
Losartan Interactions with 2-Hydroxypropyl-β-CD

 , ,
, ,  ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Results and Discussion
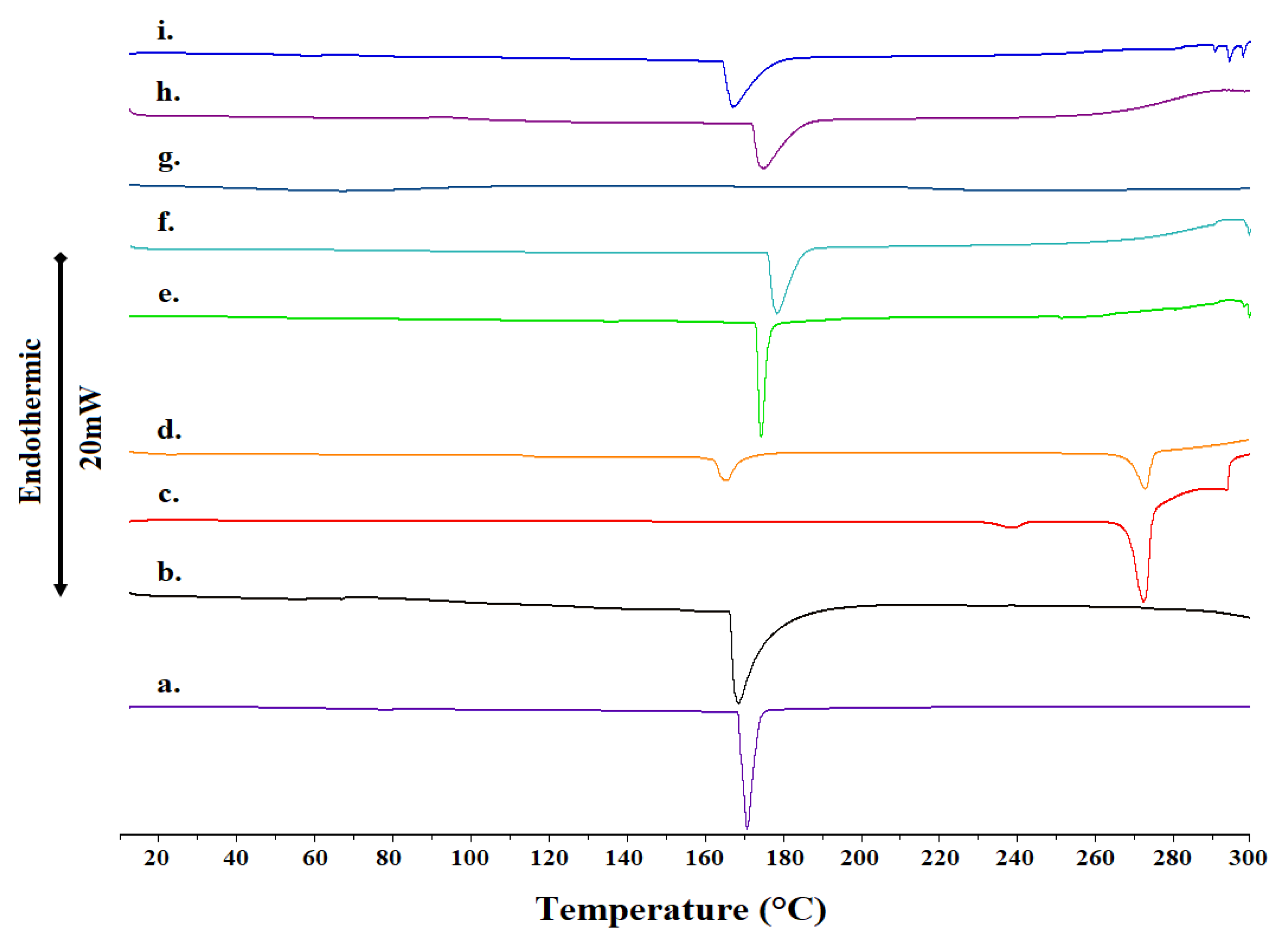
2.1. Differential Scanning Calorimetry
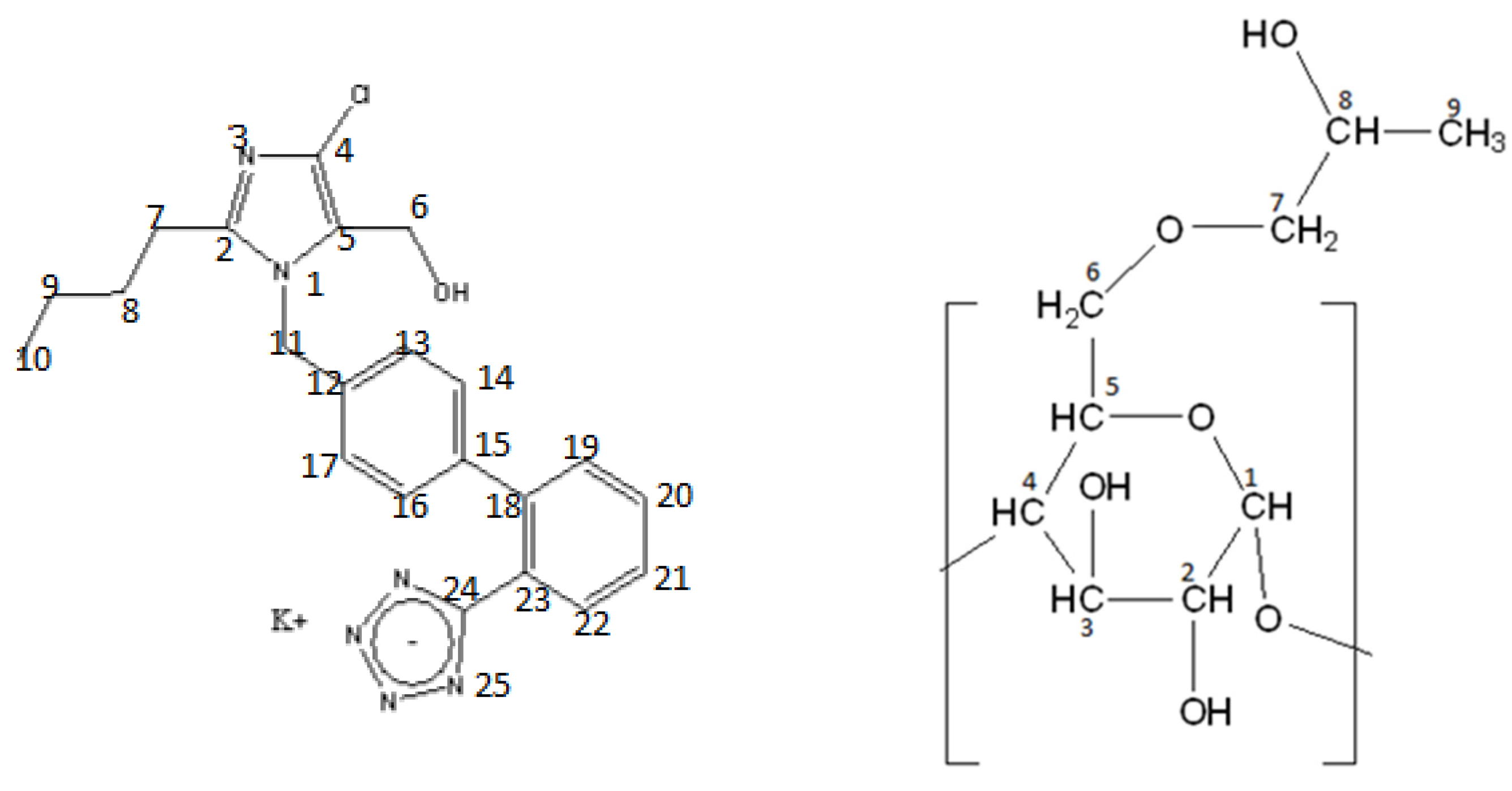
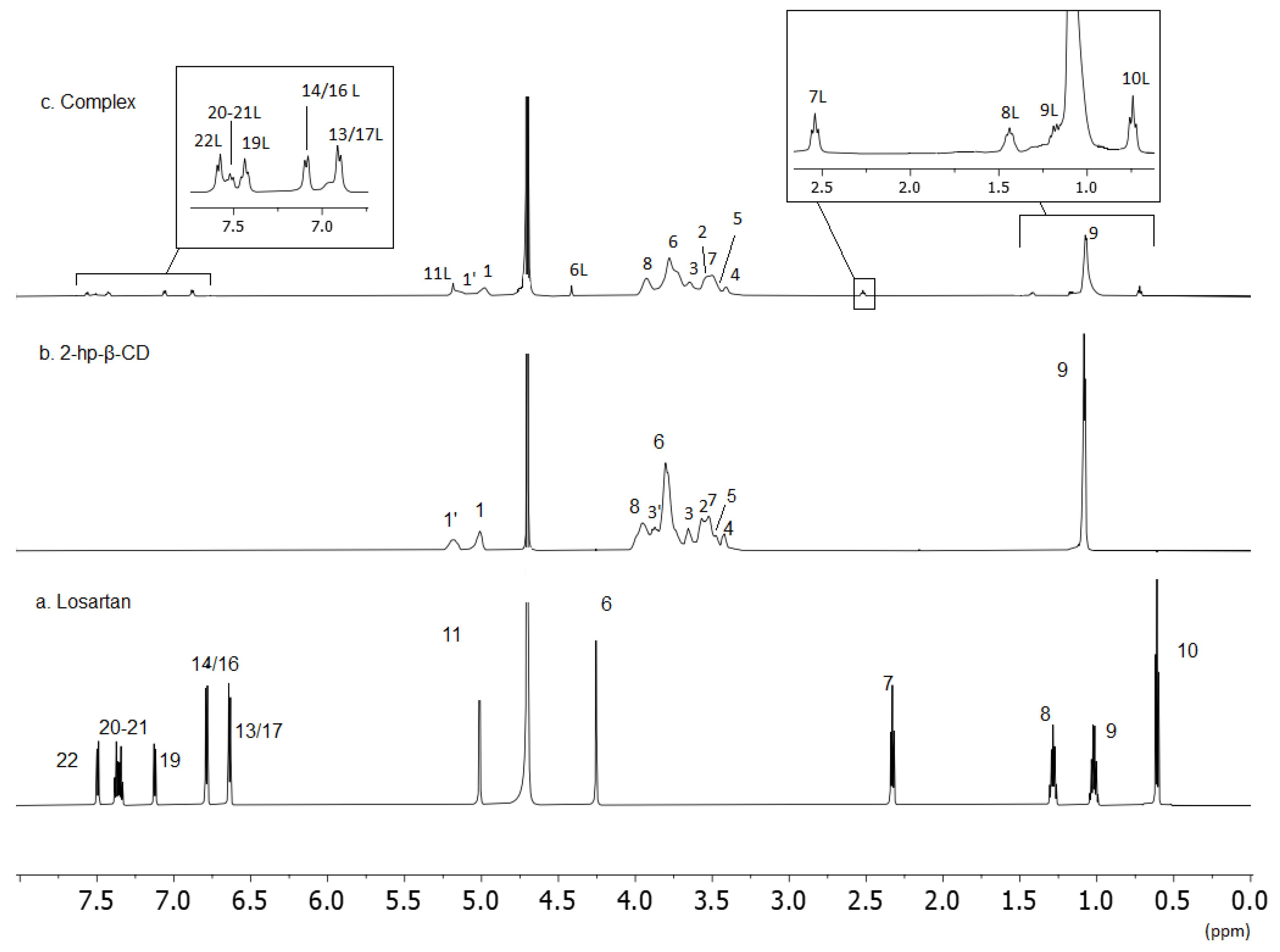
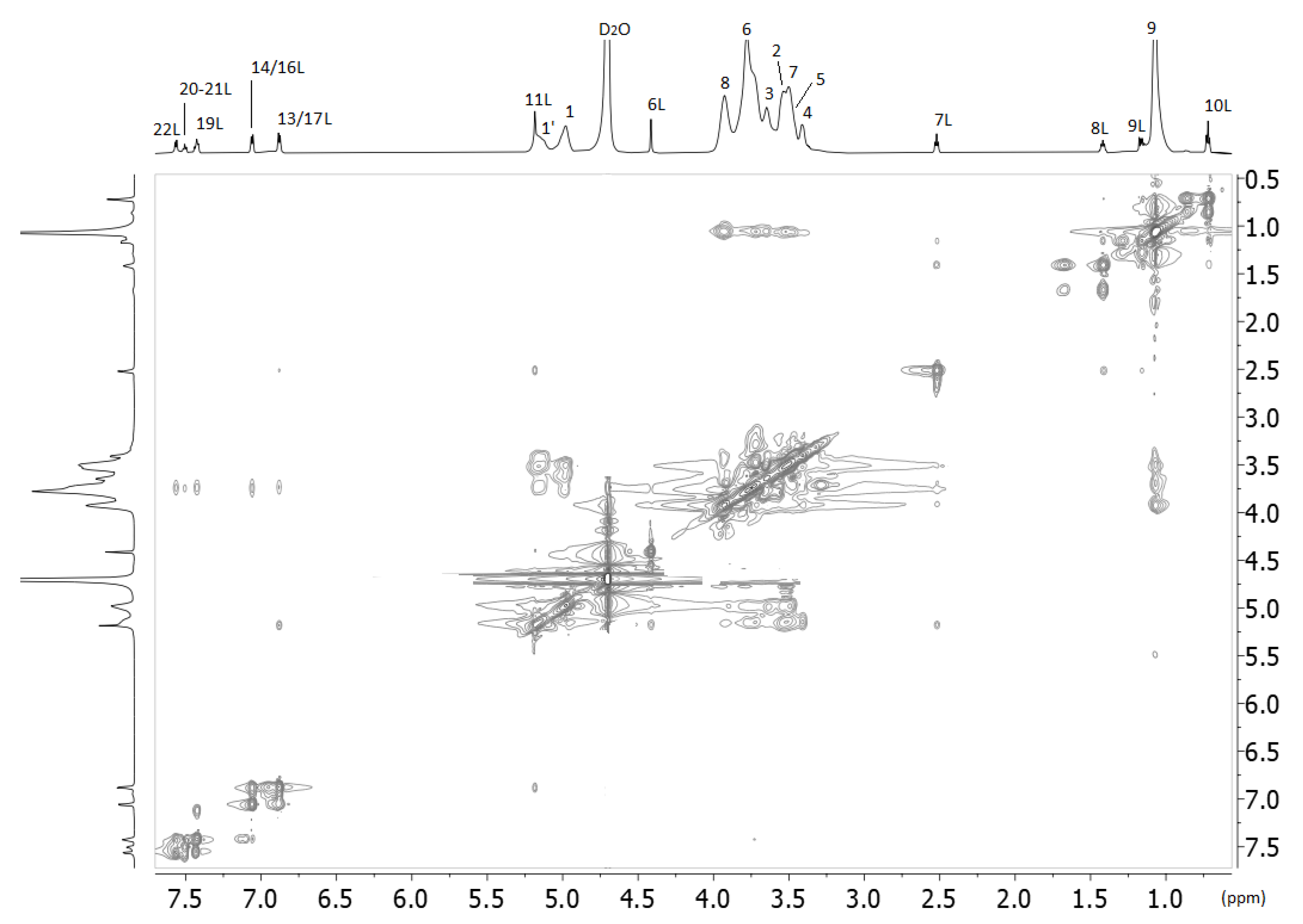
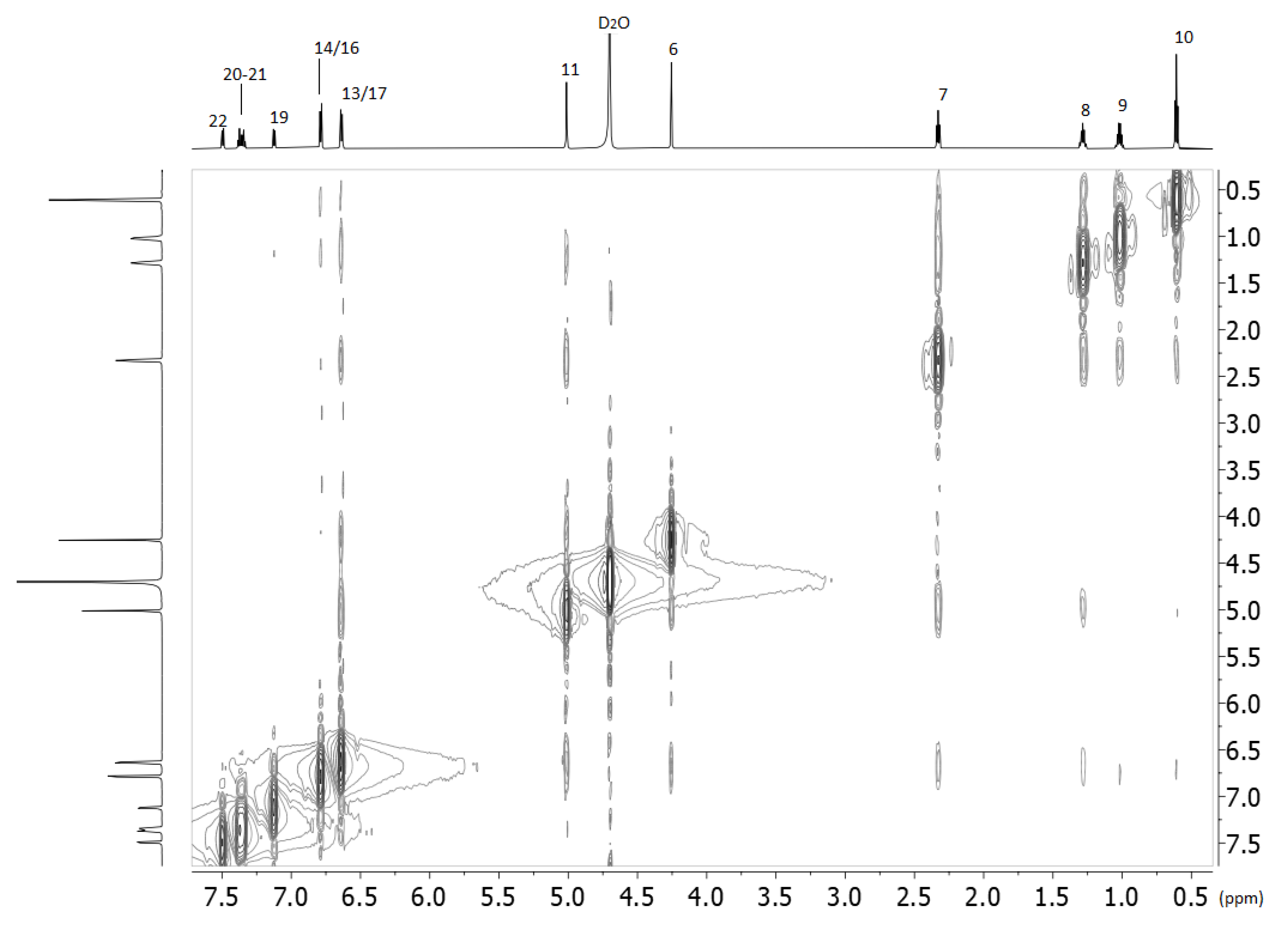
2.2. Nuclear Magnetic Resonance
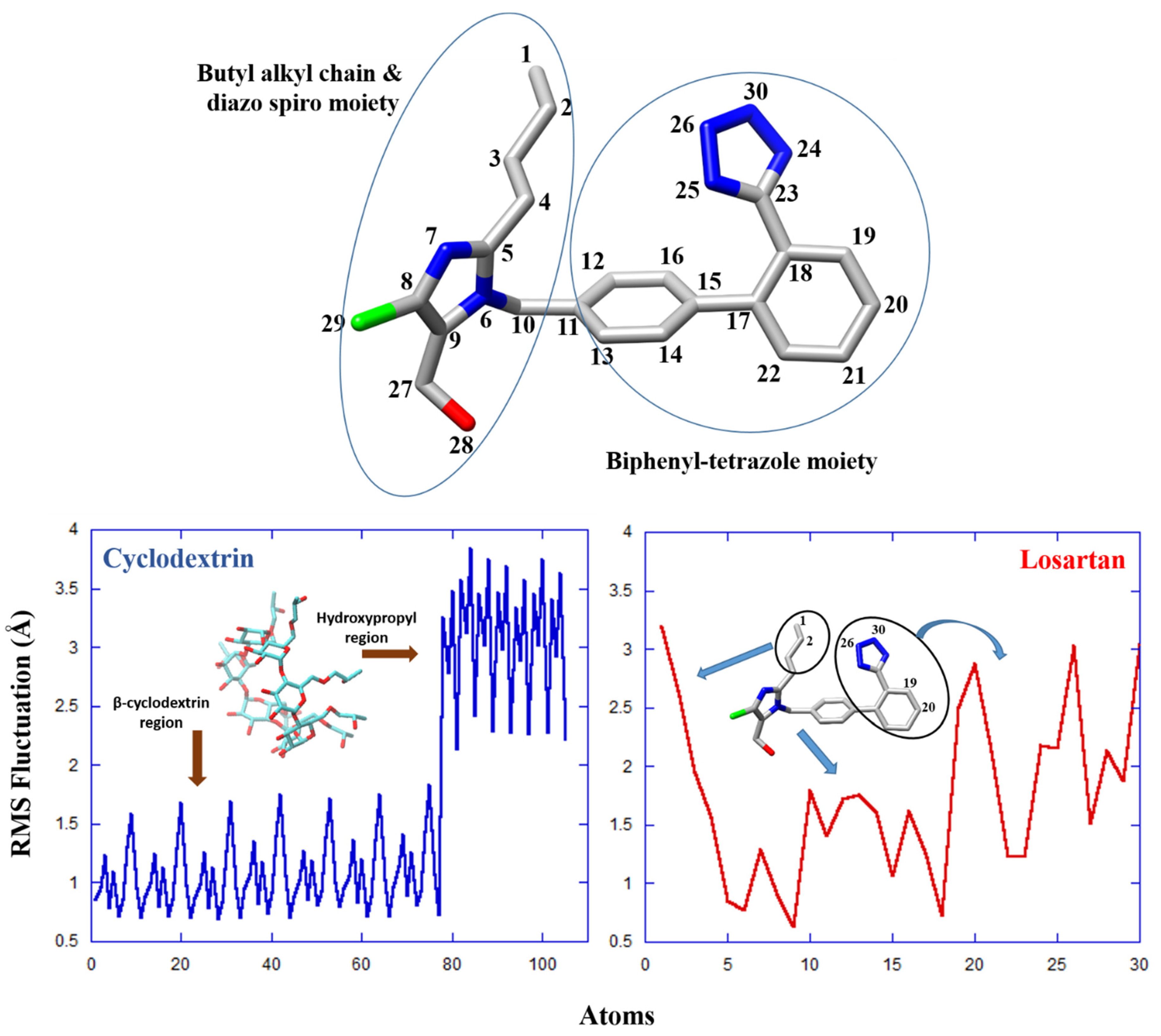
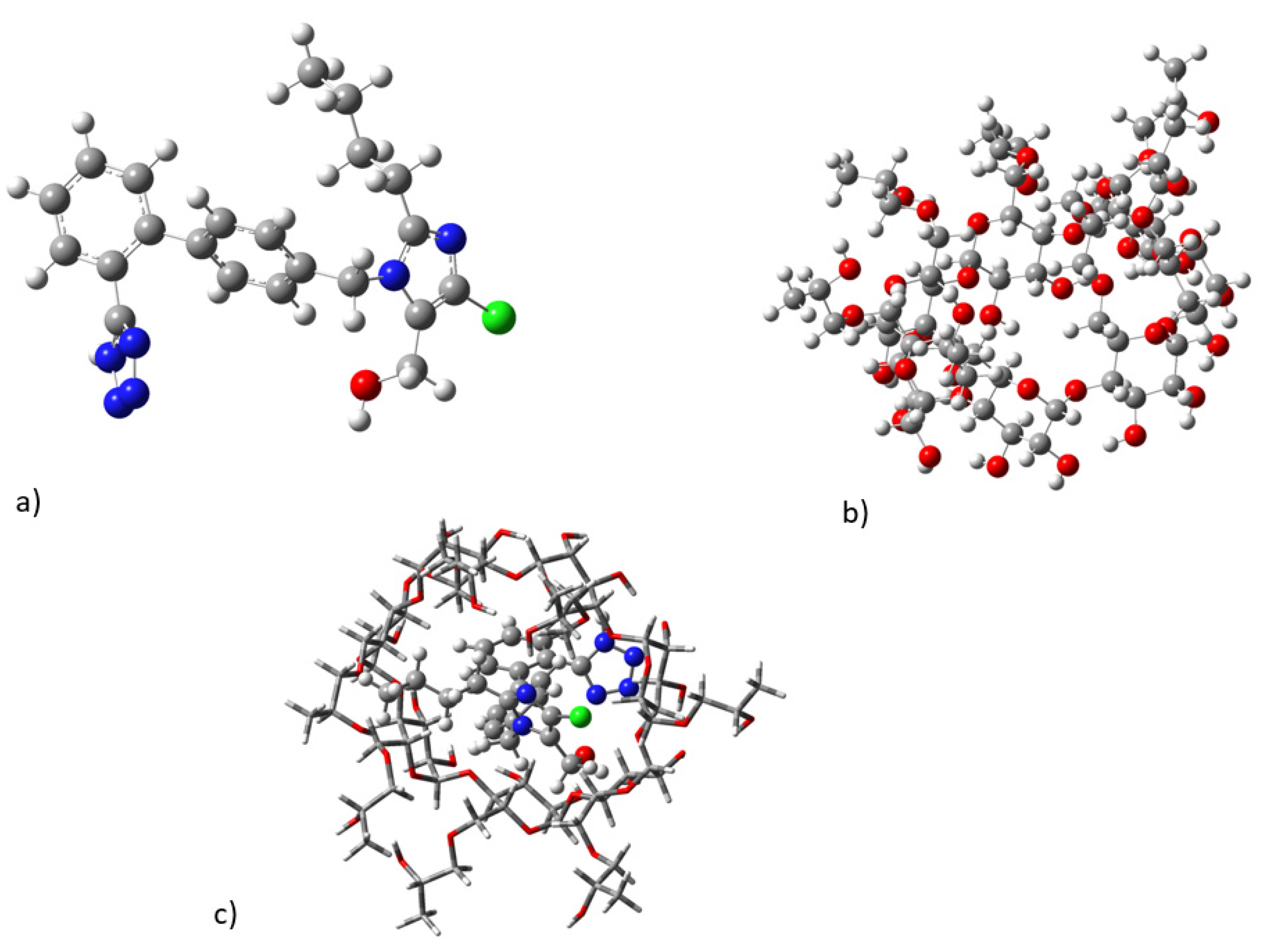
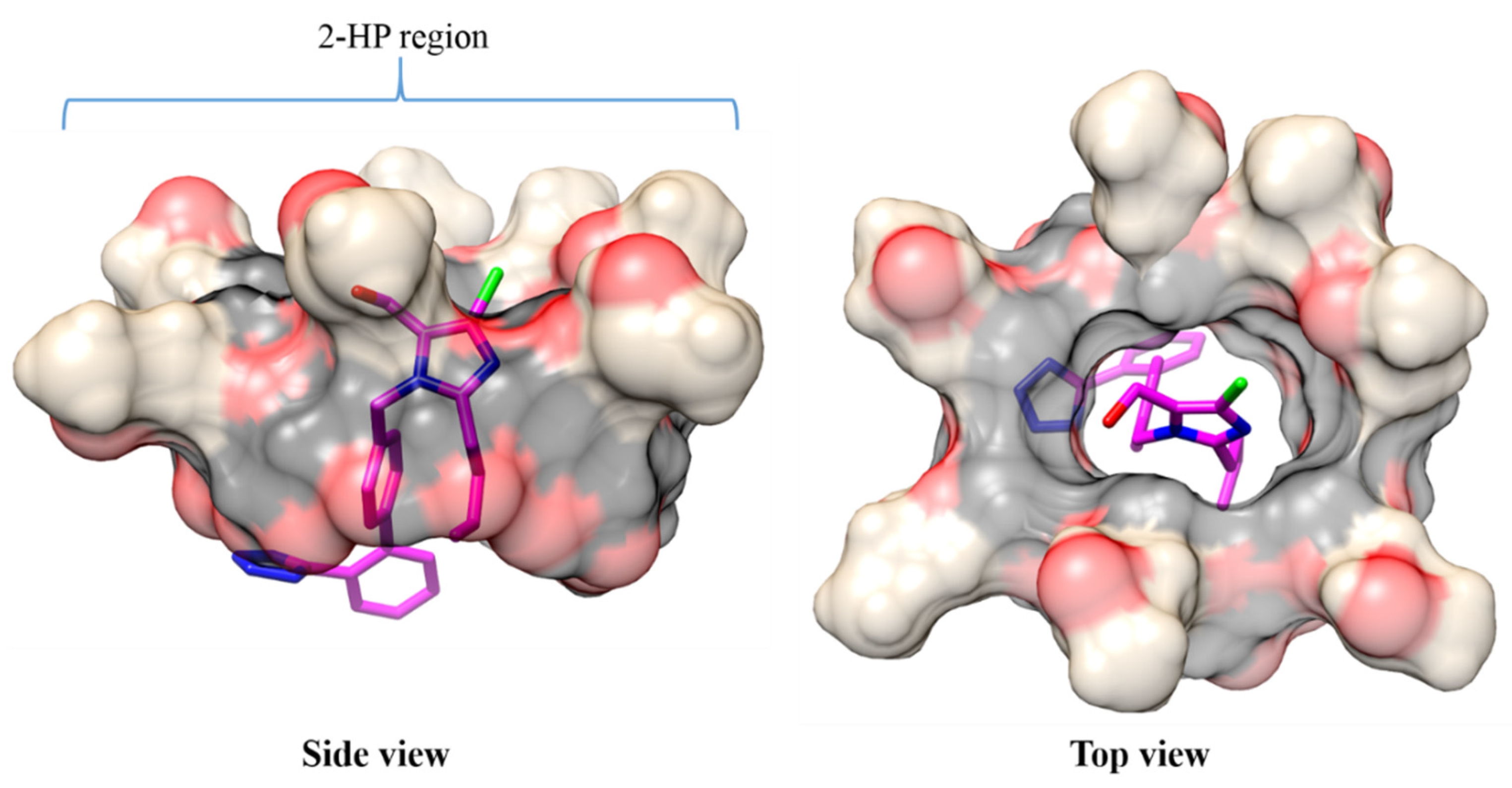
2.3. Molecular Dynamics
2.4. Conformational Analysis
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Sample Preparation
3.2.2. Differential Scanning Calorimetry
3.2.3. NMR Spectroscopy
3.2.4. DFT Quantum Mechanics
3.2.5. Structures Preparation and Molecular Docking
3.2.6. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kellici, T.F.; Tzakos, A.G.; Mavromoustakos, T. Rational Drug Design and Synthesis of Molecules Targeting the Angiotensin II Type 1 and Type 2 Receptors. Molecules 2015, 20, 3868–3897. [Google Scholar] [CrossRef] [PubMed]
- Kellici, T.F.; Liapakis, G.; Tzakos, A.G.; Mavromoustakos, T. Pharmaceutical Compositions for Antihypertensive Treatments: A Patent Review. Expert Opin. Ther. Pat. 2015, 25, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Nejat, R.; Sadr, A.S. Are losartan and imatinib effective against SARS-CoV2 pathogenesis? A pathophysiologic-based in silico study. Silico Pharmacol. 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Chroni, A.; Mavromoustakos, T.; Pispas, S. Nano-Assemblies from Amphiphilic PnBA-b-POEGA Copolymers as Drug Nanocarriers. Polymers 2021, 13, 1164. [Google Scholar] [CrossRef] [PubMed]
