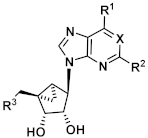
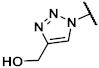
4.4. Synthetic Procedures
(1R,2R,3S,4S,5S)-1-{[(tert-Butyldiphenylsilyl)oxy]methyl-2,3-O-isopropylidenebicyclo [3.1.0]hexan-2,3,4-triol (
4). The procedure was modified according to reference [
16]. The (1
S,2
S,3
R)-4-{[(
tert-Butyldiphenylsilyl)oxy]methyl}-2,3-
O-isopropylidene-4-cyclopenten-1,2,3-triol (1.01 g, 2.37 mmol) was dissolved in dry CH
2Cl
2 (13 mL) under a nitrogen atmosphere. The reaction was cooled down to −18 °C with an ice/salt bath. Diethylzinc (1 mol/L in hexane, 2.60 mL, 2.60 mmol, 1.1 eq.) was added dropwise, and the mixture was stirred for 15 min. Diidomethane (0.22 mL, 2.73 mmol, 1.15 eq.) in dry CH
2Cl
2 (1.6 mL) was also added dropwise and the reaction was stirred for another 15 min. Both steps were repeated a second time. Then diethylzinc (1 mol/L in hexane, 2.60 mL, 2.60 mmol, 1.1 eq.) was added for the third time. After stirring for 15 min at −18 °C, the reaction was allowed to warm to rt and stirred overnight. The reaction was quenched with saturated NH
4Cl-solution and was extracted five times with CH
2Cl
2. The organic phase was dried over anh. Na
2SO
4 and concentrated in vacuo. The residue was purified by fc (cyclohexane:ethyl acetate = 7:1, Ø = 5 cm, l = 22 cm, V = 30 mL) to afford the product
4 as a colorless oil (R
f = 0.20, cyclohexane: ethyl acetate = 5:1), yield 0.90 g (86%). C
26H
34O
4Si (438.64 g/mol). Purity (HPLC: method B): > 99% (t
R = 18.94 min). Exact mass (APCI):
m/
z calculated for C
23H
27O
2Si [M-OH, -CO(CH
3)
2]
+ 363.1775, found 363.1777.
1H-NMR (600 MHz, CDCl
3) δ (ppm) = 7.66–7.60 (m, 4H, 2, 6-C
HPh), 7.46–7.34 (m, 6H, 3, 4, 5C
HPh), 5.00 (dd,
J = 6.9, 1.2 Hz, 1H, 2-C
H), 4.54 (td,
J = 6.9, 0.8 Hz, 1H, 3-C
H), 4.45 (dt,
J = 9.6, 6.1 Hz, 1H, 4C
H), 4.12 (q,
J = 7.2 Hz, 0.2H, C
H2, solvent: ethyl acetate), 4.07 (d,
J = 11.0 Hz, 1H, OC
HH), 3.29 (d,
J = 11.0 Hz, 1H, OCH
H), 2.33 (d,
J = 9.7 Hz, 1H, O
H), 2.04 (s, 0.3H, OC
H3, solvent: ethyl acetate), 1.61 (dt,
J = 9.3, 4.9 Hz, 1H, 5C
H), 1.54 (s, 3H, C(C
H3)
2), 1.31 (s, 3H, C(C
H3)
2), 1.26 (t,
J = 7.1 Hz, 0.5H, CH
2C
H3, solvent: ethyl acetate), 1.09 (t,
J = 5.0 Hz, 1H, 6-C
HH), 1.05 (s, 9H, C(C
H3)
3), 0.54 (ddt,
J = 8.8, 5.3, 1.1 Hz, 1H, 6-CH
H).
13C-NMR (151 MHz, CDCl
3) δ (ppm) = 135.7 (4C,
C-2, 6
Ph), 133.8, 133.7 (2C,
C-1
Ph), 129.9 (2C,
C-4
Ph), 127.8 (4C,
C-3, 5
Ph), 113.0 (1C,
C(CH
3)
2), 81.3 (1C,
C2), 79.9 (1C,
C3), 71.2 (1C,
C4), 65.4 (1C, O
CH
2), 35.7 (1C,
C-1), 33.0 (1C,
C-5), 27.0 (3C, C(
CH
3)
3), 26.3 (1C, C(
CH
3)
2), 24.8 (1C, C(
CH
3)
2), 19.4 (1C,
C(CH
3)
3), 10.5 (1C,
C-6). FT-IR (neat)
ṽ (cm
−1) = 2932, 2859 (C-H
aliphat.), 1470 (C = C
aromat.), 1107, 1080, 1042 (CO), 741, 702 (CH
aromat., out of plane).
7-Chloro-5-nitro-3H-imidazo[4,5-b]pyridine (7). An amount of 6-chloro-1-deazapurine (1.01 g, 6.5 mmol) and di-tert-butyl dicarbonate (3.02 g, 16.6 mmol, 2.5 eq.) were suspended in CH2Cl2 (20 mL). DMAP (0.04 g, 0.3 mmol, 0.1 eq.) was added and the mixture was stirred for 1.5 h. The reaction was quenched with silica gel and filtered through a pad of Celite®®. The mixture was concentrated in vacuo and the residue was redissolved in CH2Cl2 (20 mL). Tetrabutylammonium nitrate (3.07 g, 10.1 mmol, 1.5 eq.) was added and the mixture cooled to 0 °C with an ice bath. Trifluoroacetic anhydride (1.8 mL, 10.1 mmol, 1.5 eq.) was added dropwise and the reaction stirred for 2.5 h at rt. The solvent was evaporated, and the residue was dissolved in CH3OH (40 mL). The solution was refluxed overnight. The mixture was concentrated until the product was precipitating but was still properly suspended. After cooling the suspension in the fridge for 1 h, the solid was filtered off, washed with ice cold CH3OH, and dried in vacuo to afford the product 77 as a beige solid (Rf = 0.43, CH2Cl2: CH3OH = 9:1), yield 1.02 g (78%). C6H3ClN4O2 (198.57 g/mol). Melting point: 295.9 °C. Purity (HPLC: method B): > 99% (tR = 5.67 min). Exact mass (APCI): m/z calculated for C6H4ClN4O2 [M + H]+ 199.0007, found 199.0017. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.90 (s, 1H, 2-CH), 8.36 (s, 1H, 6-CH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 151.5 (1C, C-5), 149.6 (1C, C-2), 112.8 (1C, C-6); C-3a, C-7 and C-7a were not visible. FT-IR (neat) ṽ (cm−1) = 3098, 3013 (v C-Haromat.), 2743, 2677, 2612, 2554 (N-H), 1543, 1501 (C = Caromat.), 833, 880 (C-Haromat., out of plane).
(1R,2R,3S,4R,5S)-4-(5-Amino-7-chloro-3H-imidazo[4,5-b]pyridin-3-yl)-1-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,3-O-isopropylidenebicyclo[3.1.0]hexane-2,3-diol (10). The deazapurine 7 (0.29g, 1.48 mmol, 1.3 eq.) and triphenylphospane (0.61 g, 2.31 mmol, 2.1 eq.) were dissolved in THF (10 mL) under nitrogen atmosphere. DIAD (0.42 mL, 2.14 mmol, 1.9 eq.) was added dropwise at 0 °C. The mixture was stirred for 15 min at rt. A solution of the alcohol 4 (0.49 g, 1.11 mmol) in THF (13 mL) was added and the mixture was stirred at 70 °C for 1 h in the microwave at a power of 200 W. DIAD (0.42 mL, 2.14 mmol, 1.9 eq.) was added and the mixture was stirred again at 70 °C for 1 h in the microwave at a power of 200 W. DIAD (0.42 mL, 2.14 mmol, 1.9 eq.) was added and the mixture was stirred at 70 °C for 1 h in the microwave at a power of 200 W for the third time. The solvent was evaporated and the residue was purified by fc (cyclohexane: ethyl acetate = 5:1, Ø = 5 cm, l = 20 cm, V = 30 mL), but the intermediate was still heavily contaminated with DIAD and was directly dissolved in CH3OH (40 mL). Na2S2O4 (1.51 g, 8.64 mmol, 7.8 eq.) and 10 mL H2O were added. The mixture was stirred for 3d at rt. The solvent was evaporated and the residue was purified by fc (cyclohexane: ethyl acetate = 5:1 ⭢ 4:1 ⭢ 2:1, Ø = 5 cm, l = 20 cm, V = 30 mL) to afford the product 10 as colorless solid (Rf = 0.42, cyclohexane: ethyl acetate = 1:1), yield 0.138 g (21%). C32H37ClN4O3Si (589.21 g/mol). Melting point: 88.9 °C. Purity (HPLC: method B): > 99% (tR = 21.36 min).
