
Semiempirical Potential in Kinetics Calculations on the HC3N + CN Reaction

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
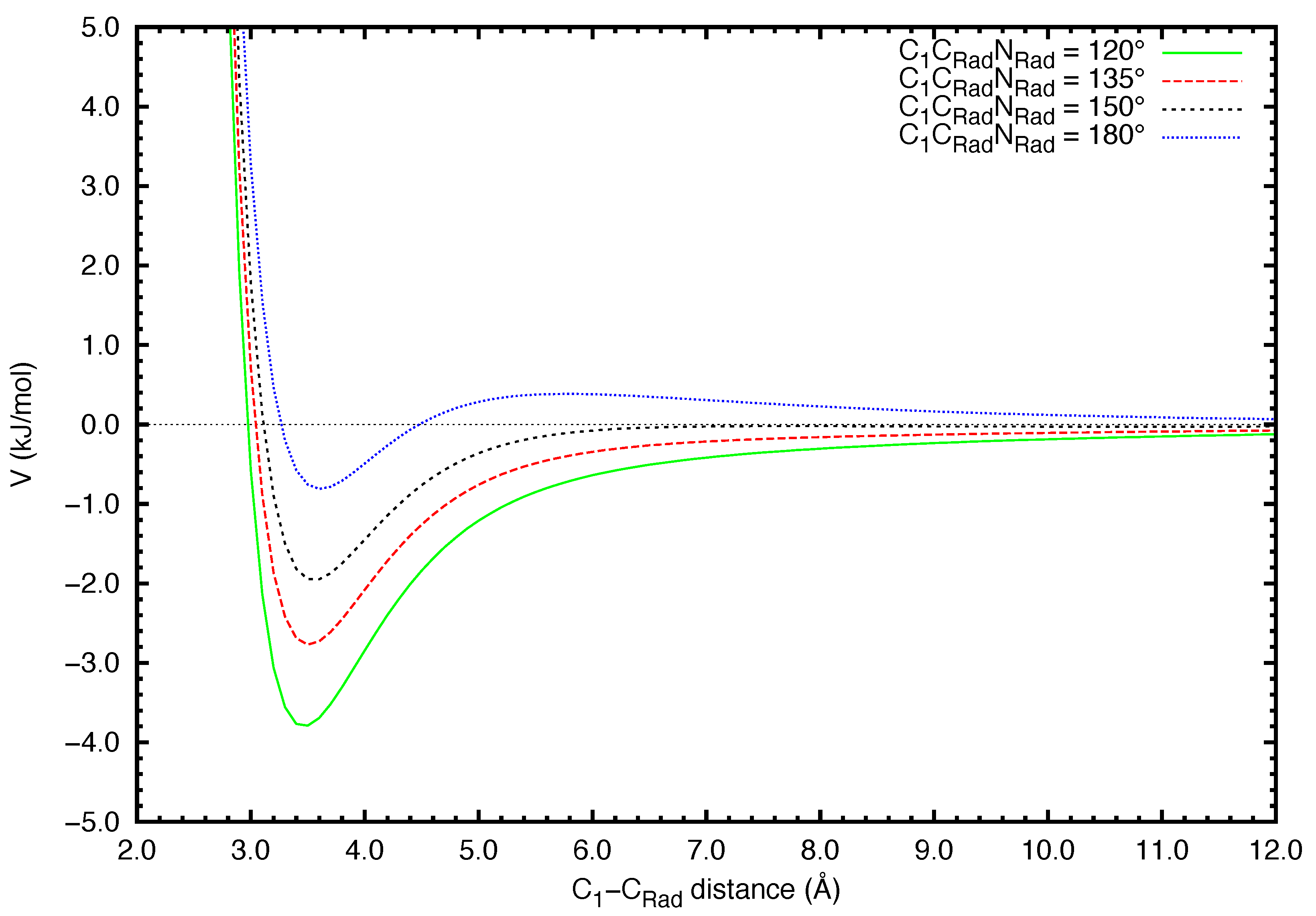
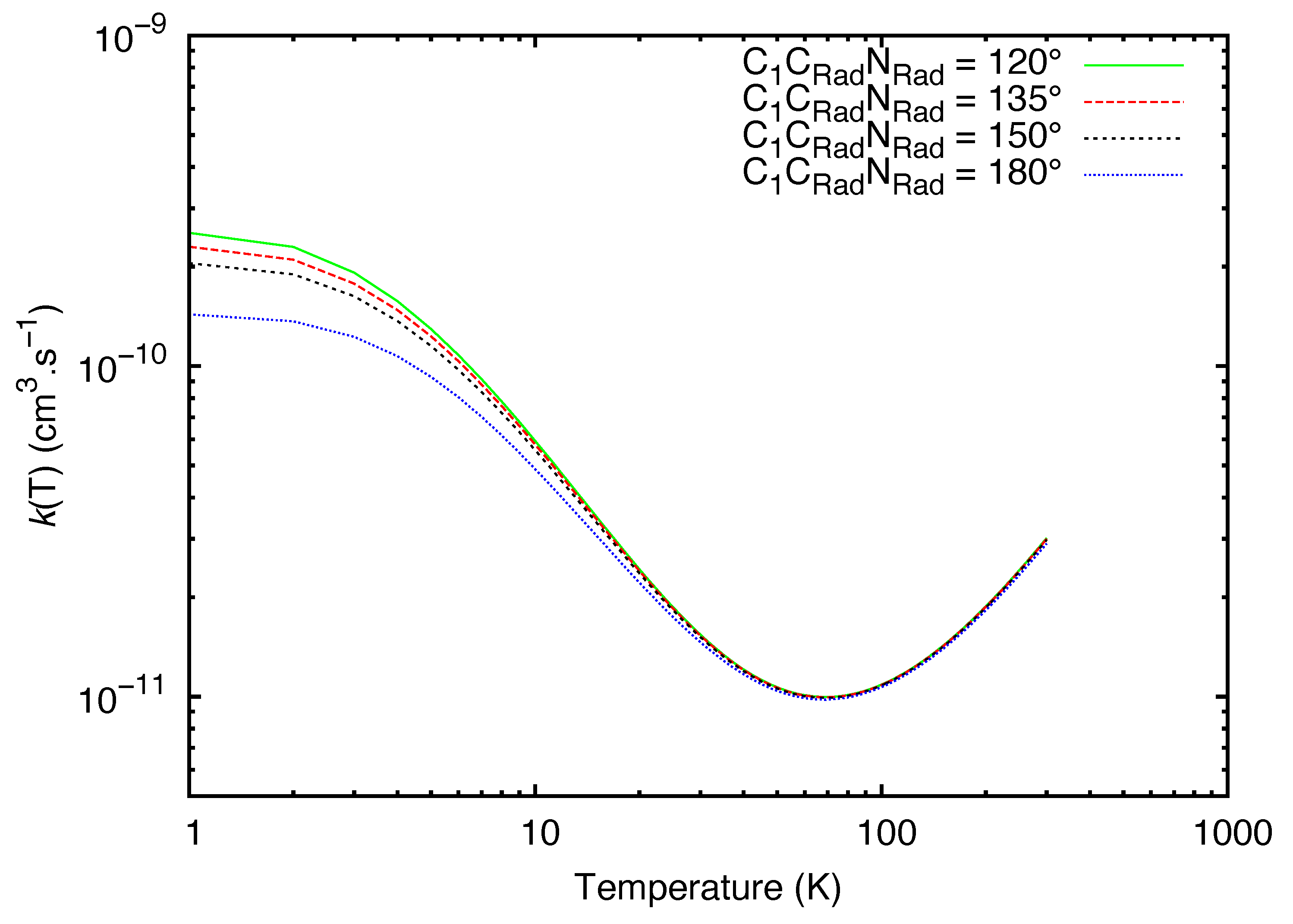
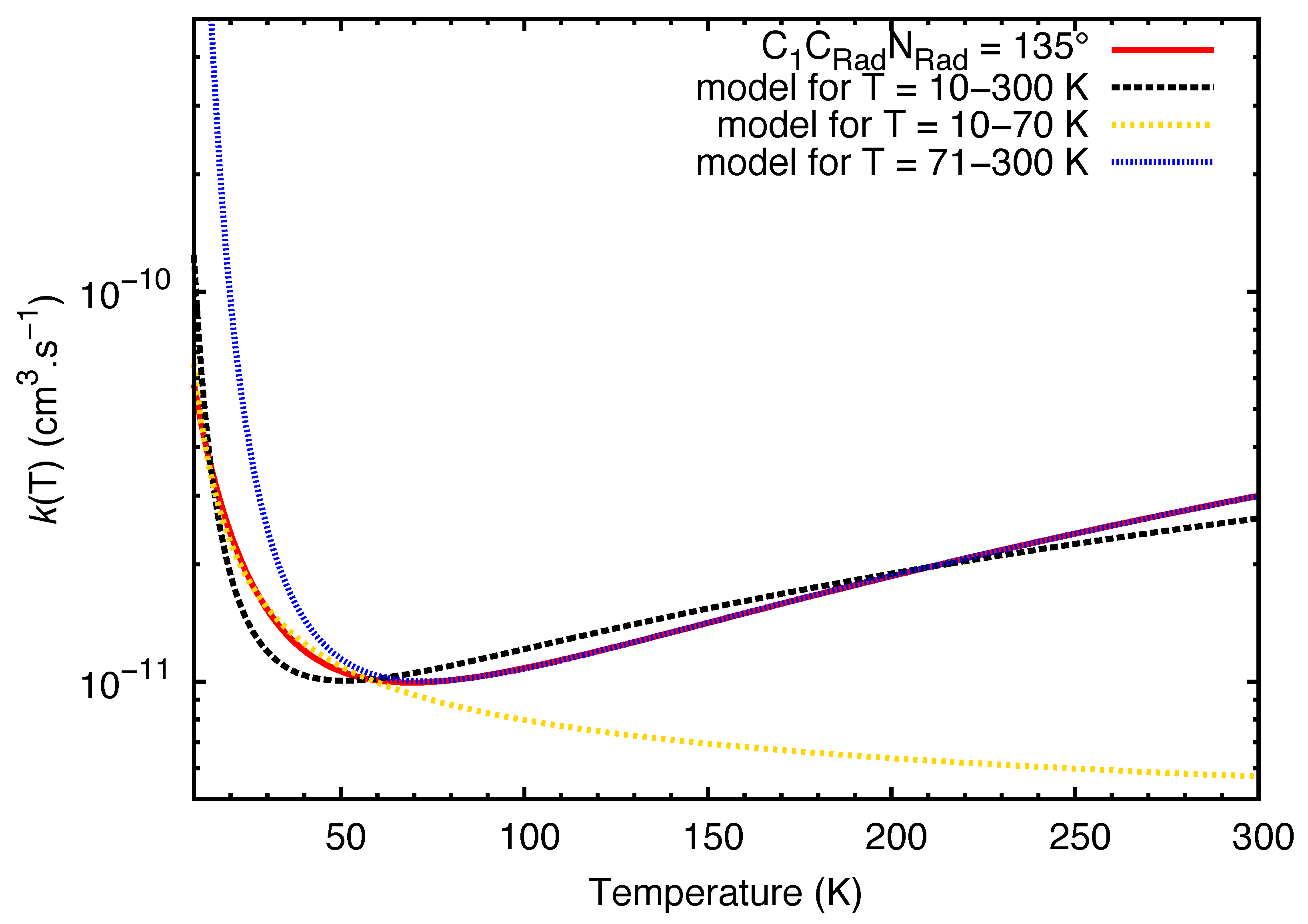
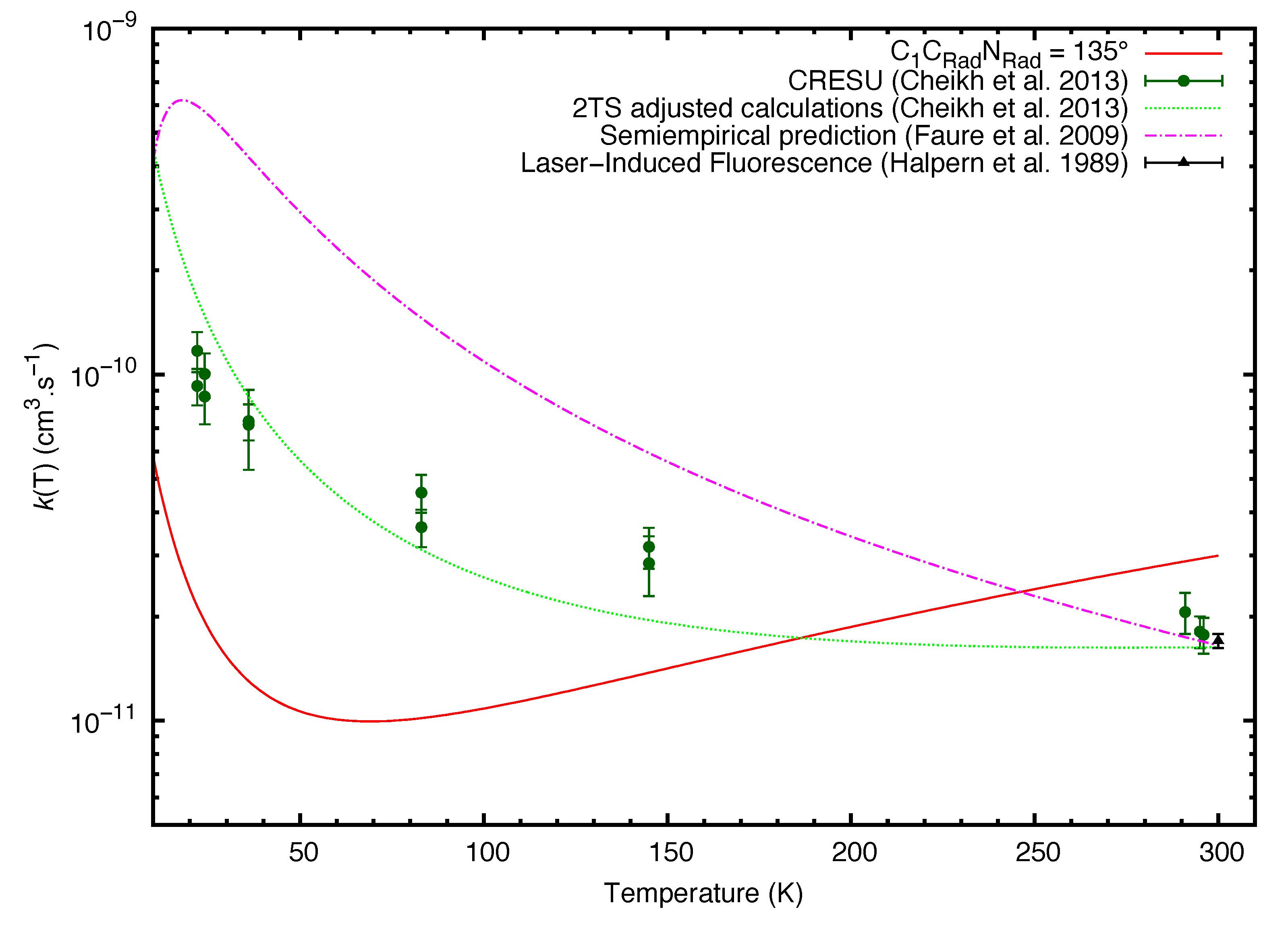
2. Results
3. Discussion
4. Theoretical Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Swings, P.; Rosenfeld, L. Considerations regarding interstellar molecules. Astrophys. J. 1937, 86, 483–486. [Google Scholar] [CrossRef]
- McKellar, A. Evidence for the molecular origin of some hitherto unidentified interstellar lines. Publ. Astron. Soc. Pac. 1940, 52, 187–192. [Google Scholar] [CrossRef]
- Adams, W.S. Some Results with the COUDÉ Spectrograph of the Mount Wilson Observatory. ApJ 1941, 93, 11. [Google Scholar] [CrossRef]
- McGuire, B.A. 2021 Census of Interstellar, Circumstellar, Extragalactic, Protoplanetary Disk, and Exoplanetary Molecules. arXiv 2021, arXiv:2109.13848. [Google Scholar] [CrossRef]
- López-Sepulcre, A.; Balucani, N.; Ceccarelli, C.; Codella, C.; Dulieu, F.; Theule, P. Interstellar formamide (NH2CHO), a key prebiotic precursor. ACS Earth Space Chem. 2019, 3, 2122–2137. [Google Scholar] [CrossRef] [Green Version]
- Caselli, P.; Ceccarelli, C. Our astrochemical heritage. Astron. Astrophys. Rev. 2012, 20, 1–68. [Google Scholar] [CrossRef] [Green Version]
- Balucani, N.; Casavecchia, P. Crossed molecular beam studies of astronomically relevant bimolecular reactions. Rend. Lincei 2011, 22, 173–181. [Google Scholar] [CrossRef]
- Rowe, B.R.; Dupeyrat, G.; Marquette, J.B.; Gaucherel, P. Study of the reactions N2+ + 2N2→ N4+ + N2 and O2+ + 2O2→ O4+ + O2 from 20 to 160 K by the CRESU technique. J. Chem. Phys. 1984, 80, 4915–4921. [Google Scholar] [CrossRef]
- Skouteris, D.; Balucani, N.; Ceccarelli, C.; Faginas Lago, N.; Codella, C.; Falcinelli, S.; Rosi, M. Interstellar dimethyl ether gas-phase formation: A quantum chemistry and kinetics study. MNRAS 2019, 482, 3567–3575. [Google Scholar] [CrossRef] [Green Version]
