang, C.; van Gemert, F.; Ma, X.; Gao, D.M.; Wang, H.; Zhang, S.; Lieftink, C.; et al. Targeting CDC7 potentiates ATR-CHK1 signaling inhibition through induction of DNA replication stress in liver cancer. Genome Med. 2021, 13, 166–180. [Google Scholar] [CrossRef]
- Szydzik, J.; Lind, D.E.; Arefin, B.; Kurhe, Y.; Umapathy, G.; Siaw, J.T.; Claeys, A.; Gabre, L.J.; Eynden, J.V.; Hallberg, B.; et al. ATR inhibition enables complete tumour regression in ALK-driven NB mouse models. Nat. Commun. 2021, 12, 6813–6830. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.-W.; Jiang, S.; Yuan, Y.-H.; Duan, J.; Mao, N.-D.; Hui, Z.; Bai, R.; Xie, T.; Ye, X.-Y. Recent Advances in Synergistic Antitumor Effects Exploited from the Inhibition of Ataxia Telangiectasia and RAD3-Related Protein Kinase (ATR). Molecules 2022, 27, 2491. https://doi.org/10.3390/molecules27082491
Wang L-W, Jiang S, Yuan Y-H, Duan J, Mao N-D, Hui Z, Bai R, Xie T, Ye X-Y. Recent Advances in Synergistic Antitumor Effects Exploited from the Inhibition of Ataxia Telangiectasia and RAD3-Related Protein Kinase (ATR). Molecules. 2022; 27(8):2491. https://doi.org/10.3390/molecules27082491
Chicago/Turabian StyleWang, Li-Wei, Songwei Jiang, Ying-Hui Yuan, Jilong Duan, Nian-Dong Mao, Zi Hui, Renren Bai, Tian Xie, and Xiang-Yang Ye. 2022. "Recent Advances in Synergistic Antitumor Effects Exploited from the Inhibition of Ataxia Telangiectasia and RAD3-Related Protein Kinase (ATR)" Molecules 27, no. 8: 2491. https://doi.org/10.3390/molecules27082491
APA StyleWang, L.-W., Jiang, S., Yuan, Y.-H., Duan, J., Mao, N.-D., Hui, Z., Bai, R., Xie, T., & Ye, X.-Y. (2022). Recent Advances in Synergistic Antitumor Effects Exploited from the Inhibition of Ataxia Telangiectasia and RAD3-Related Protein Kinase (ATR). Molecules, 27(8), 2491. https://doi.org/10.3390/molecules27082491