Exact mass (LC-MS-ESI): m/z calculated for C32H38ClN4O3Si [M + H]+ 589.2396, found 589.2401. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.15 (s, 1H, 2-CHimidazopyridine), 7.63–7.58 (m, 4H, 2, 6CHPh), 7.47–7.43 (m, 2H, 4-CHPh), 7.41–7.37 (m, 4H, 3, 5-CHPh), 6.52 (s, 1H, 6-CHimidazopyridine), 6.26 (s, 2H, NH2), 5.33 (dd, J = 7.1, 1.3 Hz, 1H, 2-CH), 4.89 (s, 1H, 4-CH), 4.65 (dd, J = 7.1, 1.5 Hz, 1H, 3-CH), 4.10 (d, J = 10.8 Hz, 1H, OCHH), 3.64 (d, J = 10.8 Hz, 1H, OCHH), 1.66 (ddd, J = 9.2, 4.4, 1.5 Hz, 1H, 5CH), 1.44 (s, 3H, C(CH3)2), 1.19 (s, 3H, C(CH3)2), 1.02 (s, 9H, C(CH3)3), 0.97 (t, J = 4.7 Hz, 1H, 6CHH), 0.88 (ddd, J = 9.1, 5.0, 1.5 Hz, 1H, 6-CHH); the 1HNMR spectrum displayed small impurities in the range of about 5%. 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 157.4 (1C, C-5imidazopyridine), 145.9 (1C, C3aimidazopyridine), 138.3 (1C, C-2imidazopyridine), 135.1 (4C, C-2, 6Ph), 134.1 (1C, C7imidazopyridine), 132.7 (2C, C-1Ph), 129.9 (2C, C4Ph), 127.9 (4C, C3, 5Ph), 124.5 (1C, C7aimidazopyridine), 111.2 (1C, C(CH3)2), 104.1 (1C, C6imidazopyridine), 88.0 (1C, C3), 80.7 (1C, C2), 64.7 (1C, OCH2), 57.8 (1C, C-4), 38.1 (1C, C1), 29.9 (1C, C-5), 26.7 (3C, C(CH3)3), 25.9 (1C, C(CH3)2), 24.3 (1C, C(CH3)2), 18.8 (1C, C(CH3)3), 11.9 (1C, C-6); the 13C-NMR spectrum displayed small impurities in the range of about 5%. FT-IR (neat) ṽ (cm−1) = 3333 (N-H), 2932 (C-Haliphat.), 1601, 1570 (C = Caromat.), 1107, 1061, 1038 (C-O), 741, 702 (CHaromat., out of plane).
(1R,2R,3S,4R,5S)-4-(5-Amino-7-chloro-3H-imidazo[4,5-b]pyridin-3-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (11). Compound 10 (0.068 g, 0.12 mmol) was dissolved in CH3OH (1.6 mL), trifluoroacetic acid (0.20 mL) and H2O (0.20 mL) were added. The mixture was heated to 70 °C for 1 d. The solvent was evaporated and the residue was purified by semi-preparative HPLC (method B) to afford the alcohol 11 as a colorless solid (Rf = 0.2, CH2Cl2: CH3OH = 9:1), yield 0.012 g (34%). C13H15ClN4O3 (310.74 g/mol). Purity (HPLC: method D): 99% (tR = 6.97 min). Exact mass (LC-MS-ESI): m/z calculated for C13H16ClN4O3 [M + H]+ 311.0905, found 311.0908. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.32 (s, 1H, 2-CHimidazopyridine), 6.49 (s, 1H, 6-CHimidazopyridine), 6.23 (s, 2H, NH2), 5.02 (s, 2H, CH2OH, 3-OH), 4.71 (s, 1H, 4-CH), 4.57 (d, J = 6.3 Hz, 2H, 2-CH, 2-OH), 4.08 (d, J = 11.3 Hz, 1H, OCHH), 3.64 (d, J = 6.3 Hz, 1H, 3-CH), 3.12 (d, J = 11.3 Hz, 1H, OCHH), 1.41 (ddd, J = 8.7, 3.9, 1.4 Hz, 1H, 5-CH), 1.32 (t, J = 4.3 Hz, 1H, 6-CHH), 0.78 (ddd, J = 8.6, 4.7, 1.3 Hz, 1H, 6-CHH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 157.2 (1C, C-5imidazopyridine), 146.1 (1C, C-3aimidazopyridine), 138.5 (1C, C-2imidazopyridine), 133.9 (1C, C-7imidazopyridine), 124.4 (1C, C-7aimidazopyridine), 104.0 (1C, C-6imidazopyridine), 76.1 (1C, C-3), 70.2 (1C, C-2), 62.2 (1C, OCH2), 60.2 (1C, C-4), 36.3 (1C, C-1), 23.4 (1C, C-5), 11.1 (1C, C-6). FT-IR (neat) ṽ (cm−1) = 3321, 3206 (O-H), 2920 (C-Haliphat.), 1601, 1574 (C = Caromat.), 1061, 1003 (C-O).
(1R,2R,3S,4R,5S)-1-{[(tert-Butyldiphenylsilyl)oxy]methyl}-4-(5,7-dichloro-3H-imidazo[4,5-b]pyridin-3-yl)-2,3-O-isopropylidenebicyclo[3.1.0]hexane-2,3-diol (9). An amount of 2,6-dichloro-1-deazapurine (6, 0.16 g, 0.86 mmol, 1.2 eq.) and triphenylphospane (0.36 g, 1.37 mmol, 1.9 eq.) were dissolved in THF (7 mL) under nitrogen atmosphere. DIAD (0.27 mL, 1.38 mmol, 1.9 eq.) was added dropwise at 0 °C. The mixture was stirred for 30 min at rt. A solution of the alcohol 4 (0.32 g, 0.73 mmol) in THF (7 mL) was added and the mixture was stirred at 70 °C for 1 h in the microwave at a power of 200 W. DIAD (0.27 mL, 1.38 mmol, 1.9 eq.) was added and the mixture was stirred at 70 °C for 1 h in the microwave at a power of 200 W again. DIAD (0.27 mL, 1.38 mmol, 1.9 eq.) was added and the mixture was stirred at 70 °C for 1 h in the microwave at a power of 200 W for the third time. The solvent was evaporated and the residue was purified by fc (cyclohexane: ethyl acetate = 7:1, Ø = 5 cm, l = 20 cm, V = 30 mL) to afford the product 87 as a colorless solid (Rf = 0.26, cyclohexane: ethyl acetate = 5:1), yield 0.40 g (89%). C32H35Cl2N3O3Si (608.64 g/mol). Melting point: 75.7 °C. Purity (HPLC: method C): 98% (tR = 17.28 min). Exact mass (LC-MS-ESI): m/z calculated for C32H36Cl2N3O3Si [M + H]+ 608.1898, found 608.1899. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.67 (s, 1H, 2-CHimidazopyridine), 8.16 (s, 1H, 6CHimidazopyridine), 7.59 (ddt, J = 10.6, 6.8, 1.4 Hz, 4H, 2, 6CHPh), 7.46–7.40 (m, 2H, 4CHPh), 7.40–7.37 (m, 2H, 3, 5-CHPh), 7.36–7.32 (m, 2H, 3, 5-CHPh), 5.23 (dd, J = 7.1, 1.3 Hz, 1H, 2CH), 5.04 (s, 1H, 4CH), 4.77 (dd, J = 7.1, 1.6 Hz, 1H, 3-CH), 4.02 (d, J = 10.6 Hz, 1H, OCHH), 3.90 (d, J = 10.6 Hz, 1H, OCHH), 1.72 (ddd, J = 9.2, 4.5, 1.6 Hz, 1H, 5CH), 1.46 (s, 3H, C(CH3)2), 1.18 (s, 3H, C(CH3)2), 1.01 (s, 10H, 6-CHH, C(CH3)3), 0.96 (ddd, J = 9.2, 5.1, 1.5 Hz, 1H, 6-CHH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 145.7 (1C, C-3aimidazopyridine), 144.9 (1C, C2imidazopyridine), 144.2 (1C, C-5imidazopyridine), 135.2 (1C, C-7imidazopyridine), 135.1 (4C, C2, 6Ph), 132.8 (2C, C-1Ph), 132.2 (1C, C7aimidazopyridine), 129.8 (2C, C4Ph), 127.8 (2C, C3, 5Ph), 127.8 (2C, C3, 5Ph), 118.1 (1C, C6imidazopyridine), 111.4 (1C, C(CH3)2), 87.9 (1C, C3), 81.6 (1C, C2), 64.5 (1C, OCH2), 59.3 (1C, C-4), 38.3 (1C, C1), 29.4 (1C, C-5), 26.7 (3C, C(CH3)3), 25.9 (1C, C(CH3)2), 24.2 (1C, C(CH3)2), 18.8 (1C, C(CH3)3), 12.0 (1C, C-6). FT-IR (neat) ṽ (cm−1) = 2978, 2932 (C-Haliphat.), 1589, 1562 (C = Caromat.), 1065, 1042 (CO), 741, 702 (CHaromat., out of plane).