- Skouteris, D.; Balucani, N.; Faginas-Lago, N.; Falcinelli, S.; Rosi, M. Dimerization of methanimine and its charged species in the atmosphere of Titan and interstellar/cometary ice analogs. Astron. Astrophys. 2015, 584, A76. [Google Scholar] [CrossRef] [Green Version]
- Balucani, N.; Skouteris, D.; Ceccarelli, C.; Codella, C.; Falcinelli, S.; Rosi, M. A theoretical investigation of the reaction between the amidogen, NH, and the ethyl, C2H5, radicals: A possible gas-phase formation route of interstellar and planetary ethanimine. Mol. Astrophys. 2018, 13, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Marcus, R.A. Unimolecular Dissociations and Free Radical Recombination Reactions. J. Chem. Phys 1952, 20, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Pirani, F.; Brizi, S.; Roncaratti, L.F.; Casavecchia, P.; Cappelletti, D.; Vecchiocattivi, F. Beyond the Lennard–Jones model: A simple and accurate potential function probed by high resolution scattering data useful for molecular dynamics simulations. Phys. Chem. Chem. Phys. 2008, 10, 5489–5503. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.E. Detection of interstellar cyanoacetylene. ApJ 1971, 163, L35–L39. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Ohishi, M.; Ishikawa, S.I.; Kaifu, N. Detection of isocyanoacetylene HCCNC in TMC-1. Astrophys. J. 1992, 386, L51–L53. [Google Scholar] [CrossRef]
- Langer, W.; Schloerb, F.; Snell, R.; Young, J. Detection of deuterated cyanoacetylene in the interstellar cloud TMC 1. Astrophys. J. 1980, 239, L125–L128. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Kasai, Y.; Ishikawa, S.I.; Ohishi, M.; Kaifu, N.; Amano, T. Detection of a new molecular ion HC3NH (+) in TMC-1. Astrophys. J. 1994, 420, L95–L97. [Google Scholar] [CrossRef]
- Bergner, J.B.; Öberg, K.I.; Garrod, R.T.; Graninger, D.M. Complex organic molecules toward embedded low-mass protostars. ApJ 2017, 841, 120. [Google Scholar] [CrossRef] [Green Version]
- Bergner, J.B.; Guzmán, V.G.; Öberg, K.I.; Loomis, R.A.; Pegues, J. A Survey of CH3CN and HC3N in Protoplanetary Disks. ApJ 2018, 857, 69. [Google Scholar] [CrossRef] [Green Version]
