
Bioactivities and Mode of Actions of Dibutyl Phthalates and Nocardamine from Streptomyces sp. H11809

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Characterization and Classification of H11809
2.2. Bioassay-Guided Fractionation and Compound Identification

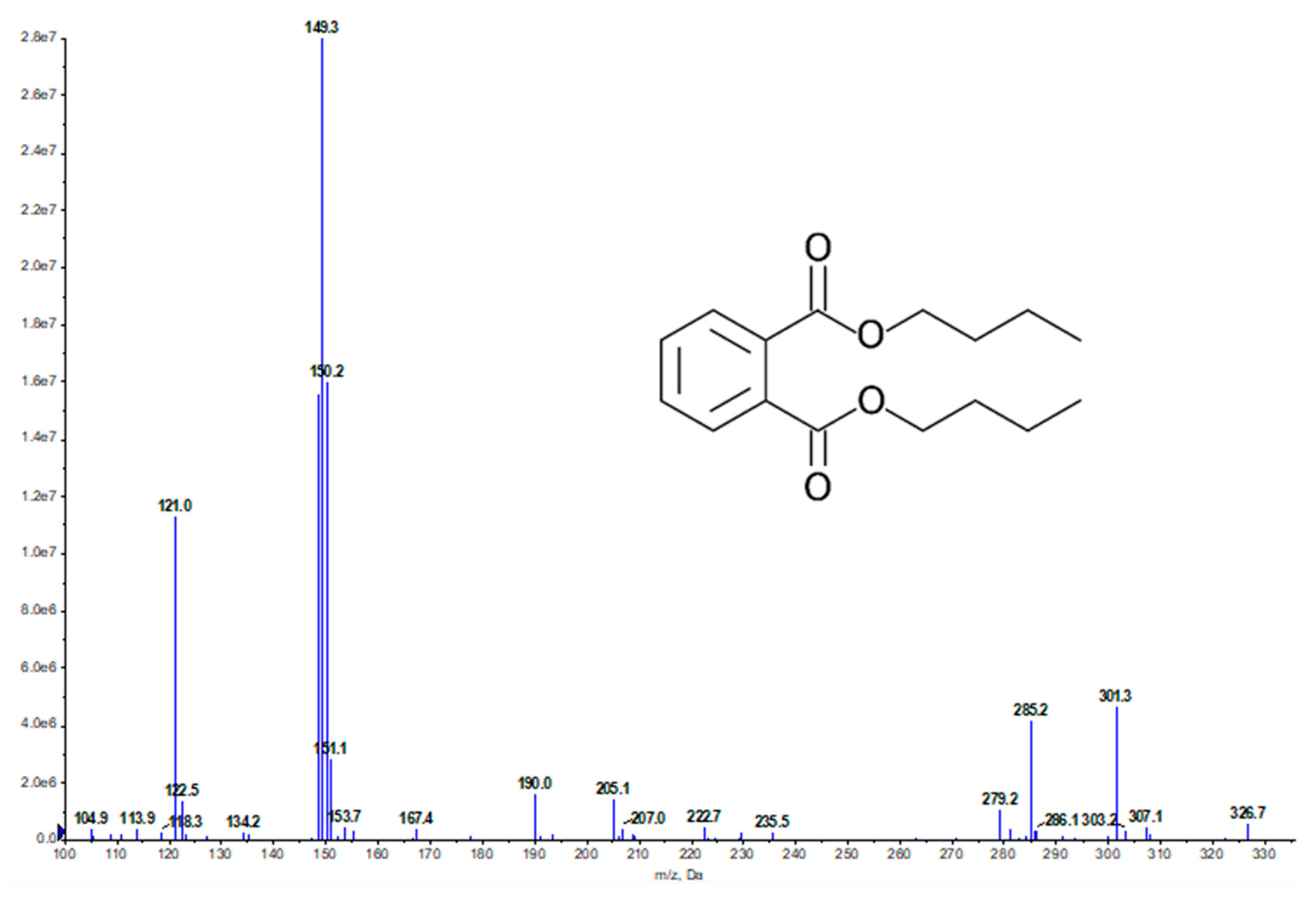{kind=link}
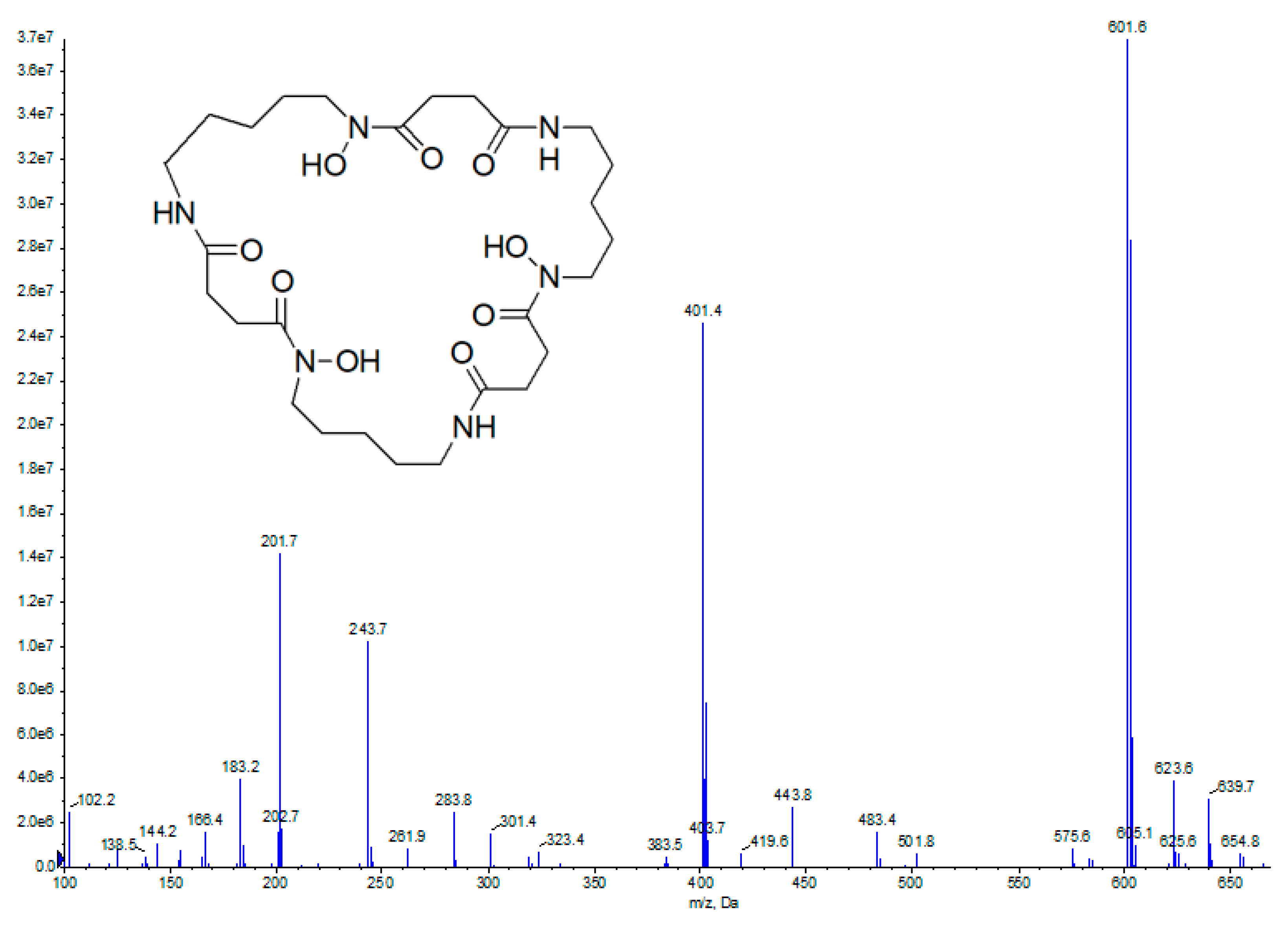{kind=link}
{kind=link}
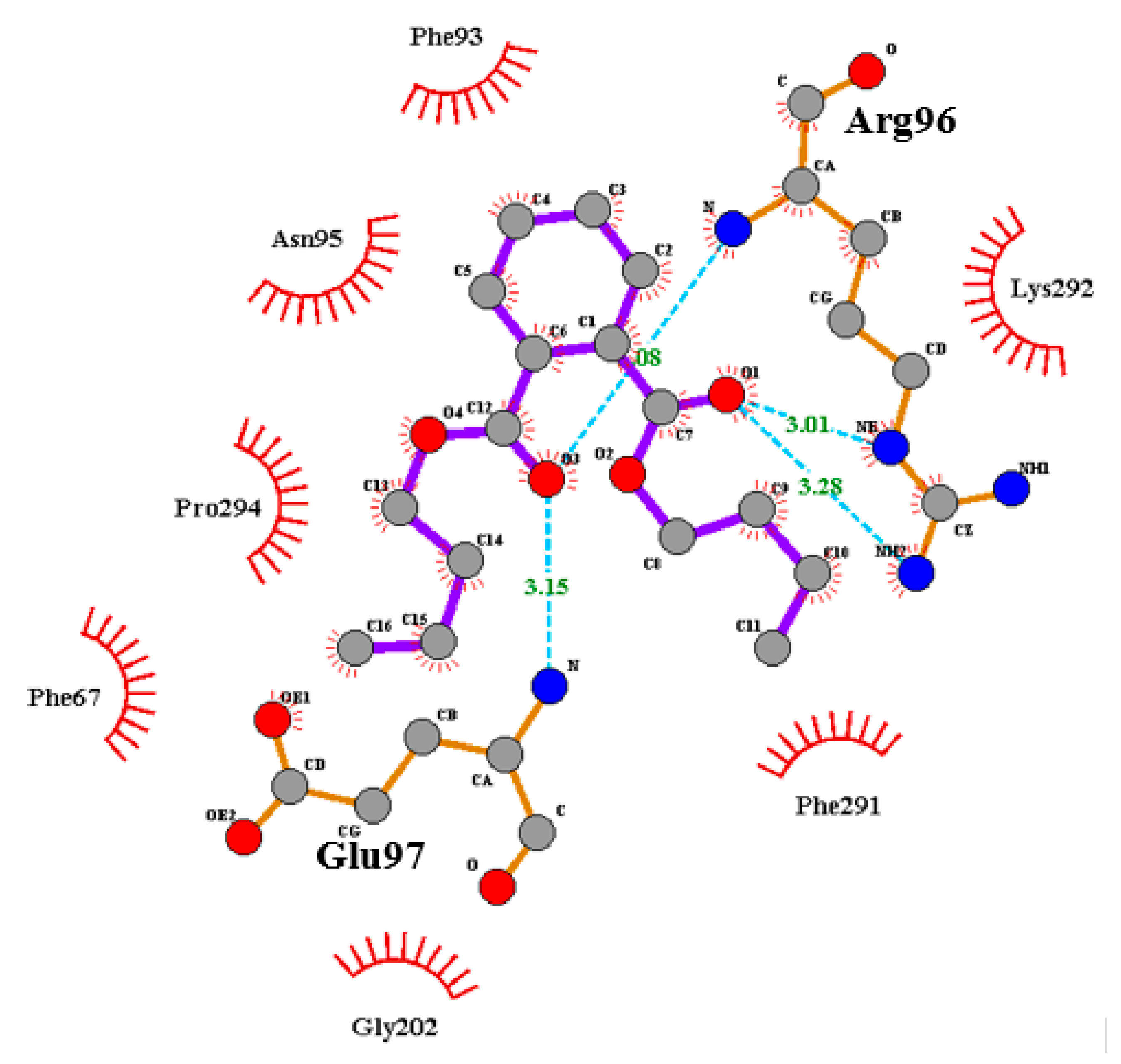{kind=link}
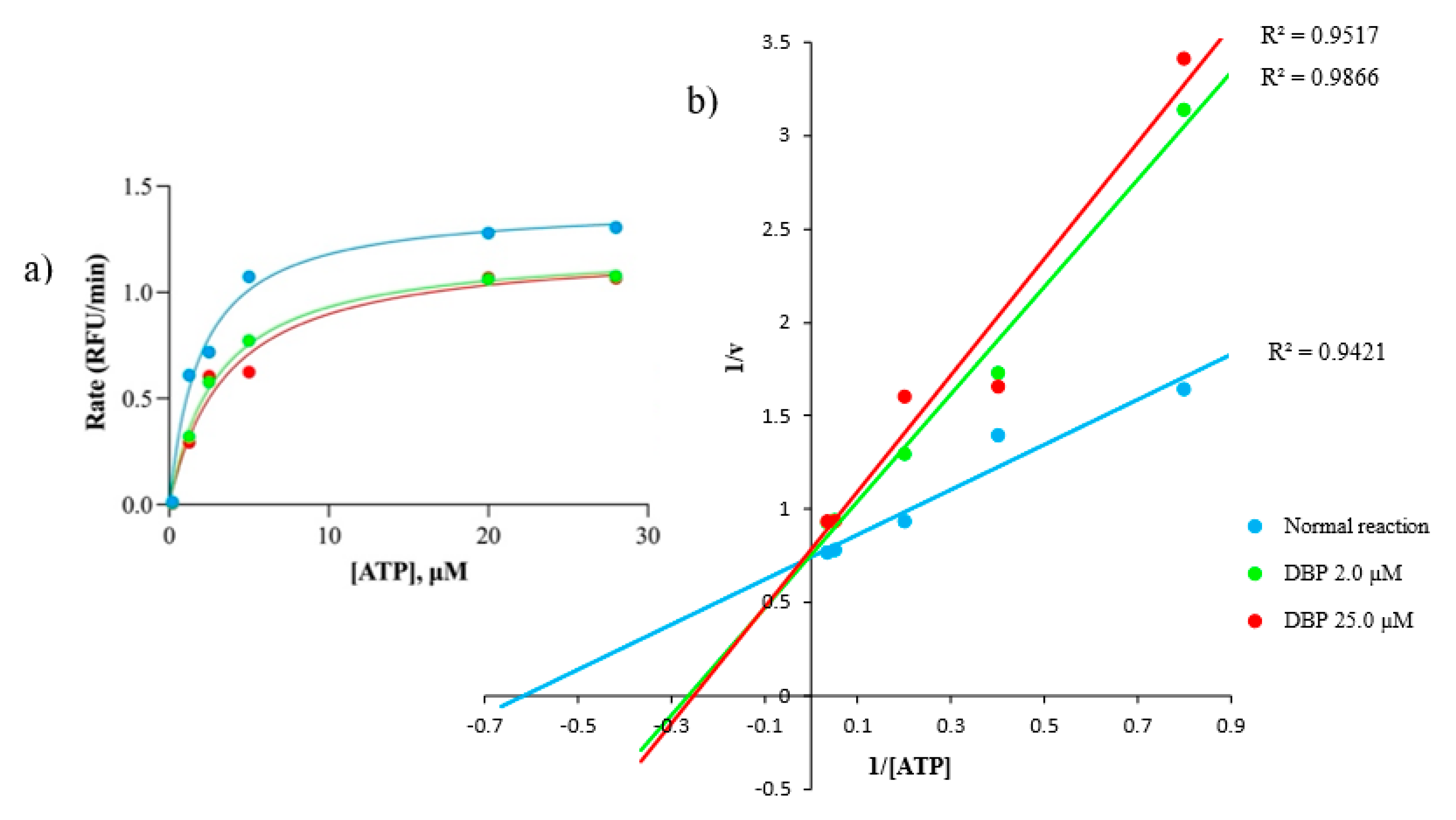{kind=link}
| Sample | Inhibition Zone, mm (±SD) | Remarks | |
|---|---|---|---|
| 28 °C | 37 °C | ||
| H11809-crude extract | 12.67 ± 0.58 | 15.0 ± 0.0 | Non-selective inhibition |
| H11809-chloroform extract | 7.0 ± 0.0 | 8.0 ± 0.0 | Non-selective inhibition |
| H11809-CCF1 | 0.0 ± 0.00 | 0.0 ± 0.00 | No activity |
| H11809-CCF2 | 0.0 ± 0.00 | 0.0 ± 0.00 | No activity |
| H11809-CCF3 | 0.0 ± 0.00 | 0.0 ± 0.00 | No activity |
| H11809-CCF4 | 8.0 ± 0.0 | 8.0 ± 0.58 | Non-selective inhibition |
| H11809-CCF5 | 18.0 ± 0.58 | 7.0 ± 0.58 | Cytotoxic |
| H11809-CCF6 | 10.0 ± 0.58 | 10.0 ± 0.0 | Cytotoxic |
| H11809-CCF7 | 8.0 ± 0.0 | 10.0 ± 0.0 | Non-selective inhibition |
| H11809-CCF8 | 0.0 ± 0.0 | 10.0 ± 0.0 | Selective inhibition |
| H11809-CCF9 | 0.0 ± 0.00 | 0.0 ± 0.0 | No activity |
| Streptomyces H7667 1 | 13.0 ± 0.0 | 14.0 ± 0.0 | Non-selective inhibition |

| Tested Conc. µg/mL | H11809-CCF5 | H11809-CCF6 | H11809-CCF7 | H11809-CCF8 | H11809-CCF9 |
|---|---|---|---|---|---|
| 500 | +++ | ++ | +++ | +++ | +++ |
| 50 | ++ | + | - | - | +++ |
| 5 | - | - | - | - | ++ |
| 0.5 | - | - | - | - | - |
2.3. DBP Inhibits Hs GSK-3β Substrate Binding Site via Mixed Inhibition
2.4. Antimalarial Mode of Action Study
2.4.1. Mutant Resistance Generation against DBP
2.4.2. Iron-Dependency of Nocardamine to Exert Antimalarial Activity
3. Discussion
4. Materials and Methods
4.1. Isolation, Cultivation, Microscopic Characterization, and Molecular Identification of H11809
4.2. Secondary Metabolites Production from H11809
4.3. Bioassay-Guided Fractionation of DBP and Nocardamine
4.3.1. Yeast-Based Assay Expressing Hs GSK-3β
4.3.2. Antimalarial Assay against Plasmodium Falciparum 3D7 (Pf 3D7)
4.4. Mode of Action Study
4.4.1. In Vitro Enzymatic Assay against Hs GSK-3β and Its Preliminary In Silico Analysis
4.4.2. Antimalarial Mode of Action Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Silva, L.J.; Crevelin, E.J.; Souza, D.T.; Lacerda-Júnior, G.V.; de Oliveira, V.M.; Ruiz, A.L.T.G.; Rosa, L.H.; Moraes, L.A.B.; Melo, I.S. Actinobacteria from Antarctica as a source for anticancer discovery. Sci. Rep. 2020, 10, 13870. [Google Scholar] [CrossRef] [PubMed]