(1R,2R,3S,4R,5S)-4-(5,7-Dichloro-3H-imidazo[4,5-b]pyridin-3-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (12). Compound 9 (0.036 g, 0.06 mmol) was dissolved in CH3OH (1.1 mL), trifluoroacetic acid (0.14 mL) and H2O (0.14 mL) were added. The mixture was heated to 70 °C for 2 d. The solvent was evaporated and the residue was purified by fc (CH3CN: H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the alcohol 12 as a colorless solid (Rf = 0.34, CH2Cl2: CH3OH = 9:1), yield 0.016 g (84%). C13H13Cl2N3O3 (330.17 g/mol). Purity (HPLC: method B): 98% (tR = 5.95 min). Exact mass (APCI): m/z calculated for C13H14Cl2N3O3 [M + H]+ 330.0407, found 330.0406. 1H-NMR (600 MHz, CD3OD) δ (ppm) = 8.93 (s, 1H, 2-CHimidazopyridine), 7.47 (s, 1H, 6-CHimidazopyridin), 5.00 (s, 1H, 4-CH), 4.78 (dd, J = 6.6, 1.7 Hz, 1H, 2-CH), 4.28 (dd, J = 11.5, 0.9 Hz, 1H, OCHH), 3.91 (dt, J = 6.6, 1.3 Hz, 1H, 3-CH), 3.36 (d, J = 11.5 Hz, 1H, OCHH), 1.65 (ddd, J = 8.7, 3.9, 1.5 Hz, 1H, 5-CH), 1.58 (dd, J = 5.2, 3.9 Hz, 1H, 6-CHH), 0.78 (ddd, J = 8.7, 5.2, 1.8 Hz, 1H, 6-CHH). 13C-NMR (151 MHz, CD3OD) δ (ppm) = 147.2 (1C, C-3aimidazopyridine), 147.0 (1C, C-5imidazopyridine), 145.6 (1C, C-2imidazopyridine), 137.1 (1C, C-7imidazopyridine), 133.1 (1C, C-7aimidazopyridine), 119.9 (1C, C-6imidazopyridine), 77.5 (1C, C-3), 72.3 (1C, C-2), 64.3 (1C, OCH2), 63.6 (1C, C-4), 37.9 (1C, C-1), 24.5 (1C, C-5), 12.2 (1C, C-6). FT-IR (neat) ṽ (cm−1) = 3244 (O-H), 2978 (C-Haliphat.), 1593, 1562 (C = Caromat.), 1064, 1006 (C-O).
(1R,2R,3S,4R,5S)-4-[7-(Benzylamino)-5-chloro-3H-imidazo[4,5-b]pyridin-3-yl]-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (13). Compound 9 (0.26 g, 0.43 mmol) was dissolved in N-methyl-2-pyrrolidone (NMP, 5.5 mL). Benzylamine (0.78 mL, 7.14 mmol, 17 eq.) and N,N-diisopropylethylamine (DIPEA, 0.52 mL, 3.06 mmol, 7 eq.) were added. The mixture was stirred at 200 °C for 1 h in the microwave at a power of 200 W. The solution was directly purified by fc (CH3CN: H2O = 30:70 ⭢ 100:0, 50 mL/min, Biotage®® SNAP C18, 120 g, V = 20 mL) to afford the protected product as a colorless solid (Rf = 0.44, cyclohexane: ethyl acetate = 1:1), yield 0.25 g (86%). C39H43ClN4O3Si (679.33 g/mol). Melting point: 81.4 °C. Purity (HPLC: method C): 97% (tR = 17.58 min). Exact mass (LC-MS-ESI): m/z calculated for C39H44ClN4O3Si [M + H]+ 678.2866, found 679.2898.1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 8.25 (s, 1H, 2-CHimidazopyridine), 7.92 (t, J = 6.5 Hz, 1H, NH), 7.64–7.57 (m, 4H, 2, 6-CHPh), 7.47–7.28 (m, 10H, 3, 4, 5-CHPh, 2, 3, 5, 6-CHbenzyl), 7.26–7.19 (m, 1H, 4-CHbenzyl), 6.29 (s, 1H, 6-CHimidazopyridine), 5.26 (dd, J = 7.1, 1.4 Hz, 1H, 2-CH), 4.92 (s, 1H, 4-CH), 4.65 (dd, J = 7.2, 1.5 Hz, 1H, 3-CH), 4.62 (s, 2H, CH2 benzyl), 4.05 (d, J = 10.7 Hz, 1H, OCHH), 3.76 (d, J = 10.7 Hz, 1H, OCHH), 1.65 (ddd, J = 9.3, 4.4, 1.5 Hz, 1H, 5-CH), 1.44 (s, 3H, C(CH3)2), 1.18 (s, 3H, C(CH3)2), 1.01 (s, 10H, 6-CHH, C(CH3)3), 0.90 (ddd, J = 9.2, 5.1, 1.4 Hz, 1H, 6-CHH). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 147.8 (1C, C-7imidazopyridine), 146.0 (1C, C-5imidazopyridine), 144.8 (1C, C-3aimidazopyridine), 139.2 (1C, C-1benzyl), 138.7 (1C, C-2imidazopyridine), 135.0 (4C, C-2, 6Ph), 132.8 (2C, C-1Ph), 129.8 (2C, C-4Ph), 128.4 (2C, C-3, 5benzyl), 127.8 (4C, C-3, 5Ph), 126.9 (2C, C-2, 6benzyl), 126.8 (1C, C-4benzyl), 122.2 (1C, C-7aimidazopyridine), 111.3 (1C, C(CH3)2), 98.0 (1C, C-6imidazopyridine), 88.2 (1C, C-3), 81.2 (1C, C-2), 64.7 (1C, OCH2), 58.2 (1C, C-4), 45.3 (1C, CH2 benzyl), 38.2 (1C, C-1), 29.8 (1C, C-5), 26.7 (3C, C(CH3)3), 25.9 (1C, C(CH3)2), 24.2 (1C, C(CH3)2), 18.8 (1C, C(CH3)3), 12.0 (1C, C-6). FT-IR (neat) ṽ (cm−1) = 2978, 2932 (C-Haliphat.), 1608, 1582 (C = Caromat.), 1111, 1069, 1038 (C-O), 737, 698 (C-Haromat., out of plane).