- Chroni, A.; Mavromoustakos, T.; Pispas, S. Biocompatible PEO-b-PCL Nanosized Micelles as Drug Carriers: Structure and Drug–Polymer Interactions. Nanomaterials 2020, 10, 1872. [Google Scholar] [CrossRef]
- De Paula, W.X.; Denadai, M.L.; Santoro, M.M.; Braga, A.N.G.; Santos, R.A.S.; Sinisterra, R.D. Supramolecular interactions between losartan and hydroxypropyl-β-CD: ESI mass-spectrometry, NMR techniques, phase solubility, isothermal titration calorimetry and anti-hypertensive studies. Int. J. Pharm. 2011, 404, 116–123. [Google Scholar] [CrossRef]
- De Melo, C.M.; Da Silva, A.L.; de Melo, K.R.; Da Silva, P.C.D.; de Souza, M.L.; de Sousa, A.L.M.D.; Rabello, M.M.; Véras, L.M.C.; Rolim, L.A.; Neto, P.J.R. In silico and in vitro study of epiisopiloturine/ hydroxypropyl-β-cyclodextrin inclusion complexes obtained by different methods. J. Drug Deliv. Sci. Technol. 2021, 65, 102658. [Google Scholar] [CrossRef]
- Mura, P. Analytical techniques for characterization of cyclodextrin complexes in the solid state: A review. J. Pharm. Biomed. Anal. 2015, 113, 226–238. [Google Scholar] [CrossRef]
- Kratz, J.M.; Teixeira, M.R.; Ferronato, K.; Teixeira, H.F.; Koester, L.S.; Simões, C.M.O. Preparation, Characterization, and In Vitro Intestinal Permeability Evaluation of Thalidomide–Hydroxypropyl-β-Cyclodextrin Complexes. AAPS PharmSciTech 2012, 13, 118–124. [Google Scholar] [CrossRef]
- Talegaonkar, S.; Yakoob Khan, A.; Kishan Khar, R.; Jalees Ahmad, F.; Khan, Z. Development and Characterization of Paracetamol Complexes with Hydroxypropyl-?-Cyclodextrin. Iran. J. Pharm. Res. 2010, 2, 95–99. [Google Scholar] [CrossRef]