- Culp, E.J.; Yim, G.; Waglechner, N.; Wang, W.; Pawlowski, A.C.; Wright, G.D. Hidden antibiotics in actinomycetes can be identified by inactivation of gene clusters for common antibiotics. Nat. Biotechnol. 2019, 37, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.L.; Chan, K.W.; Bernadr, T.Z.; Lim, S.H.; Ngao, W.C.; Tong, M.L.; Teh, S.C. Screening for potential microbial inhibitors against prokaryotic and eukaryotic signal transduction and isocitrate lyase in Mycobacterium from Danum Valley, Sabah. Sabah Soc. J. 2009, 26, 1–91. [Google Scholar]
- Lai, N.S.; Voo, C.; Yung, L.; Daim, S.; Wai, L.O.C.; Ling, K.E.E.C.; Koon, L.E.E.B.; Ching, L.O.W.A.; Chahil, J.K.; Janim, J.; et al. Screening for eukaryotic signal transduction and Mycobacterium isocitrate lyase inhibitor from actinomycetes and fungi of dipterocarp rain forests at Imbak Valley, Sabah, Malaysia. J. Trop. Biol. Conserv. 2009, 5, 87–117. [Google Scholar]
- Mahmud, F.; Maili, S.; Abdul Aziz, D.N.A.F.; Marajin, L.; Jembun, M.; How, S.E.; Gansau, J.A.; Sidek, H.M.; Embi, N.; Lee, P.-C. The Isolation Rate of Culturable Actinomycetes from Malaysian Borneo Forests and Their Activity Against Mammalian GSK-3β. Curr. Appl. Sci. Technol. 2021, 22, 33–46. [Google Scholar]
- Li, Y.; Baptista, R.P.; Kissinger, J.C. Noncoding RNAs in Apicomplexan Parasites: An Update. Trends Parasitol. 2020, 36, 835–849. [Google Scholar] [CrossRef]
- Wu, X.; Stenson, M.; Abeykoon, J.; Nowakowski, K.; Zhang, L.; Lawson, J.; Wellik, L.; Li, Y.; Krull, J.; Wenzl, K.; et al. Targeting glycogen synthase kinase 3 for therapeutic benefit in lymphoma. Blood 2019, 134, 363–373. [Google Scholar] [CrossRef]
- Griebel, G.; Stemmelin, J.; Lopez-Grancha, M.; Boulay, D.; Boquet, G.; Slowinski, F.; Pichat, P.; Beeské, S.; Tanaka, S.; Mori, A.; et al. The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer’s disease in rodents. Sci. Rep. 2019, 9, 18045. [Google Scholar] [CrossRef]
- June, C.C.; Wen, L.H.; Sani, H.A.; Latip, J.; Gansau, J.A.; Chin, L.P.; Embi, N.; Sidek, H.M. Hypoglycemic effects of Gynura procumbens fractions on streptozotocin- induced diabetic rats involved phosphorylation of GSK3β (Ser-9) in liver. Sains Malays. 2012, 41, 969–975. [Google Scholar]
- Ghazanfari, D.; Noori, M.S.; Bergmeier, S.C.; Hines, J.V.; McCall, K.D.; Goetz, D.J. A novel GSK-3 inhibitor binds to GSK-3β via a reversible, time and Cys-199-dependent mechanism. Bioorg. Med. Chem. 2021, 40, 116179. [Google Scholar] [CrossRef]
- De Simone, A.; Tumiatti, V.; Andrisano, V.; Milelli, A. Glycogen Synthase Kinase 3β: A New Gold Rush in Anti-Alzheimer’s Disease Multitarget Drug Discovery? J. Med. Chem. 2021, 64, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Osolodkin, D.I.; Zakharevich, N.V.; Palyulin, V.A.; Danilenko, V.N.; Zefirov, N.S. Bioinformatic analysis of glycogen synthase kinase 3: Human versus parasite kinases. Parasitology 2011, 138, 725–735. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Masch, A.; Kunick, C. Selective inhibitors of Plasmodium falciparum glycogen synthase-3 (PfGSK-3): New antimalarial agents? Biochim. Biophys. Acta Prot. Proteom. 2015, 1854, 1644–1649. [Google Scholar] [CrossRef] [PubMed]