Next, the compound (0.080 g, 0.12 mmol) was dissolved in CH3OH (2.5 mL), trifluoroacetic acid (0.32 mL) and H2O (0.32 mL) were added. The mixture was heated to 70 °C for 2 d. The solvent was evaporated and the residue was purified by fc (CH3CN: H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the alcohol 13 as a colorless solid (Rf = 0.20, CH2Cl2: CH3OH = 95:5), yield 0.034 g (72%). C20H21ClN4O3 (400.86 g/mol). Purity (HPLC: method B): 99% (tR = 9.53 min). Exact mass (LC-MS-ESI): m/z calculated for C20H22ClN4O3 [M + H]+ 401.1375, found 401.1367. 1H-NMR (600 MHz, CD3OD) δ (ppm) = 8.47 (s, 1H, 2-CHimidazopyridine), 7.39–7.35 (m, 2H, 2, 6-CHbenzyl), 7.32 (dd, J = 8.5, 6.8 Hz, 2H, 3, 5-CHbenzyl), 7.28–7.23 (m, 1H, 4-CHbenzyl), 6.36 (s, 1H, 6-CHimidazopyridine), 5.48 (s, 0.2H, CH2Cl2, solvent: dichloromethane), 4.83 (s, 1H, 4-CH), 4.76 (dd, J = 6.7, 1.7 Hz, 1H, 2-CH), 4.57 (s, 2H, CH2 benzyl), 4.27 (dd, J = 11.6, 0.9 Hz, 1H, OCHH), 3.84 (dt, J = 6.7, 1.2 Hz, 1H, 3-CH), 3.32 (d, J = 11.5 Hz, 1H, OCHH), 1.62 (ddd, J = 8.8, 3.9, 1.5 Hz, 1H, 5-CH), 1.55 (dd, J = 5.2, 3.9 Hz, 1H, 6-CHH), 0.74 (ddd, J = 8.7, 5.2, 1.8 Hz, 1H, 6-CHH). 13C-NMR (151 MHz, CD3OD) δ (ppm) = 149.2 (1C, C-7imidazopyridine), 148.6 (1C, C-5imidazopyridine), 146.1 (1C, C-3aimidazopyridine), 140.3 (1C, C-2imidazopyridine), 139.4 (1C, C-1benzyl), 129.7 (2C, C-3, 5benzyl), 128.4 (2C, C-2, 6benzyl), 128.3 (1C, C-4benzyl), 123.4 (1C, C-7aimidazopyridine), 99.7 (1C, C-6imidazopyridine), 77.6 (1C, C-3), 72.3 (1C, C-2), 64.4 (1C, OCH2), 63.3 (1C, C-4), 47.5 (1C, CH2 benzyl), 38.0 (1C, C-1), 24.4 (1C, C-5), 12.2 (1C, C-6). FT-IR (neat) ṽ (cm−1) = 3325 (O-H), 2978 (C-Haliphat.), 1608, 1578 (C = Caromat.), 1119, 1072 (C-O), 733, 694 (C-Haromat., out of plane).
(1R,2R,3S,4R,5S)-4-[7-(Benzylamino)-5-methylthio-3H-imidazo[4,5-b]pyridin-3-yl]-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (14). Compound 13 (0.10 g, 0.15 mmol) was dissolved in DMF (5.5 mL). NaSCH3 (0.21 g, 3.04 mmol, 20 eq.) was added. The mixture was stirred at 90 °C for 2 h in the microwave at a power of 200 W. The solvent was evaporated and the residue was purified by fc (CH3CN: H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL). The purified intermediate was dissolved in CH3OH (1 mL), trifluoroacetic acid (0.12 mL) and H2O (0.12 mL) were added. The mixture was heated to 70 °C for 4 h. The solvent was evaporated and the residue was purified by fc (CH3CN: H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the product 93 as a colorless solid alongside an impurity due to the incomplete conversion during formation of the methylthio ether. The mixture was dissolved in DMF (0.5 mL) and NaSCH3 (0.041 g, 0.58 mmol, 4 eq.) was added. The mixture was stirred at 90 °C for 1 h in the microwave at a power of 200 W. The solvent was evaporated and the residue was purified by fc (CH3CN: H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the product 93 as a beige solid with a small contamination with starting material (Rf = 0.29, CH2Cl2: CH3OH = 9:1), yield 0.010 g (16%). C21H24N4O3S (412.51 g/mol). Purity (HPLC: method B): 93% (tR = 9.78 min). Exact mass (LC-MS-ESI): m/z calculated for C21H25N4O3S [M + H]+ 413.1642, found 413.1643. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 8.28 (s, 1H, 2-CHimidazopyridine), 7.41 (t, J = 6.4 Hz, 1H, NH), 7.38–7.27 (m, 4H, 2, 3, 5, 6-CHbenzyl), 7.25–7.19 (m, 1H, 4CHbenzyl), 6.12 (s, 1H, 6CHimidazopyridine), 5.10 (s, 1H, 3-OH), 4.98 (s, 1H, CH2OH), 4.79 (s, 1H, 4-CH), 4.58 (s, 3H, 2CH, CH2 benzyl), 4.46 (s, 1H, 2-OH), 4.06 (d, J = 11.3 Hz, 1H, OCHH), 4.03 (q, J = 7.1 Hz, 0.3H, CH2, solvent: ethyl acetate), 3.68 (d, J = 6.5 Hz, 1H, 3-CH), 3.15 (d, J = 11.4 Hz, 1H, OCHH), 2.45 (s, 3H, SCH3), 2.08 (s, 0.1H, CH3CN, solvent: acetonitrile), 1.99 (s, 0.3H, OCH3, solvent: ethyl acetate), 1.43 (ddd, J = 8.7, 4.6, 1.4 Hz, 1H, 5-CH), 1.33 (t, J = 4.3 Hz, 1H, 6CHH), 1.17 (t, J = 7.1 Hz, 0.1H, CH2CH3, solvent: ethyl acetate), 0.59 (ddd, J = 8.6, 4.7, 1.6 Hz, 1H, 6-CHH); the 1HNMR spectrum displayed small impurities in the range of about 5% assigned to compound 13. 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 153.9 (1C, C-5imidazopyridine), 146.1 (1C, C7imidazopyridine), 146.0 (1C, C-3aimidazopyridine), 139.7 (1C, C-1benzyl), 137.2 (1C, C2imidazopyridine), 128.3 (2C, C-3, 5benzyl), 127.0 (2C, C-2, 6benzyl), 126.7 (1C, C4benzyl), 120.9 (1C, C7aimidazopyridine), 95.9 (1C, C6imidazopyridine), 76.2 (1C, C3), 70.3 (1C, C2), 62.3 (1C, OCH2), 60.2 (1C, C-4), 45.4 (1C, CH2 benzyl), 36.4 (1C, C-1), 23.3 (1C, C-5), 13.1 (1C, SCH3), 11.1 (1C, C-6); the 13C-NMR spectrum displayed small impurities in the range of about 5% assigned to compound 13. FT-IR (neat) ṽ (cm−1) = 3302 (O-H), 2978 (C-Haliphat.), 1605, 1582 (C = Caromat.), 1115, 1072, 1006 (C-O), 737, 698 (CHaromat., out of plane).
(1R,2R,3S,4R,5S)-4-{5-Chloro-7-[(4-methoxy)benzylamino]-3H-imidazo[4,5-b]pyridin-3-yl}-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (15). Compound 9 (0.41 g, 0.68 mmol) was dissolved in NMP (8 mL). 4-methoxybenzylamine (1.3 mL, 9.95 mmol, 15 eq.) and DIPEA (0.56 mL, 3.29 mmol, 4.5 eq.) were added. The mixture was stirred at 200 °C for 5 min in the microwave at a power of 200 W and was directly purified by fc (CH3CN: H2O = 5:95 ⭢ 100:0, 50 mL/min, Biotage®® SNAP C18, 120 g, V = 20 mL) to afford the acetonide-protected product (1R,2R,3S,4R,5S)-4-(7-Amino-5-chloro-3H-imidazo[4,5-b]pyridin-3-yl)-1-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,3-O-isopropylidenebicyclo[3.1.0]hexane-2,3-diol as a colorless solid (Rf = 0.35, cyclohexane: ethyl acetate = 1:1), yield 0.39 g (80%). C40H45ClN4O4Si (708.29 g/mol). Melting point: 82.3 °C. Purity (HPLC: method C): 96% (tR = 17.51 min). Exact mass (LC-MS-ESI): m/z calculated for C40H46ClN4O4Si [M + H]+ 709.2971, found 709.2958. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 8.24 (s, 1H, 2-CHimidazopyridine), 7.85 (t, J = 5.2 Hz, 1H, NH), 7.637.56 (m, 4H, 2, 6-CHPh), 7.46–7.32 (m, 6H, 3, 4, 5-CHPh), 7.32–7.27 (m, 2H, 2, 6CHbenzyl), 6.91–6.86 (m, 2H, 3, 5-CHbenzyl), 6.29 (s, 1H, 6CHimidazopyridine), 5.25 (dd, J = 7.3, 1.2 Hz, 1H, 2CH), 4.91 (s, 1H, 4CH), 4.65 (dd, J = 7.1, 1.5 Hz, 1H, 3-CH), 4.53 (s, 2H, CH2 benzyl), 4.04 (d, J = 10.7 Hz, 1.2H, OCHH, CH2, solvent: ethyl acetate), 3.76 (d, J = 10.7 Hz, 1H, OCHH), 3.71 (s, 3H, OCH3), 2.07 (s, 0.1H, CH3CN, solvent: acetonitrile), 1.99 (s, 0.2H, OCH3, solvent: ethyl acetate), 1.65 (ddd, J = 9.3, 4.5, 1.5 Hz, 1H, 5CH), 1.45 (s, 3H, C(CH3)2), 1.18 (s, 3.2H, C(CH3)2, CH2CH3, solvent: ethyl acetate), 1.02 (s, 9H, C(CH3)3), 0.98 (t, J = 4.8 Hz, 1H, 6-CHH), 0.90 (ddd, J = 9.2, 5.2, 1.4 Hz, 1H, 6-CHH); the 1HNMR spectrum displayed small impurities in the range of about 5%.13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 158.2 (1C, C4benzyl), 147.8 (1C, C7imidazopyridine), 146.0 (1C, C5imidazopyridine), 144.7 * (1C, C-3aimidazopyridine), 138.6 (1C, C2imidazopyridine), 135.0 (4C, C-2, 6Ph), 132.8 (2C, C-1Ph), 131.0 * (1C, C-1benzyl), 129.8 (2C, C4Ph), 128.3 (2C, C2, 6benzyl), 127.8 (4C, C3, 5Ph), 122.2 (1C, C7aimidazopyridine), 113.8 (2C, C3imidazopyridine, 5benzyl), 111.3 (1C, C(CH3)2), 98.0 * (1C, C6imidazopyridine), 88.2 (1C, C3), 81.2 (1C, C2), 64.8 (1C, OCH2, 58.2 (1C, C-4), 55.0 (1C, OCH3), 44.7 (1C, CH2 benzyl), 38.2 (1C, C-1), 29.8 (1C, C-5), 26.7 (3C, C(CH3)3), 25.9 (1C, C(CH3)2), 24.3 (1C, C(CH3)2), 18.8 (1C, C(CH3)3), 12.0 (1C, C-6); *: C-3aimidazopyridine, C-6imidazopyridine and C-1benzyl could only be seen in 2D NMR spectra. FT-IR (neat) ṽ (cm−1) = 3071 (v C-Haromat.), 2932 (C-Haliphat.), 1609, 1582 (C = Caromat.), 1111, 1065, 1038 (C-O), 741, 702 (CHaromat., out of plane).