- Chapillon, E.; Dutrey, A.; Guilloteau, S.; Piétu, V.; Wakelam, V.; Hersant, F.; Gueth, F.; Henning, T.; Launhardt, R.; Schreyer, K.; et al. Chemistry in Disks. VII. First Detection of HC3N in Protoplanetary Disks. ApJ 2012, 756, 58. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.; Turner, B.; Palmer, P.; Zuckerman, B. Cyanoacetylene in dense interstellar clouds. Astrophys. J. 1976, 205, 82–93. [Google Scholar] [CrossRef]
- Kunde, V.; Aikin, A.; Hanel, R.; Jennings, D.; Maguire, W.; Samuelson, R. C4H2, HC3N and CH2NH2 in Titan’s atmosphere. Nature 1981, 292, 686–688. [Google Scholar] [CrossRef]
- Cordiner, M.; Nixon, C.; Teanby, N.; Irwin, P.; Serigano, J.; Charnley, S.B.; Milam, S.N.; Mumma, M.J.; Lis, D.; Villanueva, G.; et al. ALMA Measurements of the HNC and HC3N Distributions in Titan’s Atmosphere. ApJL 2014, 795, L30. [Google Scholar] [CrossRef] [Green Version]
- Broten, N.W.; Oka, T.; Avery, L.W.; MacLeod, J.M.; Kroto, H.W. The detection of HC9N in interstellar space. ApJL 1978, 223, L105–L107. [Google Scholar] [CrossRef]
- Cordiner, M.A.; Charnley, S.B.; Kisiel, Z.; McGuire, B.A.; Kuan, Y.J. Deep K-band Observations of TMC-1 with the Green Bank Telescope: Detection of HC7O, Nondetection of HC11N, and a Search for New Organic Molecules. ApJ 2017, 850, 187. [Google Scholar] [CrossRef]
- Balucani, N.; Asvany, O.; Huang, L.; Lee, Y.; Kaiser, R.; Osamura, Y.; Bettinger, H. Formation of nitriles in the interstellar medium via reactions of cyano radicals, CN (X2Σ+), with unsaturated hydrocarbons. Astrophys. J. 2000, 545, 892. [Google Scholar] [CrossRef]
- Chastaing, D.; James, P.L.; Sims, I.R.; Smith, I.W. Neutral–neutral reactions at the temperatures of interstellar clouds Rate coefficients for reactions of C2H radicals with O2, C2H2, C2H4 and C3H6 down to 15 K. Faraday Discuss. 1998, 109, 165–181. [Google Scholar] [CrossRef]
- Imanaka, H.; Smith, M.A. Formation of nitrogenated organic aerosols in the Titan upper atmosphere. Proc. Natl. Acad. Sci. USA 2010, 107, 12423–12428. [Google Scholar] [CrossRef] [Green Version]
- Israël, G.; Szopa, C.; Raulin, F.; Cabane, M.; Niemann, H.; Atreya, S.; Bauer, S.; Brun, J.F.; Chassefière, E.; Coll, P.; et al. Complex organic matter in Titan’s atmospheric aerosols from in situ pyrolysis and analysis. Nature 2005, 438, 796–799. [Google Scholar] [CrossRef]