- Da Silva, L.F.J.S.; do Carmo, F.A.; de Almeida Borges, V.R.; Monteiro, L.M.; Rodrigues, C.R.; Cabral, L.M.; de Sousa, V.P. Preparation and Evaluation of Lidocaine Hydrochloride in Cyclodextrin Inclusion Complexes for Development of Stable Gel in Association with Chlorhexidine Gluconate for Urogenital Use. Int. J. Nanomed. 2011, 6, 1143. [Google Scholar] [CrossRef][Green Version]
- Khandai, M.; Chakraborty, S.; Ghosh, A.K. Losartan Potassium Loaded Bioadhesive Micro-Matrix System: An Investigation on Effects of Hydrophilic Polymeric Blend on Drug Release. Pharm. Anal. Acta 2014, 2153–2435. [Google Scholar]
- Liu, M.; Cao, W.; Sun, Y.; He, Z. Preparation, characterization and in vivo evaluation of formulation of repaglinide with hydroxypropyl-β-cyclodextrin. Int. J. Pharm. 2014, 477, 159–166. [Google Scholar] [CrossRef] [PubMed]
- De Freitas, M.R.; Rolim, L.A.; Soares, M.F.D.L.R.; Rolim-Neto, P.J.; de Albuquerque, M.M.; Soares-Sobrinho, J.L. Inclusion complex of methyl-β-cyclodextrin and olanzapine as potential drug delivery system for schizophrenia. Carbohydr. Polym. 2012, 89, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Van Duijneveldt, F.B.; van Duijneveldt-van de Rijdt, J.G.; van Lenthe, J.H. State of the Art in Counterpoise Theory. Chem. Rev. 1994, 94, 1873–1885. [Google Scholar] [CrossRef]
- Christodoulou, E.; Ntountaniotis, D.; Leonis, G.; Mavromoustakos, T.; Valsami, G. Application of Neutralization and Technique for the Preparation of the Beneficial in Drug 2-Hydroxypropyl-β-Cyclodextrin with. In Supramolecules in Drug Discovery and Drug Delivery; Humana: New York, NY, USA, 2021; pp. 1–11. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Curtiss, L.A.; McGrath, M.P.; Blaudeau, J.; Davis, N.E.; Binning, R.C.; Radom, L. Extension of Gaussian-2 theory to molecules containing third-row atoms Ga–Kr. J. Chem. Phys. 1995, 103, 6104–6113. [Google Scholar] [CrossRef]