- Dahari, D.E.D.E.; Mohamad Salleh, R.; Mahmud, F.; Ping Chin, L.; Embi, N.; Mohd Sidek, H. Anti-malarial activities of two soil actinomycete isolates from sabah via inhibition of glycogen synthase kinase 3β. Trop. Life Sci. Res. 2016, 27, 53–71. [Google Scholar] [CrossRef]
- Brown, D.G.; Wobst, H.J. Opportunities and challenges in phenotypic screening for neurodegenerative disease research. J. Med. Chem. 2020, 63, 1823–1840. [Google Scholar] [CrossRef]
- Yin, P.; Chen, H.; Liu, X.; Wang, Q.; Jiang, Y.; Pan, R. Mass Spectral Fragmentation Pathways of Phthalate Esters by Gas Chromatography–Tandem Mass Spectrometry. Anal. Lett. 2014, 47, 1579–1588. [Google Scholar] [CrossRef]
- National Institute of Advanced Industrial Science and Technology (AIST). Spectral Database for Organic Compounds (SDBS). Available online: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/landingpage?sdbsno=1932 (accessed on 18 February 2022).
- Cheenpracha, S.; Zhang, H.; Mar, A.M.N.; Foss, A.P.; Sek, H.F.; Ngit, S.L.; Jap, M.J.; Heng, F.S.; Coy, C.H.; Leng, C.C. Yeast glycogen synthase kinase-3β pathway inhibitors from an organic extract of Streptomyces sp. J. Nat. Prod. 2009, 72, 1520–1523. [Google Scholar] [CrossRef]
- Ueki, M.; Suzuki, R.; Takamatsu, S.; Takagi, H.; Uramoto, M.; Ikeda, H.; Osada, H. Nocardamin Production by Streptomyces avermitilis. Actinomycetologica 2009, 23, 34–39. [Google Scholar] [CrossRef]
- Lopez, J.A.V.; Nogawa, T.; Futamura, Y.; Shimizu, T.; Osada, H. Nocardamin glucuronide, a new member of the ferrioxamine siderophores isolated from the ascamycin-producing strain Streptomyces sp. 80H647. J. Antibiot. 2019, 72, 991–995. [Google Scholar] [CrossRef]
- Abdul Aziz, D.N.A.F.N.A.F.; Mahmud, F.; Sidek, H.; Embi, N.; Andoh, T.T.T.; Lee, P.C.C. Assessment of the inhibitory mechanism of action for a yeast cell-based screening system targeting glycogen synthase kinase-3α (GSK-3α). J. Biol. Sci. 2017, 17, 47–51. [Google Scholar] [CrossRef][Green Version]
- Kassir, Y.; Rubin-Bejerano, I.; Mandel-Gutfreund, Y. The Saccharomyces cerevisiae GSK-3 beta homologs. Curr. Drug Targets 2006, 7, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Andoh, T.; Hirata, Y.; Kikuchi, A. Yeast Glycogen Synthase Kinase 3 Is Involved in Protein Degradation in Cooperation with Bul1, Bul2, and Rsp5. Mol. Cell. Biol. 2000, 20, 6712–6720. [Google Scholar] [CrossRef]