- Yung, Y.L. An update of nitrile photochemistry on Titan. Icarus 1987, 72, 468–472. [Google Scholar] [CrossRef]
- Halpern, J.; Miller, G.; Okabe, H. The reaction of CN radicals with cyanoacetylene. Chem. Phys. Lett. 1989, 155, 347–350. [Google Scholar] [CrossRef]
- Faure, A.; Vuitton, V.; Thissen, R.; Wiesenfeld, L. A Semiempirical Capture Model for Fast Neutral Reactions at Low Temperature. J. Phys. Chem. A 2009, 113, 13694–13699. [Google Scholar] [CrossRef] [PubMed]
- Kooij, D.M. Über die Zersetzung des gasförmigen Phosphorwasserstoffs. Z. Phys. Chem. 1893, 12U, 155–161. [Google Scholar] [CrossRef]
- Woodall, J.; Agúndez, M.; Markwick-Kemper, A.J.; Millar, T.J. The UMIST database for astrochemistry 2006. Astron. Astrophys. 2007, 466, 1197–1204. [Google Scholar] [CrossRef] [Green Version]
- Wakelam, V.; Herbst, E.; Loison, J.C.; Smith, I.W.M.; Chandrasekaran, V.; Pavone, B.; Adams, N.G.; Bacchus-Montabonel, M.C.; Bergeat, A.; Béroff, K.; et al. A Kinetic Database for Astrochemistry (KIDA). ApJS 2012, 199, 21. [Google Scholar] [CrossRef]
- Cheikh Sid Ely, S.; Morales, S.B.; Guillemin, J.C.; Klippenstein, S.J.; Sims, I.R. Low Temperature Rate Coefficients for the Reaction CN + HC3N. J. Phys. Chem. A 2013, 117, 12155–12164. [Google Scholar] [CrossRef]
- Greenwald, E.E.; North, S.W.; Georgievskii, Y.; Klippenstein, S.J. A Two Transition State Model for Radical-Molecule Reactions: A Case Study of the Addition of OH to C2H4. J. Phys. Chem. A 2005, 109, 6031–6044. [Google Scholar] [CrossRef]
- Valença Ferreira de Aragão, E.; Liang, P.; Mancini, L.; Marchione, D.; Vanuzzo, G.; Faginas-Lago, N.; Rosi, M.; Skouteris, D.; Pirani, F.; Casavecchia, P.; et al. Combined Crossed Molecular Beam and Theoretical Studies on the Cyanoacetylene (HC3N) + CN(X2Σ+) Reaction; Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia: Perugia, Italy, 2022; to be submitted. [Google Scholar]
- Quantities, Units and Symbols in Physical Chemistry, 3rd ed.; The Royal Society of Chemistry: London, UK, 2007.