- Tzeli, D.; Tsoungas, P.G.; Petsalakis, I.D.; Kozielewicz, P.; Zloh, M. Intramolecular cyclization of β-nitroso-o-quinone methides. A theoretical endoscopy of a potentially useful innate ‘reclusive’ reaction. Tetrahedron 2015, 71, 359–369. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Tzeli, D.; Mavridis, A.; Xantheas, S.S. First Principles Examination of the Acetylene−Water Clusters, HCCH−(H2O)x, x = 2, 3, and 4. J. Phys. Chem. A 2002, 106, 11327–11337. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01. In Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Schrödinger. Schrödinger Release 2015-2; Maestro, LLC.: New York, NY, USA, 2015. [Google Scholar]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D.; et al. DOCK 6: Combining techniques to model RNA–small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Berryman, J.; Betz, R.M.; Cerutti, D.S.; Cheatham Iii, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. Amber 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Accounts Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into Protein–Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras–Raf and Ras–RalGDS Complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Wang, W.; Kollman, P.A. Computational study of protein specificity: The molecular basis of HIV-1 protease drug resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 14937–14942. [Google Scholar] [CrossRef] [PubMed]
- Kellici, T.F.; Chatziathanasiadou, M.V.; Diamantis, D.; Chatzikonstantinou, A.V.; Andreadelis, I.; Christodoulou, E.; Valsami, G.; Mavromoustakos, T.; Tzakos, A.G. Mapping the Interactions and Bioactivity of Quercetin-(2-Hydroxypropyl)-β-Cyclodextrin Complex. Int. J. Pharm. 