- Martinez, A.; Alonso, M.; Castro, A.; Pérez, C.; Moreno, F.J. First non-ATP competitive glycogen synthase kinase 3 β (GSK-3β) inhibitors: Thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J. Med. Chem. 2002, 45, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Palomo, V.; Soteras, I.; Perez, D.I.; Perez, C.; Gil, C.; Campillo, N.E.; Martinez, A. Exploring the binding sites of glycogen synthase kinase 3. identification and characterization of allosteric modulation cavities. J. Med. Chem. 2011, 54, 8461–8470. [Google Scholar] [CrossRef] [PubMed]
- Licht-Murava, A.; Plotkin, B.; Eisenstein, M.; Eldar-Finkelman, H. Elucidating substrate and inhibitor binding sites on the surface of GSK-3β and the refinement of a competitive inhibitor. J. Mol. Biol. 2011, 408, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormö, M.; Nilsson, Y.; Radesäter, A.C.; Jerning, E.; Markgren, P.O.; Borgegård, T.; et al. Structural Insights and Biological Effects of Glycogen Synthase Kinase 3-specific Inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef]
- Sutherland, C. What Are the bona fide GSK3 Substrates? Int. J. Alzheimers Dis. 2011, 2011, 505607. [Google Scholar]
- Silverstein, T.P. When both Km and Vmax are altered, Is the enzyme inhibited or activated? Biochem. Mol. Biol. Educ. 2019, 47, 446–449. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Appendix: Vmax and KM Can Be Determined by Double-Reciprocal Plots. In Biochemistry; W. H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Sharma, R. Enzyme Inhibition: Mechanism and Scope. In Enzyme Inhibition and Bioapplication; Sharma, R., Ed.; InTech: Rijeka, Croatia, 2012; p. 16. [Google Scholar]
- Tian, C.K.; Ni, J.R.; Chang, F.; Liu, S.T.; Xu, N.; Sun, W.L.; Xie, Y.; Guo, Y.Z.; Ma, Y.R.; Yang, Z.X.; et al. Bio-source of di-n-butyl phthalate production by filamentous fungi. Sci. Rep. 2016, 6, 8. [Google Scholar] [CrossRef]
- Wei, Y.; Zhao, Y.; Zhou, D.; Qi, D.; Li, K.; Tang, W.; Chen, Y.; Jing, T.; Zang, X.; Xie, J.; et al. A Newly Isolated Streptomyces sp. YYS-7 With a Broad-Spectrum Antifungal Activity Improves the Banana Plant Resistance to Fusarium oxysporum f. sp. cubense Tropical Race 4. Front. Microbiol. 2020, 11, 1712. [Google Scholar] [CrossRef]
- Roy, R.N.; Laskar, S.; Sen, S.K. Dibutyl phthalate, the bioactive compound produced by Streptomyces albidoflavus 321.2. Microbiol. Res. 2006, 161, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Pande, V.; Ramos, M.J. Structural basis for the GSK-3β binding affinity and selectivity against CDK-2 of 1-(4-aminofurazan-3yl)-5-dialkylaminomethyl-1H-[1,2,3] triazole-4-carboxylic acid derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 5129–5135. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Finkelman, H.; Martinez, A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.Z.; Mandelkow, E.M.; et al. Indirubins inhibit glycogen synthase kinase-3β and CDK5/P25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Selenica, M.L.; Jensen, H.S.; Larsen, A.K.; Pedersen, M.L.; Helboe, L.; Leist, M.; Lotharius, J. Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation. Br. J. Pharmacol. 2007, 152, 959–979. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- Avrahami, L.; Licht-Murava, A.; Eisenstein, M.; Eldar-Finkelman, H. GSK-3 inhibition: Achieving moderate efficacy with high selectivity. Biochim. Biophys. Acta Prot. Proteom. 2013, 1834, 1410–1414. [Google Scholar] [CrossRef]
- Meister, S.; Plouffe, D.M.; Kuhen, K.L.; Bonamy, G.M.C.; Wu, T.; Barnes, S.W.; Bopp, S.E.; Borboa, R.; Bright, A.T.; Che, J.; et al. Imaging of plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 2011, 334, 1372–1377. [Google Scholar] [CrossRef]
- Crowther, G.J.; Napuli, A.J.; Gilligan, J.H.; Gagaring, K.; Borboa, R.; Francek, C.; Chen, Z.; Dagostino, E.F.; Stockmyer, J.B.; Wang, Y.; et al. Identification of inhibitors for putative malaria drug targets among novel antimalarial compounds. Mol. Biochem. Parasitol. 2011, 175, 21–29. [Google Scholar] [CrossRef]
- Fairbairn, E.A.; Bonthius, J.; Cherr, G.N. Polycyclic aromatic hydrocarbons and dibutyl phthalate disrupt dorsal-ventral axis determination via the Wnt/β-catenin signaling pathway in zebrafish embryos. Aquat. Toxicol. 2012, 124–125, 188–196. [Google Scholar] [CrossRef]
- Cabrera, D.G.; Horatscheck, A.; Wilson, C.R.; Basarab, G.; Eyermann, C.J.; Chibale, K. Plasmodial Kinase Inhibitors: License to Cure? J. Med. Chem. 2018, 61, 8061–8077. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Jann, M.L.S.; Sudi, S.; Hassan, W.R.M.; Chin, L.P.; Embi, N.; Sidek, H.M. Anti-malarial and Anti-inflammatory Effects of Gynura procumbens are Mediated by Kaempferol via Inhibition of Glycogen Synthase Kinase-3β (GSK3β). Sains Malays. 2015, 44, 1489–1500. [Google Scholar]
- Solyakov, L.; Halbert, J.; Alam, M.M.; Semblat, J.P.; Dorin-Semblat, D.; Reininger, L.; Bottrill, A.R.; Mistry, S.; Abdi, A.; Fennell, C.; et al. Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat. Commun. 2011, 2, 512–565. [Google Scholar] [CrossRef]
- Burrows, J.N.; Duparc, S.; Gutteridge, W.E.; Hooft Van Huijsduijnen, R.; Kaszubska, W.; Macintyre, F.; Mazzuri, S.; Möhrle, J.J.; Wells, T.N.C. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017, 16, 1–29. [Google Scholar]
- Cortés-Vieyra, R.; Silva-García, O.; Gómez-García, A.; Gutiérrez-Castellanos, S.; Álvarez-Aguilar, C.; Baizabal-Aguirre, V.M. Glycogen Synthase Kinase 3β Modulates the Inflammatory Response Activated by Bacteria, Viruses, and Parasites. Front. Immunol. 2021, 12, 675751. [Google Scholar] [CrossRef]
- Grau, G.E.; Fajardo, L.F.; Piguet, P.F.; Allet, B.; Lambert, P.H.; Vassalli, P. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 1987, 237, 1210–1212. [Google Scholar] [CrossRef]