Next, the acetonide-protected compound (0.050 g, 0.07 mmol) was dissolved in CH3OH (1.6 mL), trifluoroacetic acid (0.20 mL) and H2O (0.20 mL) were added. The mixture was heated to 70 °C for 1 d. The solvent was evaporated and the residue was purified by fc (CH3CN: H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the alcohol 95 as a colorless solid (Rf = 0.19, CH2Cl2: CH3OH = 95:5), yield 0.027 g (89%). C21H23ClN4O4 (430.89 g/mol). Purity (HPLC: method B): 98% (tR = 9.67 min). Exact mass (LC-MS-ESI): m/z calculated for C21H24ClN4O4 [M + H]+ 431.1481, found 431.1486.1H-NMR (400 MHz, DMSO-d6 δ (ppm) = 8.44 (s, 1H, 2-CHimidazopyridine), 7.73 (t, J = 6.8 Hz, 1H, NH), 7.31–7.25 (m, 2H, 2, 6-CHbenzyl), 6.94–6.83 (m, 2H, 3, 5-CHbenzyl), 6.30 (s, 1H, 6-CHimidazopyridine), 5.75 (s, 0.4H, CH2Cl2, solvent: dichloromethane), 4.72 (s, 1H, 4-CH), 4.58 (dd, J = 6.5, 1.5 Hz, 3H, 2-CH, CH2 benzyl), 4.09 (d, J = 11.4 Hz, 1H, OCHH), 3.71 (s, 3H, OCH3), 3.66 (dt, J = 6.4, 1.2 Hz, 1H, 3-CH), 3.15 (d, J = 11.4 Hz, 1H, OCHH), 1.44 (ddd, J = 8.8, 3.9, 1.4 Hz, 1H, 5-CH), 1.34 (t, J = 4.3 Hz, 1H, 6-CHH), 0.60 (ddd, J = 8.6, 4.7, 1.5 Hz, 1H, 6-CHH). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 158.2 (1C, C-4benzyl), 147.7 (1C, C-7imidazopyridine), 145.9 (1C, C-5imidazopyridine), 145.0 (1C, C-3aimidazopyridine), 138.5 (1C, C-2imidazopyridine), 131.0 (1C, C-1benzyl), 128.3 (2C, C-2, 6benzyl), 121.8 (1C, C-7aimidazopyridine), 113.8 (2C, C-3, 5benzyl), 98.0 (1C, C-6imidazopyridine), 76.0 (1C, C-3), 70.3 (1C, C-2), 62.2 (1C, OCH2), 60.5 (1C, C-4), 55.0 (1C, OCH3), 54.9 (0.2C, CH2Cl2, solvent: dichloromethane), 44.9 (1C, CH2 benzyl), 36.4 (1C, C-1), 23.2 (1C, C-5), 11.0 (1C, C-6). FT-IR (neat) ṽ (cm−1) = 3318 (O-H), 2920 (C-Haliphat.), 1609 (C = Caromat.), 1119, 1080, 1030 (C-O), 737 (C-Haromat., out of plane).
(1R,2R,3S,4R,5S)-4-(7-Amino-5-chloro-3H-imidazo[4,5-b]pyridin-3-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (16). The acetonide-protected intermediate (1R,2R,3S,4R,5S)-4-(7-amino-5-chloro-3H-imidazo[4,5-b]pyridin-3-yl)-1-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,3-O-isopropylidenebicyclo[3.1.0]hexane-2,3-diol (0.26 g, 0.43 mmol) was dissolved in CH2Cl2 (2.7 mL). H2O (0.3 mL) and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 0.038 g, 0.17 mmol, 1.5 eq.) were added. The mixture was stirred at rt overnight. H2O was added and the mixture was extracted three times with CH2Cl2. The solvent was evaporated and the residue was purified by fc (CH3CN: H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP C18, 12 g, V = 20 mL) to afford the amine as a beige solid (Rf = 0.19, cyclohexane: ethyl acetate = 1:1), yield 0.049 g (74%). C32H37ClN4O3Si (588.23 g/mol). Melting point: 101.0 °C. Purity (HPLC: method B): 98% (tR = 22.35 min). Exact mass (LC-MS-ESI): m/z calculated for C32H38ClN4O3Si [M + H]+ 589.2396, found 589.2395. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.21 (s, 1H, 2-CHimidazopyridine), 7.63–7.58 (m, 4H, 2, 6CHPh), 7.47–7.42 (m, 2H, 4-CHPh), 7.41–7.37 (m, 4H, 3, 5-CHPh), 6.80 (s, 2H, NH2), 6.38 (s, 1H, 6-CHimidazopyridine), 5.75 (s, 0.1H, CH2Cl2, solvent: dichloromethane), 5.27 (dd, J = 7.0, 1.3 Hz, 1H, 2-CH), 4.90 (s, 1H, 4CH), 4.64 (dd, J = 7.2, 1.5 Hz, 1H, 3-CH), 4.04 (d, J = 10.7 Hz, 1H, OCHH), 3.74 (d, J = 10.7 Hz, 1H, OCHH), 1.65 (ddd, J = 9.3, 4.4, 1.5 Hz, 1H, 5CH), 1.45 (s, 3H, C(CH3)2), 1.19 (s, 3H, C(CH3)2), 1.02 (s, 9H, C(CH3)3), 0.98 (t, J = 4.8 Hz, 1H, 6-CHH), 0.89 (ddd, J = 9.2, 5.1, 1.5 Hz, 1H, 6CHH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 148.9 (1C, C-7imidazopyridine), 145.7 (1C, C5imidazopyridine), 145.3 (1C, C-3aimidazopyridine), 138.7 (1C, C-2imidazopyridine), 135.1 (4C, C2, 6Ph), 132.8 (2C, C-1Ph), 129.9 (2C, C4Ph), 127.9 (4C, C3, 5Ph), 122.1 (1C, C7aimidazopyridine), 111.3 (1C, C(CH3)2), 100.5 (1C, C6imidazopyridine), 88.3 (1C, C3), 81.2 (1C, C2), 64.9 (1C, OCH2), 58.1 (1C, C-4), 38.3 (1C, C-1), 29.9 (1C, C-5), 26.8 (3C, C(CH3)3), 25.9 (1C, C(CH3)2), 24.3 (1C, C(CH3)2), 18.9 (1C, C(CH3)3), 12.1 (1C, C-6). FT-IR (neat) ṽ (cm−1) = 3360 (N-H), 2978, 2932 (C-Haliphat.), 1624, 1601 (C = Caromat.), 1111, 1065, 1038 (C-O), 741, 702 (CHaromat., out of plane).