2016, 511, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Kellici, T.F.; Ntountaniotis, D.; Leonis, G.; Chatziathanasiadou, M.; Chatzikonstantinou, A.V.; Becker-Baldus, J.; Glaubitz, C.; Tzakos, A.G.; Viras, K.; Chatzigeorgiou, P.; et al. Investigation of the Interactions of Silibinin with 2-Hydroxypropyl-β-cyclodextrin through Biophysical Techniques and Computational Methods. Mol. Pharm. 2015, 12, 954–965. [Google Scholar] [CrossRef]
- Leonis, G.; Christodoulou, E.; Ntountaniotis, D.; Chatziathanasiadou, M.V.; Mavromoustakos, T.; Naziris, N.; Chountoulesi, M.; Demetzos, C.; Valsami, G.; Damalas, D.E.; et al. Antihypertensive activity and molecular interactions of irbesartan in complex with 2-hydroxypropyl-β-cyclodextrin. Chem. Biol. Drug Des. 2020, 96, 668–683. [Google Scholar] [CrossRef]
- Tzoupis, H.; Leonis, G.; Mavromoustakos, T.; Papadopoulos, M.G. A Comparative Molecular Dynamics, MM–PBSA and Thermodynamic Integration Study of Saquinavir Complexes with Wild-Type HIV-1 PR and L10I, G48V, L63P, A71V, G73S, V82A and I84V Single Mutants. J. Chem. Theory Comput. 2013, 9, 1754–1764. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Molar Ratio | Weight Ratio | Tonset (°C) | T (°C) | ΔT1/2 (°C) | ΔH (J g−1) |
|---|---|---|---|---|---|---|
| 2-HP-β-CD Raw Material | - | - | 168.14 | 169.93 | 3.08 | 190.61 |
| 2-HP-β-CD Lyophilized | - | - | 166.03 | 168.15 | 6.77 | 179.98 |
Losartan
| - | - |
|
|
|
|
Losartan Lyophilized
| - | - |
|
|
|
|
| Raw 2-HP-β-CD:Raw Losartan Mixture | 1:1 | 3.6:1 | 172.82 | 173.62 | 1.96 | 179.54 |
| Lyo 2-HP-β-CD:Raw Losartan Mixture | 1:1 | 3.6:1 | 175.79 | 178.04 | 5.06 | 202.20 |
| Raw 2-HP-β-CD:Lyo Losartan Mixture | 1:1 | 3.6:1 | 69.11 | 138.75 | 96.27 | 241.36 |
| Lyo 2-HP-β-CD:Lyo Losartan Mixture | 1:1 | 3.6:1 | 171.90 | 174.84 | 7.36 | 145.75 |