- Petrie, S.; Osamura, Y. NCCN and NCCCCN Formation in Titan’s Atmosphere: 2. HNC as a Viable Precursor. J. Phys. Chem. A 2004, 108, 3623–3631. [Google Scholar] [CrossRef]
- Troiani, A.; Rosi, M.; Garzoli, S.; Salvitti, C.; de Petris, G. Vanadium Hydroxide Cluster Ions in the Gas Phase: Bond-Forming Reactions of Doubly-Charged Negative Ions by SO2-Promoted V-O Activation. Chem. Eur. J. 2017, 23, 11752–11756. [Google Scholar] [CrossRef]
- de Petris, G.; Cartoni, A.; Rosi, M.; Troiani, A. The HSSS Radical and the HSSS− Anion. J. Phys. Chem. A 2008, 112, 8471–8477. [Google Scholar] [CrossRef]
- de Petris, G.; Rosi, M.; Troiani, A. SSOH and HSSO Radicals: An Experimental and Theoretical Study of [S2OH]0/+/− Species. J. Phys. Chem. A 2007, 111, 6526–6533. [Google Scholar] [CrossRef] [PubMed]
- Rosi, M.; Falcinelli, S.; Balucani, N.; Casavecchia, P.; Skouteris, D. A theoretical study of formation routes and dimerization of methanimine and implications for the aerosols formation in the upper atmosphere of Titan. LNCS 2013, 7971, 47–56. [Google Scholar] [CrossRef]
- Sleiman, C.; El Dib, G.; Rosi, M.; Skouteris, D.; Balucani, N.; Canosa, A. Low temperature kinetics and theoretical studies of the reaction CN + CH3NH2: A potential source of cyanamide and methyl cyanamide in the interstellar medium. Phys. Chem. Chem. Phys. 2018, 20, 5478–5489. [Google Scholar] [CrossRef] [PubMed]
- Berteloite, C.; Le Picard, S.D.; Sims, I.R.; Rosi, M.; Leonori, F.; Petrucci, R.; Balucani, N.; Wang, X.; Casavecchia, P. Low temperature kinetics, crossed beam dynamics and theoretical studies of the reaction S(1D)+ CH4 and low temperature kinetics of S(1D)+ C2H2. Phys. Chem. Chem. Phys. 2011, 13, 8485–8501. [Google Scholar] [CrossRef]