- Harawa, V.; Njie, M.; Kessler, A.; Choko, A.; Kumwenda, B.; Kampondeni, S.; Potchen, M.; Kim, K.; Jaworowski, A.; Taylor, T.; et al. Brain swelling is independent of peripheral plasma cytokine levels in Malawian children with cerebral malaria. Malar. J. 2018, 10, 1656. [Google Scholar] [CrossRef]
- Mizobuchi, H.; Fujii, W.; Isokawa, S.; Ishizuka, K.; Wang, Y.; Watanabe, S.; Sanjoba, C.; Matsumoto, Y.; Goto, Y. Exacerbation of hepatic injury during rodent malaria by myeloid-related protein 14. PLoS ONE 2018, 13, e0199111. [Google Scholar] [CrossRef]
- Ali, A.H.; Sudi, S.; Shi-Jing, N.; Hassan, W.R.M.; Basir, R.; Agustar, H.K.; Embi, N.; Sidek, H.M.; Latip, J. Dual anti-malarial and gsk3β-mediated cytokine-modulating activities of quercetin are requisite of its potential as a plant-derived therapeutic in malaria. Pharmaceuticals 2021, 14, 248. [Google Scholar] [CrossRef]
- Kesely, K.R.; Pantaleo, A.; Turrini, F.M.; Olupot-Olupot, P.; Low, P.S. Inhibition of an Erythrocyte Tyrosine Kinase with Imatinib Prevents Plasmodium falciparum Egress and Terminates Parasitemia. PLoS ONE 2016, 11, e0164895. [Google Scholar] [CrossRef]
- Chien, H.D.; Pantaleo, A.; Kesely, K.R.; Noomuna, P.; Putt, K.S.; Tuan, T.A.; Low, P.S.; Turrini, F.M. Imatinib augments standard malaria combination therapy without added toxicity. J. Exp. Med. 2021, 218, e20210724. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.A.; Goheen, M.M.; Cerami, C. Influence of host iron status on Plasmodium falciparum infection. Front. Pharmacol. 2014, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C. Iron deficiency: New insights into diagnosis and treatment. Hematology 2015, 2015, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Rubin, H.; Salem, J.S.; Li, L.S.; Yang, F.D.; Mama, S.; Wang, Z.M.; Fisher, A.; Hamann, C.S.; Cooperman, B.S. Cloning, sequence determination, and regulation of the ribonucleotide reductase subunits from Plasmodium falciparum: A target for antimalarial therapy. Proc. Natl. Acad. Sci. USA 1993, 90, 9280–9284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Wu, J.; Shah, B.N.; Greutélaers, K.C.; Ghosh, M.C.; Ollivierre, H.; Su, X.Z.; Thuma, P.E.; Bedu-Addo, G.; Mockenhaupt, F.P.; et al. Erythrocytic ferroportin reduces intracellular iron accumulation, hemolysis, and malaria risk. Science 2018, 359, 1520–1523. [Google Scholar] [CrossRef]
- Mabeza, G.F.; Loyevsky, M.; Gordeuk, V.R.; Weiss, G. Iron Chelation Therapy for Malaria: A Review. Pharmacol. Ther. 1999, 81, 53–75. [Google Scholar] [CrossRef]
- Bashri, G.A.P.; Singh, R.; Parihar, P.; Prasad, S.M. Mineral Solubilization by Microorganisms: Mitigating Strategy in Mineral Deficient Soil. In Microbial Biotechnology: Volume 1. Applications in Agriculture and Environment; Patra, J.K., Vishnuprasad, C.N., Das, G., Eds.; Springer Nature: Singapore, 2018; p. 268. [Google Scholar]