Next the acetonide-protected intermediate (0.051 g, 0.09 mmol) was dissolved in CH3OH (1.6 mL), trifluoroacetic acid (0.20 mL) and H2O (0.20 mL) were added. The mixture was heated to 70 °C for 1 d. The solvent was evaporated and the residue was purified by semi-preparative HPLC (method B) to afford the alcohol 16 as a colorless solid (Rf = 0.24, CH2Cl2: CH3OH = 9:1), yield 0.016 g (60%). C13H15ClN4O3 (310.74 g/mol). Purity (HPLC: method B): > 99% (tR = 4.11 min). Exact mass (LC-MS-ESI): m/z calculated for C13H16ClN4O3 [M + H]+ 311.0905, found 311.0905. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.45 (s, 1H, 2-CHimidazopyridine), 6.76 (s, 2H, NH2), 6.39 (s, 1H, 6-CHimidazopyridine), 4.72 (s, 1H, 4-CH), 4.57 (dd, J = 6.5, 1.6 Hz, 1H, 2-CH), 4.08 (d, J = 11.4 Hz, 1H, OCHH), 3.66 (d, J = 6.4 Hz, 1H, 3-CH), 3.14 (d, J = 11.4 Hz, 1H, OCHH), 1.44 (ddd, J = 8.8, 3.9, 1.4 Hz, 1H, 5-CH), 1.34 (t, J = 4.3 Hz, 1H, 6-CHH), 0.60 (ddd, J = 8.5, 4.7, 1.6 Hz, 1H, 6-CHH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 148.6 (1C, C-5imidazopyridine), 145.6 (1C, C-7imidazopyridine), 145.4 (1C, C-3aimidazopyridine), 138.7 (1C, C-2imidazopyridine), 121.4 (1C, C-7aimidazopyridine), 100.5 (1C, C-6imidazopyridine), 76.0 (1C, C-3), 70.3 (1C, C-2), 62.2 (1C, OCH2), 60.6 (1C, C-4), 36.4 (1C, C-1), 23.2 (1C, C-5), 11.1 (1C, C-6). FT-IR (neat) ṽ (cm−1) = 3345, 3217 (O-H), 2924 (C-Haliphat.), 1632, 1601 (C = Caromat.), 1115, 1069, 1007 (C-O).
(1R,2R,3S,4R,5S)-4-(7-Amino-5-methylthio-3H-imidazo[4,5-b]pyridin-3-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (17). An amount of (1R,2R,3S,4R,5S)-4-(7-Amino-5-chloro-3H-imidazo[4,5-b]pyridin-3-yl)-1-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,3-O-isopropylidenebicyclo[3.1.0]hexane-2,3-diol (0.030 g, 0.05 mmol) was dissolved in DMF (1.5 mL). NaSCH3 (0.078 g, 1.11 mmol, 22 eq.) was added. The mixture was stirred at 90 °C for 2 h in the microwave at a power of 200 W. H2O was added and the reaction was extracted three times with ethyl acetate. The organic phase was dried over anh. Na2SO4, filtered and concentrated in vacuo. Due to incomplete conversion, the residue was dissolved in DMF (1.5 mL), and NaSCH3 (0.054 g, 0.77 mmol, 15 eq.) was added. The mixture was stirred at 90 °C for 1 h in the microwave at a power of 200 W. H2O was added and the reaction was extracted three times with ethyl acetate. The organic phase was dried over anh. Na2SO4, filtered and concentrated in vacuo. The residue was purified by fc (CH3CN: H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP C18, 12 g, V = 20 mL) to afford the methylthio ether as a colorless oil (Rf = 0.23, CH2Cl2: CH3OH = 95:5), yield 0.011 g (58%). C17H22N4O3S (362.45 g/mol). Melting point: 109.2 °C. Purity (HPLC: method B): 94% (tR = 9.14 min). Exact mass (LC-MS-ESI): m/z calculated for C17H23N4O3S [M + H]+ 363.1485, found 363.1493. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.15 (s, 1H, 2-CHimidazopyridine), 6.39 (s, 2H, NH2), 6.27 (s, 1H, 6-CHimidazopyridine), 5.75 (s, 0.4H, CH2Cl2, solvent: dichloromethane), 5.22 (dd, J = 7.0, 1.4 Hz, 1H, 2-CH), 4.98 (t, J = 5.2 Hz, 1H, OH), 4.94 (s, 1H, 4-CH), 4.55 (dd, J = 7.1, 1.5 Hz, 1H, 3CH), 3.86 (dd, J = 11.7, 3.2 Hz, 1H, OCHH), 3.36–3.31 (m, 1H, OCHH), 2.49 (s, 3H, SCH3), 1.61 (ddd, J = 9.2, 4.4, 1.5 Hz, 1H, 5-CH), 1.44 (s, 3.3H, C(CH3)2, 1.17 (s, 3.3H, C(CH3)2, 0.98 (t, J = 4.8 Hz, 1H, 6-CHH), 0.880.84 (m, 1H, 6-CHH): 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 153.9 (1C, C-5imidazopyridine), 147.1 (1C, C7imidazopyridine), 146.1 (1C, C-3aimidazopyridine), 137.5 (1C, C-2imidazopyridine), 121.1 (1C, C7aimidazopyridine), 111.2 (1C, C(CH3)2), 98.3 (1C, C6imidazopyridine), 88.5 (1C, C3), 80.8 (1C, C2), 62.6 (1C, OCH2), 57.7 (1C, C4), 54.9 (0.2C, CH2Cl2, solvent: dichloromethane), 38.7 (1C, C1), 29.8 (1C, C5), 25.9 (1C, C(CH3)2), 24.2 (1C, C(CH3)2), 13.2 (1C, SCH3), 12.6 (1C, C6): FT-IR (neat) ṽ (cm−1) = 3352, 3202 (O-H), 2986, 2924 (C-Haliphat.), 1624, 1582 (C = Caromat.), 1057, 1026, 1015 (C-O).
Next, the acetonide-protected methylthioether intermediate (0.025 g, 0.07 mmol) was dissolved in CH3OH (1.6 mL) and trifluoroacetic acid (0.20 mL) and H2O (0.20 mL) were added. The mixture was heated to 50 °C overnight. The solvent was evaporated and the residue was purified by semi-preparative HPLC (method C) to afford the product 17 as a colorless solid (Rf = 0.22, CH2Cl2: CH3OH = 9:1), yield 0.012 g (55%). C14H18N4O3S (322.38 g/mol). Purity (HPLC: method D): 99% (tR = 10.45 min). Exact mass (LC-MS-ESI): m/z calculated for C14H19N4O3S [M + H]+ 323.1172, found 323.1167. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.43 (s, 1H, 2-CHimidazopyridine), 6.46 (s, 2H, NH2), 6.31 (s, 1H, 6-CHimidazopyridine), 4.81 (s, 1H, 4-CH), 4.57 (dd, J = 6.6, 1.5 Hz, 1H, 2-CH), 4.06 (d, J = 11.4 Hz, 1H, OCHH), 3.70 (d, J = 6.4 Hz, 1H, 3-CH), 3.15 (d, J = 11.4 Hz, 1H, OCHH), 2.54 (s, 0.4H, CH3, solvent: DMSO), 2.50 (s, 3H, SCH3), 2.07 (s, 0.1H, CH3CN, solvent: acetonitrile), 1.45 (ddd, J = 8.7, 3.9, 1.3 Hz, 1H, 5CH), 1.34 (t, J = 4.3 Hz, 1H, 6-CHH), 0.61 (ddd, J = 8.6, 4.6, 1.5 Hz, 1H, 6-CHH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 154.2 (1C, C-5imidazopyridine), 146.5 (1C, C7imidazopyridine), 145.9 (1C, C3aimidazopyridine), 137.2 (1C, C-2imidazopyridine), 119.4 (1C, C7aimidazopyridine), 98.7 (1C, C6imidazopyridine), 76.0 (1C, C3), 70.2 (1C, C2), 62.2 (1C, OCH2), 60.4 (1C, C-4), 40.5 (0.1C, CH3, solvent: DMSO), 36.4 (1C, C1), 23.2 (1C, C-5), 13.1 (1C, SCH3), 11.2 (1C, C6). FT-IR (neat) ṽ (cm−1) = 3341, 3217 (O-H), 2920 (C-Haliphat.), 1674, 1628, 1597 (C = Caromat.), 1119, 1069, 1011 (C-O).