- Rosi, M.; Mancini, L.; Skouteris, D.; Ceccarelli, C.; Lago, N.F.; Podio, L.; Codella, C.; Lefloch, B.; Balucani, N. Possible scenarios for SiS formation in the interstellar medium: Electronic structure calculations of the potential energy surfaces for the reactions of the SiH radical with atomic sulphur and S2. Chem. Phys. Lett. 2018, 695, 87–93. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Accounts 2008, 120, 215–241. [Google Scholar]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Bartlett, R.J. Many-body perturbation theory and coupled cluster theory for electron correlation in molecules. Annu. Rev. Phys. Chem. 1981, 32, 359–401. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Olsen, J.; Jørgensen, P.; Koch, H.; Balkova, A.; Bartlett, R.J. Full configuration–interaction and state of the art correlation calculations on water in a valence double-zeta basis with polarization functions. J. Chem. Phys. 1996, 104, 8007–8015. [Google Scholar] [CrossRef]
- Dunning Jr, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision A. 02, 2009; Gaussian Inc.: Wallingford, CT, USA, 2009; Volume 201. [Google Scholar]
- Vazart, F.; Latouche, C.; Skouteris, D.; Balucani, N.; Barone, V. Cyanomethanimine Isomers in Cold Interstellar Clouds: Insights from Electronic Structure and Kinetics Calculations. ApJ 2015, 810, 111. [Google Scholar] [CrossRef] [Green Version]
- Vazart, F.; Calderini, D.; Puzzarini, C.; Skouteris, D.; Barone, V. State-of-the-Art Thermochemical and Kinetic Computations for Astrochemical Complex Organic Molecules: Formamide Formation in Cold Interstellar Clouds as a Case Study. J. Chem. Theory Comput. 2016, 12, 5385–5397. [Google Scholar] [CrossRef]
- Skouteris, D.; Balucani, N.; Ceccarelli, C.; Vazart, F.; Puzzarini, C.; Barone, V.; Codella, C.; Lefloch, B. The Genealogical Tree of Ethanol: Gas-phase Formation of Glycolaldehyde, Acetic Acid, and Formic Acid. ApJ 2018, 854, 135. [Google Scholar] [CrossRef]