- Johnstone, T.C.; Nolan, E.M. Beyond iron: Non-classical biological functions of bacterial siderophores. Dalt. Trans. 2015, 44, 6320–6339. [Google Scholar] [CrossRef]
- De Villiers, K.A.; Egan, T.J. Heme Detoxification in the Malaria Parasite: A Target for Antimalarial Drug Developmen. Acc. Chem. Res. 2021, 54, 2649–2659. [Google Scholar] [CrossRef]
- Goh, L.P.W.; Mahmud, F.; Lee, P.C. Draft genome sequence data of Streptomyces sp. FH025. Data Br. 2021, 36, 107128. [Google Scholar] [CrossRef]
- Yamanaka, K.; Oikawa, H.; Ogawa, H.O.; Hosono, K.; Shinmachi, F.; Takano, H.; Sakuda, S.; Beppu, T.; Ueda, K. Desferrioxamine E produced by Streptomyces griseus stimulates growth and development of Streptomyces tanashiensis. Microbiology 2005, 151, 2899–2905. [Google Scholar] [CrossRef]
- Thipubon, P.; Uthaipibull, C.; Kamchonwongpaisan, S.; Tipsuwan, W.; Srichairatanakool, S. Inhibitory effect of novel iron chelator, 1-(N-acetyl-6-aminohexyl)-3-hydroxy-2-methylpyridin-4-one (CM1) and green tea extract on growth of Plasmodium falciparum. Malar. J. 2015, 14, 382. [Google Scholar] [CrossRef]
- Whitehead, S.; Peto, T.E. Stage-dependent effect of deferoxamine on growth of Plasmodium falciparum in vitro. Blood 1990, 76, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Lytton, S.D.; Mester, B.; Libman, J.; Shanzer, A.; Cabantchik, Z.L. Mode of action of iron (III) chelators as antimalarials: II. Evidence for differential effects on parasite iron-dependent nucleic acid synthesis. Blood 1994, 84, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Golenser, J.; Domb, A.; Mordechai-Daniel, T.; Leshem, B.; Luty, A.; Kremsner, P. Iron chelators: Correlation between effects on Plasmodium spp. and immune functions. J. Parasitol. 2006, 92, 170–177. [Google Scholar] [CrossRef]
- Beutler, E.; Bothwell, T.H.; Charlton, R.W.; Motulsky, A.G. Hereditary hemochromatosis. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3127–3162. [Google Scholar]
- Prentice, A.M.; Ghattas, H.; Doherty, C.; Cox, S.E. Iron metabolism and malaria. Food Nutr. Bull. 2007, 28, S524–S539. [Google Scholar] [CrossRef] [PubMed]
- Gabay, T.; Ginsburg, H. Hemoglobin denaturation and iron release in acidified red blood cell lysate—A possible source of iron for intraerythrocytic malaria parasites. Exp. Parasitol. 1993, 77, 261–272. [Google Scholar] [CrossRef]
- Sanchez-Lopez, R.; Haldar, K. A transferrin-independent iron uptake activity in Plasmodium falciparum-infected and uninfected erythrocytes. Mol. Biochem. Parasitol. 1992, 55, 9–20. [Google Scholar] [CrossRef]
- Lloyd, J.B.; Cable, H.; Rice-Evans, C. Evidence that desferrioxamine cannot enter cells by passive diffusion. Biochem. Pharmacol. 1991, 127, 915–934. [Google Scholar] [CrossRef]