5-Chloro-6-nitro-3H-imidazo[4,5-b]pyridine (19). 2-Chloro-1-deazapurine (0.10 g, 0.66 mmol) and di-tert-butyl dicarbonate (0.20 g, 0.91 mmol, 1.4 eq.) were suspended in CH2Cl2 (1 mL). A catalytic amount of DMAP (~1 mg) was added and the mixture was stirred for 2.5 h. The reaction was quenched with silica gel and filtered through a pad of Celite®®. The mixture was concentrated in vacuo and the residue was redissolved in CH2Cl2 (2 mL). Tetrabutylammonium nitrate (0.30 g, 0.98 mmol, 1.5 eq.) was added and the mixture cooled to 0 °C with an ice bath. Trifluoroacetic anhydride (0.14 mL, 0.99 mmol, 1.5 eq.) was added dropwise and the reaction stirred for 5 h at rt and under reflux overnight. The solvent was evaporated, and the residue was purified by fc (CH2Cl2:CH3OH = 97.5:2.5 ⭢ 96.5:3.5 ⭢ 95.5, Ø = 3 cm, l = 24 cm, V = 10 mL) to afford the product 19 as light brown solid (76%). C6H3ClN4O2 (198.57 g/mol). 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 8.87 (s, 1H, 7-CH), 8.80 (s, 1H, 2-CH), 3.17 (s, 0.4H, CH3OH, solvent: methanol). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 149.6 (1C, C-2), 139.9 (1C, C-5), 135.6 (1C, C-6), 48.6 (0.1H, CH3OH, solvent: methanol); C-3a, C-7 and C-7a were not visible.
Crystal data for C6H3ClN4O2 (M = 198.57 g/mol): orthorhombic, Pbca (No. 61), a = 11.3385(3) Å, b = 6.5407(2) Å, c = 20.0740(7) Å, V = 1488.72(8) Å3, Z = 8, 1.772 mg/m3, T = 173(2) K, μ(CuKα) = 0.479 mm−1, final R indices [I > 2σ(I)] R1 = 0.0316, wR2 = 0.0798, R indices (all data) R1 = 0.0355, wR2 = 0.0827
7-Chloro-5-nitro-3-tosyl-3H-imidazo[4,5-b]pyridine (20). Compound 7 (0.50 g, 2.5 mmol) was suspended in CH2Cl2 (20 mL). Tosyl chloride (0.97 g, 5.1 mmol, 2 eq.) and DIPEA (0.88 mL, 5.1 mmol, 2 eq.) were added and the mixture stirred for 3 h at rt. The reaction was neutralized with saturated NH4Cl solution and was extracted with CH2Cl2. The organic phase was dried over anh. Na2SO4 and concentrated in vacuo. The residue was purified by fc (cyclohexane: ethyl acetate = 5:1 ⭢ 2:1, Ø = 6 cm, l = 22 cm, V = 65 mL). The mixed fractions were purified again using fc (cyclohexane: ethyl acetate = 5:1, Ø = 6 cm, l = 22 cm, V = 65 mL) to afford the product 20 as a colorless solid (Rf = 0.23, cyclohexane: ethyl acetate = 3:1), yield 0.81 g (91%). C13H9ClN4O4S (352.75 g/mol). Melting point: 200.2 °C. Purity (HPLC: method B): > 99% (tR = 15.72 min). Exact mass (APCI): m/z calculated for C13H10ClN4O4S [M + H]+ 353.0106, found 353.0105. 1H-NMR (400 MHz, CD3CN) δ (ppm) = 8.88 (s, 1H, 2-CH), 8.39 (s, 1H, 6-CH), 8.228.18 (m, 2H, 2, 6-CHtosyl), 7.48–7.43 (m, 2H, 3, 5-CHtosyl), 2.40 (s, 3H, CH3). 13C-NMR (101 MHz, CD3CN) δ (ppm) = 153.6 (1C, C-5), 148.9 (1C, C4tosyl), 147.9 (1C, C-2), 144.0 (1C, C-3a), 139.1 (1C, C7a), 138.5 (1C, C-7), 133.9 (1C, C-1tosyl), 131.3 (2C, C3, 5tosyl), 130.0 (2C, C-2, 6tosyl), 116.7 (1C, C6), 21.8 (1C, CH3). FT-IR (neat) ṽ (cm−1) = 3117, 3102 (v C-Haromat.), 2978 (C-Haliphat.), 1598, 1555 (C = Caromat.), 1373, 1327 (NO2), 1176 (S = O), 837, 814 (CHaromat., out of plane).
N,N-Dibenzyl-N’-[4-chloro-2-(4-methylphenyl)sulfonamido-6-nitropyridin-3-yl]formimidamide (21). Compound 20 (0.074 g, 0.21 mmol) was dissolved in CH2Cl2 (2 mL). Dibenzylamine (0.40 mL, 2.08 mmol, 10 eq.) was added and the mixture stirred overnight at rt. The reaction was washed with saturated NH4Cl solution and was extracted with CH2Cl2. The organic phase was dried over anh. Na2SO4 and concentrated in vacuo. The residue was purified by fc (cyclohexane:ethylacetate:CH3OH = 25:3:2 + 1% triethylamine, Ø = 2 cm, l = 25 cm, V = 10 mL) to afford the product 21 as a red solid. Red solid (Rf = 0.36, ethyl acetate = 100%), yield 0.090 g (78%). C27H24ClN5O4S (550.03 g/mol). Melting point: 171.2 °C. Purity (HPLC: method B): > 99% (tR = 21.02 min). Exact mass (LC-MS-ESI): m/z calculated for C27H25ClN5O4S [M + H]+ 550.1310, found 550.1285. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 10.30 (s, 1H, NH), 8.44 (s, 1H, N = CH), 8.09 (d, 2H, J = 8.1 Hz, 2, 6-CHtosyl), 7.92 (s, 1H, 5-CHpyridine), 7.43–7.26 (m, 12H, 3, 5CHtosyl, 2, 3, 4, 5, 6CHbenzyl), 4.68 (s, 2H, CH2 benzyl), 4.46 (s, 2H, CH2 benzyl), 2.34 (s, 3H, CH3); the 1HNMR spectrum displayed small impurities in the range of about 5%. 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 157.6 (1C, C-1), 147.1 (1C, C-6pyridine), 142.9 (1C, C4tosyl), 139.8 (1C, C-4pyridine), 136.5 (3C, C-1tosyl, C-1benzyl), 132.8 (1C, C-3pyridine), 128.6 (2C, C3, 5tosyl), 128.5 (4C, C3, 5benzyl), 128.4 (2C, C-2, 6tosyl), 128.2 (2C, C2, 6benzyl), 128.0 (2C, C2, 6benzyl), 127.8 (1C, C4benzyl), 127.2 (1C, C4benzyl), 112.8 (1C, C5pyridine), 53.6 (1C, CH2 benzyl), 47.1 (1C, CH2 benzyl), 21.0 (1C, CH3); the signal for C-4pyridine could not be seen in 13CNMR spectrum. FT-IR (neat) ṽ (cm−1) = 3251 (N-H), 2978, 2924 (C-Haliphat.), 1616 (C = Caromat.), 1327(NO2), 1161 (S = O), 829, 814, 748, 737 (CHaromat., out of plane).