- Maitland, G.; Smith, E. A simplified representation of intermolecular potential energy. Chem. Phys. Lett. 1973, 22, 443–446. [Google Scholar] [CrossRef]
- Pirani, F.; Albertí, M.; Castro, A.; Moix Teixidor, M.; Cappelletti, D. Atom-bond pairwise additive representation for intermolecular potential energy surfaces. Chem. Phys. Lett. 2004, 394, 37–44. [Google Scholar] [CrossRef]
- Bartolomei, M.; Cappelletti, D.; de Petris, G.; Teixidor, M.M.; Pirani, F.; Rosi, M.; Vecchiocattivi, F. The intermolecular potential in NO–N2 and (NO–N2)+ systems: Implications for the neutralization of ionic molecular aggregates. Phys. Chem. Chem. Phys. 2008, 10, 5993–6001. [Google Scholar] [CrossRef]
- Cappelletti, D.; Pirani, F.; Bussery-Honvault, B.; Gomez, L.; Bartolomei, M. A bond–bond description of the intermolecular interaction energy: The case of weakly bound N2–H2 and N2–N2 complexes. Phys. Chem. Chem. Phys. 2008, 10, 4281–4293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacifici, L.; Verdicchio, M.; Faginas-Lago, N.; Lombardi, A.; Costantini, A. A high-level ab initio study of the N2 + N2 reaction channel. J. Comput. Chem. 2013, 34, 2668–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
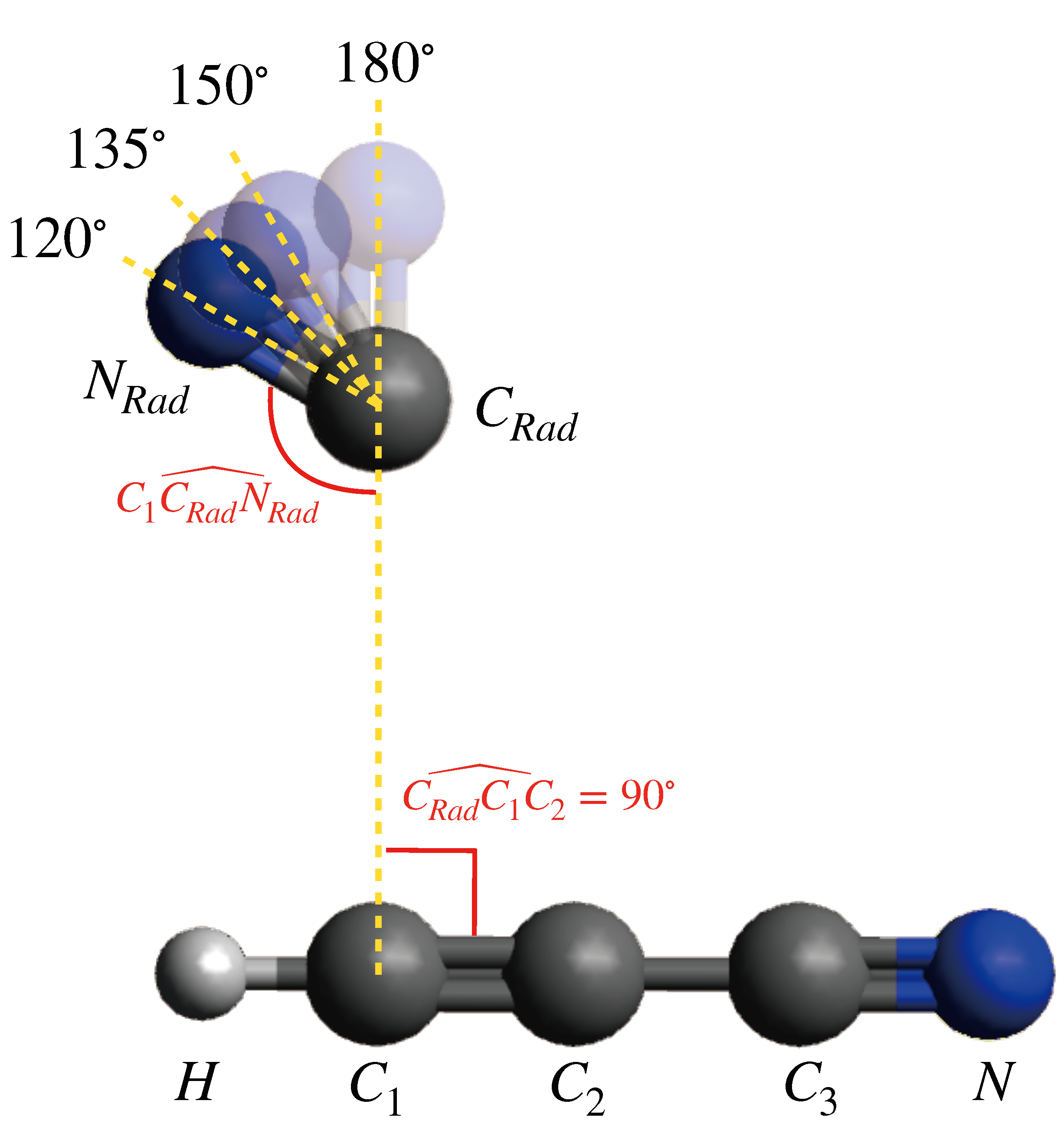
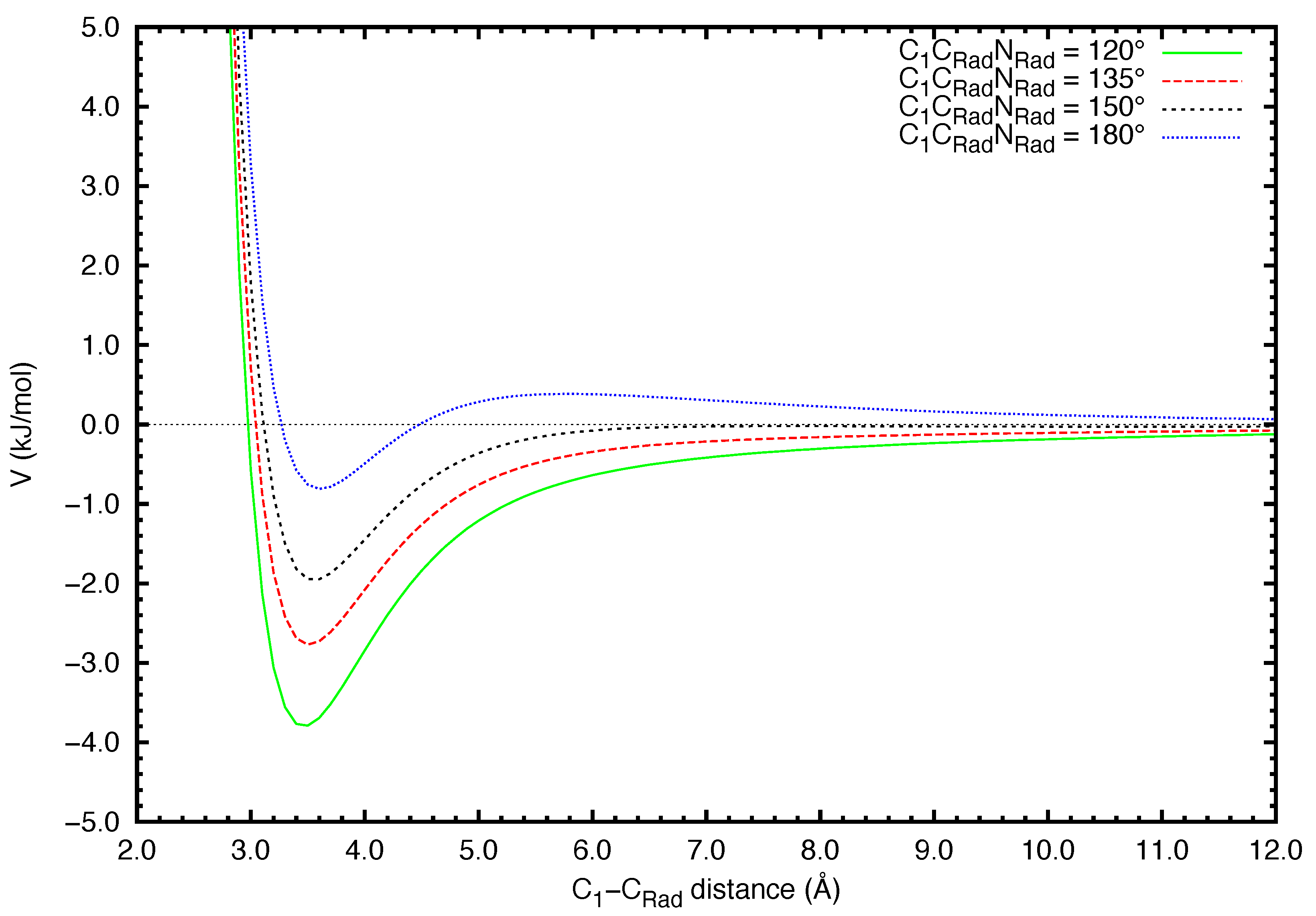

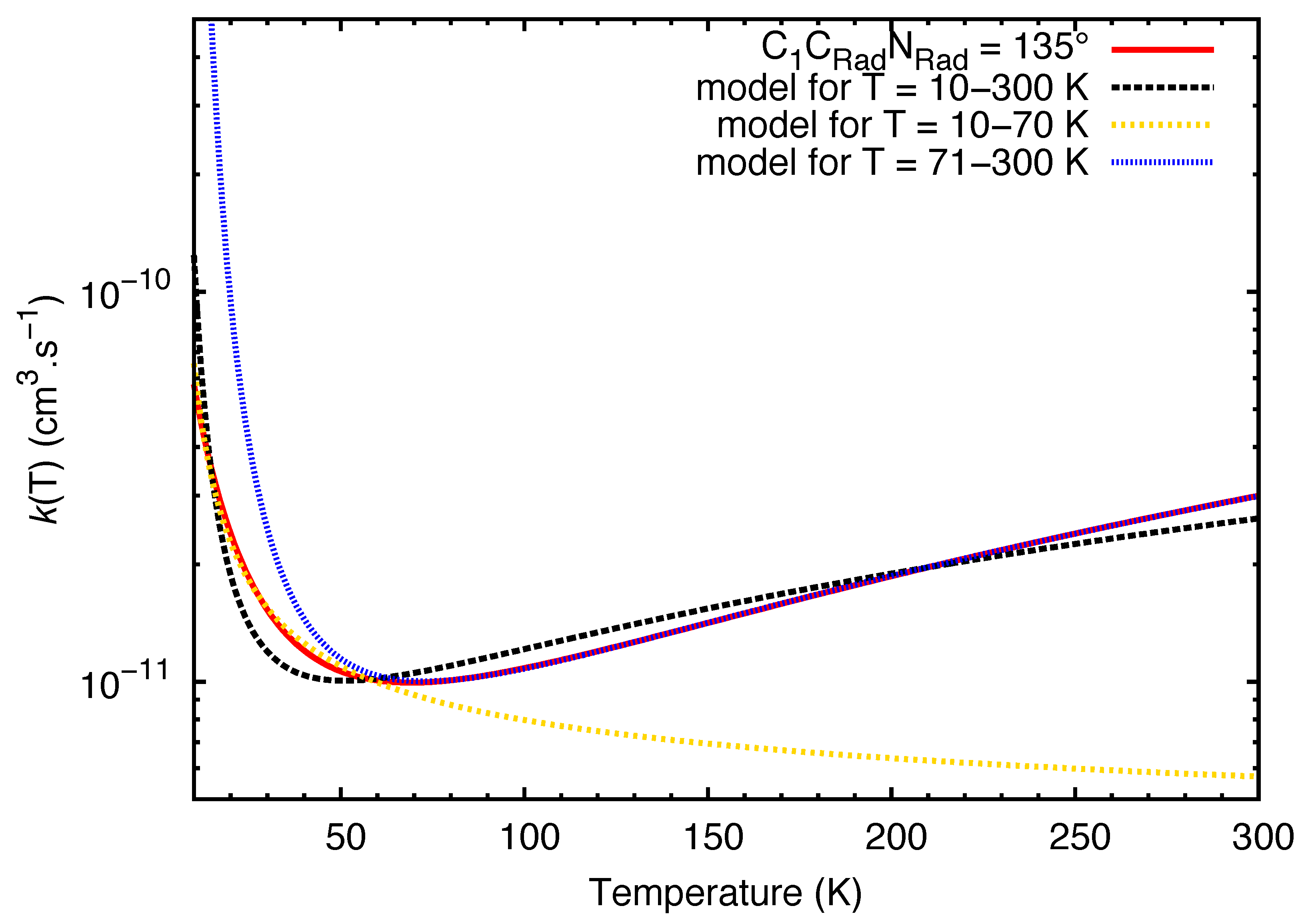


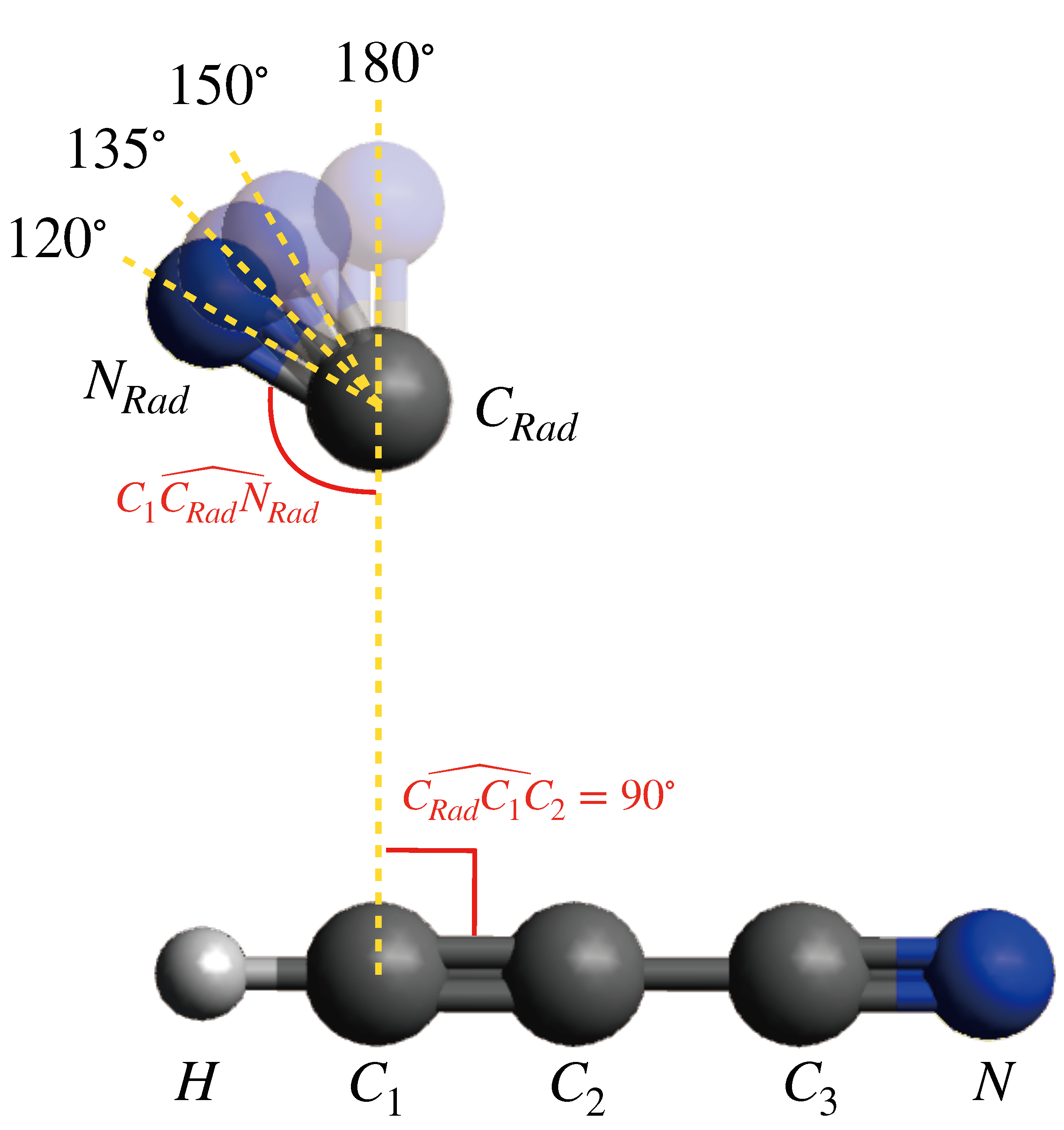{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| () | () | (kJmol) | |
|---|---|---|---|
| HCN + CN | −261.8584480 | 0.032858 | 0.0 |
| vdW | −261.8604704 | 0.033320 | −4.1 |
| TS-vdW-INT | −261.8587898 | 0.033110 | −0.2 |
| INT | −261.9504299 | 0.036495 | −231.9 |
| TS-INT-Product | −261.8702081 | 0.028342 | −42.7 |
| CN + H | −261.8795165 | 0.027620 | −69.1 |
| C (EÅ) | b (E) | |
|---|---|---|
| 120° | 4.67 | −9.60 × 10 |
| 135° | 3.37 | −4.66 × 10 |
| 150° | 2.31 | 7.07 × 10 |