- Vejanan, V.; Latip, J.; Chin, L.P.; Embi, N.; Sidek, H.M. In vitro and in vivo anti-plasmodial activities of Gynura procumbens. Sains Malays. 2012, 41, 1535–1542. [Google Scholar]
- Kiss, R.; Sandor, M.; Szalai, F.A. http://Mcule.com: A public web service for drug discovery. J. Cheminform. 2012, 4, P17. [Google Scholar] [CrossRef]
- Tombuloglu, G.; Tombuloglu, H.; Cevik, E.; Sabit, H. Genome-wide identification of Lysin-Motif Receptor-Like Kinase (LysM-RLK) gene family in Brachypodium distachyon and docking analysis of chitin/LYK binding. Physiol. Mol. Plant Pathol. 2019, 106, 217–225. [Google Scholar] [CrossRef]
- Vaezi, M.; Behbehani, G.R.; Gheibi, N.; Farasat, A. Thermodynamic, kinetic and docking studies of some unsaturated fatty acids-quercetin derivatives as inhibitors of mushroom tyrosinase. AIMS Biophys. 2020, 7, 393–410. [Google Scholar] [CrossRef]




| P1 (ATP-Binding Site) | P2 (Substrate Binding Site) |
|---|---|
| Ala83, Arg141, Asp133, Asp200, Cys199, Gln185, Glu137, Gly63, Ile62, Leu132, Leu188, Lys85, Pro136, Thr138, Tyr134, Val70, Val110, Val135 Binding affinity = −6.1 kcal/mol | Ala204, (Arg96), Arg180, Asn95, Gln89, (Glu97), Gly202, Ile217, Lys85, Lys94, Phe67, Phe93, Ser203, Tyr216, Val87, Val214 Binding affinity = −6.9 kcal/mol |
| Rate | Normal Reaction | DBP, μM | Remarks | |
|---|---|---|---|---|
| 2.0 | 25.0 | |||
| Vmax, RFU/min | 1.4 | 1.3 | 1.2 | Decreasing Vmax and increasing Km as DBP increased, indicating mixed inhibition |
| Km, µM | 1.6 | 3.8 | 4.0 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmud, F.; Lai, N.S.; How, S.E.; Gansau, J.A.; Mustaffa, K.M.F.; Leow, C.H.; Osman, H.; Sidek, H.M.; Embi, N.; Lee, P.-C. Bioactivities and Mode of Actions of Dibutyl Phthalates and Nocardamine from Streptomyces sp. H11809. Molecules 2022, 27, 2292. https://doi.org/10.3390/molecules27072292
Mahmud F, Lai NS, How SE, Gansau JA, Mustaffa KMF, Leow CH, Osman H, Sidek HM, Embi N, Lee P-C. Bioactivities and Mode of Actions of Dibutyl Phthalates and Nocardamine from Streptomyces sp. H11809. Molecules. 2022; 27(7):2292. https://doi.org/10.3390/molecules27072292
Chicago/Turabian StyleMahmud, Fauze, Ngit Shin Lai, Siew Eng How, Jualang Azlan Gansau, Khairul Mohd Fadzli Mustaffa, Chiuan Herng Leow, Hasnah Osman, Hasidah Mohd Sidek, Noor Embi, and Ping-Chin Lee. 2022. "Bioactivities and Mode of Actions of Dibutyl Phthalates and Nocardamine from Streptomyces sp. H11809" Molecules 27, no. 7: 2292. https://doi.org/10.3390/molecules27072292
APA StyleMahmud, F., Lai, N. S., How, S. E., Gansau, J. A., Mustaffa, K. M. F., Leow, C. H., Osman, H., Sidek, H. M., Embi, N., & Lee, P.-C. (2022). Bioactivities and Mode of Actions of Dibutyl Phthalates and Nocardamine from Streptomyces sp. H11809. Molecules, 27(7), 2292. https://doi.org/10.3390/molecules27072292