| 2-HP-β-CD:Losartan Complex | 1:1 | 3.6:1 | 164.08 | 166.85 | 7.20 | 221.85 |
| Position | Losartan | Complex of Losartan: 2HP-β-CD |
|---|---|---|
| 1H (ppm) | 1H (ppm) | |
| 10 | 0.61 | 0.72 |
| 9 | 1.02 | 1.17 |
| 8 | 1.28 | 1.42 |
| 7 | 2.33 | 2.52 |
| 6 | 4.26 | 4.41 |
| 11 | 5.01 | 5.18 |
| 13–17 | 6.64 | 6.88–6.87 |
| 14–16 | 6.79 | 7.06–7.05 |
| 19 | 7.12 | 7.42 |
| 21 | 7.34 | 7.50 |
| 20 | 7.36 | 7.51 |
| 22 | 7.49 | 7.56 |
| Position | 2HPβCD | Complex of losartan: 2HP-β-CD |
| 1H (ppm) | 1H (ppm) | |
| 9 | 1.07 | 1.07 |
| 2, 4, 5, 7 | 3.38–3.60 | 3.37–3.58 |
| 3 | 3.65 | 3.65 |
| 6 | 3.80 | 3.78 |
| 3, 6 | 3.85–3.90 | 3.81 |
| 8 | 3.95 | 3.93 |
| 1 | 5.01 | 4.98 |
| 1′ | 5.18 | 5.01 |
| Spatial Interactions | Distances Expressed in Å Obtained through MD Calculations | Qualitative Characterization of the Spatial Interactions |
|---|---|---|
| H10 (LOSARTAN)-H6(2-HΡ-β-CD) | 5.33 | w |
| H10 (LOSARTAN)-H8(2-HP-β-CD) | 5.18 | w |
| H10 (LOSARTAN)-H7(2-HP-β-CD) | 5.22 | w |
| H13 (LOSARTAN)-H3(2-HP-β-CD) | 4.70 | w |
| H17 (LOSARTAN)-H1(2-HP-β-CD) | 5.02 | w |
| H17(LOSARTAN)-H3(2-HP-β-CD) | 4.19 | m |
| H16(LOSARTAN)-H3(2-HP-β-CD) | 2.64 | s |
| H14(LOSARTAN)-H1(2-HP-β-CD) | 4.91 | w |
| H14(LOSARTAN)-H3(2-HP-β-CD) | 2.98 | s |
| H19(LOSARTAN)-H3(2-HP-β-CD) | 2.57 | s |
| H19(LOSARTAN)-H6(2-HP-β-CD) | 4.55 | w |
| H19(LOSARTAN)-H1(2-HP-β-CD) | 4.82 | w |
| H19(LOSARTAN)-H2(2-HP-β-CD) | 5.20 | w |
| H20(LOSARTAN)-H3(2-HP-β-CD) | 3.88 | m |
| H20(LOSARTAN)-H2(2-HP-β-CD) | 4.31 | m |
| H21(LOSARTAN)-H3(2-HP-β-CD) | 4.25 | m |
| H22(LOSARTAN)-H3(2-HP-β-CD) | 5.22 | w |
| H11(LOSARTAN)-H7(2-HP-β-CD) | 5.01 | w |
| H11(LOSARTAN)-H6(2-HP-β-CD) | 4.08 | m |
| H11(LOSARTAN)-H5(2-HP-β-CD) | 3.25 | s |