| 180° | 0.73 | 8.64 × 10 |
| Temperature Range (K) | (cms) | |||
|---|---|---|---|---|
| 10−70 | 120° | 5.37 × 10 | −0.19 | −18.80 |
| 135° | 5.37 × 10 | −0.19 | −18.47 | |
| 150° | 5.36 × 10 | −0.20 | −17.97 | |
| 180° | 5.41 × 10 | −0.20 | −16.39 | |
| 71−300 | 120° | 2.01 × 10 | 1.67 | −121.77 |
| 135° | 2.00 × 10 | 1.66 | −121.16 | |
| 150° | 1.99 × 10 | 1.65 | −120.31 | |
| 180° | 1.96 × 10 | 1.62 | −117.34 |
| Atom | ESP Partial Charges (a.u.) |
|---|---|
| C | −0.234 |
| C | 0.120 |
| C | 0.478 |
| N | −0.424 |
| H | 0.300 |
| C | 0.339 |
| N | −0.339 |
| Interacting Pair | (Å) | (meV) |
|---|---|---|
| C-C | 3.80 | 6.16 |
| N-C | 3.73 | 6.07 |
| H-C | 3.54 | 2.43 |
| C-N | 3.71 | 5.95 |
| N-N | 3.63 | 6.07 |
| H-N | 3.41 | 2.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valença Ferreira de Aragão, E.; Mancini, L.; Faginas-Lago, N.; Rosi, M.; Skouteris, D.; Pirani, F. Semiempirical Potential in Kinetics Calculations on the HC3N + CN Reaction. Molecules 2022, 27, 2297. https://doi.org/10.3390/molecules27072297
Valença Ferreira de Aragão E, Mancini L, Faginas-Lago N, Rosi M, Skouteris D, Pirani F. Semiempirical Potential in Kinetics Calculations on the HC3N + CN Reaction. Molecules. 2022; 27(7):2297. https://doi.org/10.3390/molecules27072297
Chicago/Turabian StyleValença Ferreira de Aragão, Emília, Luca Mancini, Noelia Faginas-Lago, Marzio Rosi, Dimitrios Skouteris, and Fernando Pirani. 2022. "Semiempirical Potential in Kinetics Calculations on the HC3N + CN Reaction" Molecules 27, no. 7: 2297. https://doi.org/10.3390/molecules27072297
APA StyleValença Ferreira de Aragão, E., Mancini, L., Faginas-Lago, N., Rosi, M., Skouteris, D., & Pirani, F. (2022). Semiempirical Potential in Kinetics Calculations on the HC3N + CN Reaction. Molecules, 27(7), 2297. https://doi.org/10.3390/molecules27072297