| H6(LOSARTAN)-H9(2-HP-β-CD) | 4.57 | w |
| H6(LOSARTAN)-H8(2-HP-β-CD) | 4.55 | w |
| H6(LOSARTAN)-H6(2-HP-β-CD) | 3.66 | m |
| H6(LOSARTAN)-H5(2-HP-β-CD) | 3.72 | m |
| H7(LOSARTAN)-H6(2-HP-β-CD) | 3.73 | m |
| H7(LOSARTAN)-H7(2-HP-β-CD) | 4.46 | m |
| H7(LOSARTAN)-H5(2-HP-β-CD) | 4.07 | m |
| 2-hp-β-CD | |||
| Free 2HP-β-CD | Complexed 2HP-β-CD | ||
| Chemical Shifts (ppm) | T1 (s) | Chemical Shifts | T1 (s) |
| 1.07 (9) | 0.63 | 1.07 (9) | 0.59 |
| 3.80 (6) | 0.66 | 3.78 (6) | 0.54 |
| 3.95 (8) | 1.10 | 3.93 | 0.76 |
| 5.01 (1) | 0.94 | 4.98 | 0.71 |
| Losartan | |||
| Free Losartan | Complexed Losartan | ||
| Chemical Shifts | T1 (s) | Chemical Shifts | T1 (s) |
| 0.61 (10) | 0.65 | 0.72 (10) | 1.19 |
| 1.02 (9) | 0.58 | 1.17 (9) | 0.52 |
| 1.28 (8) | 0.53 | 1.42 (8) | 0.48 |
| 2.33 (7) | 0.47 | 2.52 (7) | |
| 4.26 (6) | 0.44 | 4.41 (6) | 0.47 |
| 5.01 (11) | 0.40 | 5.18 (11) | 0.54 |
| 6.64 (13/17) | 0.60 | 6.88–6.87 (13/17) | 0.79 |
| 6.79 (14/16) | 0.66 | 7.06–7.05 (14/16) | 0.84 |
| 7.12 (19) | 0.62 | 7.42 (19) | 0.83 |
| 7.34 (21) | 0.66 | 7.50 (21) | 0.93 |
| 7.36 (20) | 0.67 | 7.51 (20) | 0.94 |
| 7.49 (22) | 0.82 | 7.56 (22) | 1.02 |
| Spatial Interactions | Distances Expressed in Å | Qualitative Characterization of the Spatial Interactions |
|---|---|---|
| H10-H13/H17 | 5.20 | w |
| H9-H13/H17 | 3.78 | m |
| H8-H13/H17 | 3.70–3.77 | m |
| H7-H13/H17 | 4.64 | w |
| H6-H11 | 2.19–3.67 | s |
| H11-H7 | 2.28 (3.75) | s |
| H8-H11 | 3.92 | m |
| H7-H14 | 4.74 | w |
| H7-H16 | 5.88 | w |
| H7-H19 | 5.84 | w |
| H14-H19 | 2.65 | s |
| Energy (kcal mol−1) | MM–PBSA | MM–GBSA |
|---|---|---|
| ΔEvdW | −33.19 ± 0.09 a | −33.19 ± 0.09 |
| ΔEelec | −5.30 ± 0.25 | −5.30 ± 0.25 |
| ΔEMM, gas | −38.49 ± 0.28 | −38.49 ± 0.28 |
| ΔGPB/GB | 15.53 ± 0.18 | 14.53 ± 0.20 |
| ΔGelec(tot) b | 10.23 ± 0.26 | 9.23 ± 0.13 |
| ΔGNP | −3.00 ± 0.00 | −3.88 ± 0.01 |
| ΔGsolv | 12.52 ± 0.18 | 10.67 ± 0.20 |
| ΔH(MM + solv) | −25.97 ± 0.13 | −27.83 ± 0.11 |
| −TΔStot | 21.15 ± 0.12 | 21.15 ± 0.12 |
| ΔGMM–PB(GB)SA | −4.82 ± 0.13 c | −6.68 ± 0.11 c |
| PM6 a | B3LYP b | B3LYP a | |
|---|---|---|---|
| BE | −3.06 | −6.61 | −5.47 |
| BEBSSE | --- | 12.40 | 12.10 |
| BEr | −12.21 | −28.53 | −20.85 |
| DefL | −4.38 | 5.83 | 4.76 |
| DefCD | --- | 16.09 | 10.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palli, V.; Leonis, G.; Zoupanou, N.; Georgiou, N.; Chountoulesi, M.; Naziris, N.; Tzeli, D.; Demetzos, C.; Valsami, G.; Marousis, K.D.; et al. Losartan Interactions with 2-Hydroxypropyl-β-CD. Molecules 2022, 27, 2421. https://doi.org/10.3390/molecules27082421
Palli V, Leonis G, Zoupanou N, Georgiou N, Chountoulesi M, Naziris N, Tzeli D, Demetzos C, Valsami G, Marousis KD, et al. Losartan Interactions with 2-Hydroxypropyl-β-CD. Molecules. 2022; 27(8):2421. https://doi.org/10.3390/molecules27082421
Chicago/Turabian StylePalli, Vasiliki, Georgios Leonis, Nikoletta Zoupanou, Nikitas Georgiou, Maria Chountoulesi, Nikolaos Naziris, Demeter Tzeli, Costas Demetzos, Georgia Valsami, Konstantinos D. Marousis, and et al. 2022. "Losartan Interactions with 2-Hydroxypropyl-β-CD" Molecules 27, no. 8: 2421. https://doi.org/10.3390/molecules27082421
APA StylePalli, V., Leonis, G., Zoupanou, N., Georgiou, N., Chountoulesi, M., Naziris, N., Tzeli, D., Demetzos, C., Valsami, G., Marousis, K. D., Spyroulias, G. A., & Mavromoustakos, T. (2022). Losartan Interactions with 2-Hydroxypropyl-β-CD. Molecules, 27(8), 2421. https://doi.org/10.3390/molecules27082421