Crystal data for C27H24ClN5O4S (M = 550.02 g/mol): monoclinic, P21/n (No. 14), a = 13.4033(2) Å, b = 9.4786(2) Å, β = 98.623(1)°, c = 20.9689(4) Å, V = 2633.87(8) Å3, Z = 4, 1.387 mg/m3, T = 173(2), μ(CuKα) = 0.268 mm−1, Final R indices [I > 2σ(I)]R1 = 0.0424, wR2 = 0.0934, R indices (all data) R1 = 0.0502, wR2 = 0.0992
(1R,2R,3S,4R,5S)-4-[6-(Dibenzylamino)-9H-purin-9-yl]-1-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,3-O-isopropylidenebicyclo[3.1.0]hexane-2,3-diol (24). An amount of 6-chloropurine (22, 1.00 g, 6.47 mmol) was suspended in isopropanol (60 mL). Dibenzylamine (5.0 mL, 26.0 mmol, 4 eq.) was added. The mixture was stirred at 90 °C under reflux for 7 h. The solvent was evaporated and the residue was purified by fc (CH2Cl2:CH3OH = 59:1 ⭢ 29:1 + 0.5% HCOOH, Ø = 6 cm, l = 20 cm, V = 65 mL) to afford the N,N-dibenzyladenine as a colorless solid (Rf = 0.36, ethyl acetate = 100%), yield 1.91 g (94%). C19H17N5 (315.38 g/mol). Melting point: 186.4 °C. Purity (HPLC: method B): > 99% (tR = 15.31 min). Exact mass (APCI): m/z calculated for C19H18N5 [M + H]+ 316.1557, found 316.1568. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 13.14 (s, 1H, 9-NH), 8.28 (s, 1H, 2-CH), 8.14 (s, 1H, 8-CH), 7.46–7.34 (m, 10H, 2, 3, 4, 5, 6-CHbenzyl), 5.50 (s, 2H, CH2 benzyl), 4.94 (s, 2H, CH2 benzyl). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 154.1 (1C, C-6), 151.9 (1C, C-2), 151.6 (1C, C-4), 138.5 (1C, C-8), 138.0 (1C, C1benzyl), 128.5 (4C, C3, 5benzyl), 127.4 (4C, C2, 6benzyl), 127.0 (2C, C4benzyl), 118.5 (1C, C-5), 50.6 (1C, CH2 benzyl), 48.5 (1C, CH2 benzyl). FT-IR (neat) ṽ (cm−1) = 3059 (v C-Haromat.), 2978, (C-Haliphat.), 1574 (C = Caromat.), 752, 737, 698 (CHaromat., out of plane).
The N,N-dibenzyladenine (0.47 g, 1.48 mmol, 1.3 eq.) and triphenylphospane (0.47 g, 1.78 mmol, 1.6 eq.) were dissolved in THF (10 mL) under nitrogen atmosphere. Diisopropyl azodicarboxylate (DIAD, 0.34 mL, 1.73 mmol, 1.5 eq.) was added dropwise at 0 °C. The mixture was stirred for 30 min at rt. A solution of the alcohol 4 (0.50 g, 1.15 mmol) in THF (10 mL) was added and the solution was stirred overnight. The solvent was evaporated and the residue was purified by fc (cyclohexane:ethyl acetate = 19:1 ⭢ 9:1, Ø = 6 cm, l = 20 cm, V = 65 mL) to afford the product 24 as a colorless solid (Rf = 0.29, cyclohexane:ethyl acetate = 9:1), yield 0.73 g (86%). C45H49N5O3Si (736.00 g/mol). Melting point: 77.6 °C. Purity (HPLC: method C): >99% (tR = 18.82 min). Exact mass (APCI): m/z calculated for C45H50N5O3Si [M + H]+ 736.3677, found 736.3707. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.30 (s, 1H, 8-CHpurine), 8.25 (s, 1H, 2-CHpurine), 7.60–7.55 (m, 4H, 2, 6-CHPh), 7.42–7.37 (m, 2H, 4-CHPh), 7.36–7.32 (m, 4H, 3, 5-CHPh), 7.31–7.24 (m, 10H, 2, 3, 4, 5, 6-CHbenzyl), 5.60 (s, 1H, CHHbenzyl), 5.47 (s, 1H, CHHbenzyl), 5.28 (dd, J = 7.0, 1.3 Hz, 1H, 2-CH), 4.98 (s, 1H, 4-CH), 4.96 (s, 1H, CHHbenzyl), 4.88 (s, 1H, CHHbenzyl), 4.76 (dd, J = 7.1, 1.4 Hz, 1H, 3-CH), 4.04 (d, J = 10.7 Hz, 1H, OCHH), 3.72 (d, J = 10.8 Hz, 1H, OCHH), 1.67 (ddd, J = 9.3, 4.5, 1.6 Hz, 1H, 5-CH), 1.46 (s, 3H, C(CH3)2), 1.39 (s, 1.6H, CH2, solvent: cyclohexane), 1.20 (s, 3H, C(CH3)2), 1.01 (t, J = 4.8 Hz, 1H, 6-CHH), 0.98 (s, 9H, C(CH3)3), 0.90 (ddd, J = 9.1, 5.1, 1.5 Hz, 1H, 6-CHH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 154.1 (1C, C-6purine), 152.0 (1C, C-2purine), 150.0 (1C, C-4purine), 138.5 (1C, C-8purine), 137.9 (2C, C-1benzyl), 135.1 (4C, C-2, 6Ph), 132.8 (2C, C-1Ph), 129.8 (2C, C-4Ph), 128.5 (4C, C-3, 5benzyl), 127.8 (4C, C-3, 5Ph), 127.4 (4C, C-2, 6benzyl), 127.1 (2C, C-4benzyl), 119.0 (1C, C-5purine), 111.3 (1C, C(CH3)2), 87.9 (1C, C-2), 81.1 (1C, C-3), 64.7 (1C, OCH2), 58.4 (1C, C-4), 50.8 (1C, CH2 benzyl), 48.6 (1C, CH2 benzyl), 38.1 (1C, C-1), 29.9 (1C, C-5), 26.7 (3C, C(CH3)3), 26.3 (s, 0.8C, CH2, solvent: cyclohexane), 25.9 (1C, C(CH3)2), 24.3 (1C, C(CH3)2), 18.8 (1C, C(CH3)3), 12.2 (1C, C-6). FT-IR (neat) ṽ (cm−1) = 2978 (C-Haliphat.), 1574 (C = Caromat.), 1107, 1064, 1037 (C-O), 737, 698 (C-Haromat., out of plane).
(1R,2R,3S,4R,5S)-4-[6-(Dibenzylamino)-9H-purin-9-yl]-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (26). Compound 24 (0.125 g, 0.17 mmol) was dissolved in CH3OH (3.6 mL) and trifluoroacetic acid (0.40 mL) and H2O (0.40 mL) were added. The mixture was heated to 70 °C for 2 d. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the alcohol 106 as a colorless solid (Rf = 0.24, ethyl acetate = 100%), yield 0.051 g (65%). C26H27N5O3 (457.53 g/mol). Purity (HPLC: method B): > 99% (tR = 11.99 min). Exact mass (LC-MS-ESI): m/z calculated for C26H28N5O3 [M + H]+ 458.2187, found 458.2187. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.49 (s, 1H, 8-CHpurine), 8.31 (s, 1H, 2CHpurine), 7.347.29 (m, 4H, 3, 5-CHbenzyl), 7.29–7.23 (m, 6H, 2, 4, 6-CHbenzyl), 5.63 (s, 1H, CHHbenzyl), 5.42 (s, 1H, CHHbenzyl), 5.25 (s, 1H, 3-OH), 5.03 (t, J = 5.0 Hz, 1H, CH2OH), 4.97 (s, 1H, CHHbenzyl), 4.82 (s, 2H, CHHbenzyl, 4-CH), 4.58 (t, J = 5.2 Hz, 1H, 2-CH), 4.49 (t, J = 6.8 Hz, 1H, 2-OH), 4.07 (dd, J = 11.4, 4.9 Hz, 1H, OCHH), 3.72 (d, J = 6.4 Hz, 1H, 3CH), 3.13 (dd, J = 11.4, 4.1 Hz, 1H, OCHH), 1.49 (ddd, J = 8.7, 3.9, 1.4 Hz, 1H, 5CH), 1.37 (dd, J = 4.7, 3.9 Hz, 1H, 6CHH), 0.61 (ddd, J = 8.5, 4.7, 1.6 Hz, 1H, 6CHH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 154.1 (1C, C-6purine), 151.9 (1C, C-2purine), 150.1 (1C, C-4purine), 138.3 (1C, C-8purine), 137.9 (2C, C-1benzyl), 128.5 (4C, C3, 5benzyl), 127.4 (4C, C2, 6benzyl), 127.1 (2C, C-4benzyl), 118.9 (1C, C-5purine), 75.9 (1C, C-3), 70.2 (1C, C2), 62.3 (1C, OCH2), 60.8 (1C, C-4), 50.7 (1C, CH2 benzyl), 48.6 (1C, CH2 benzyl), 36.4 (1C, C-1), 23.1 (1C, C5), 11.2 (1C, C-6). FT-IR (neat) ṽ (cm−1) = 3310 (O-H), 3028 (v C-Haromat.), 2978, 2920 (C-Haliphat.), 1578 (C = Caromat.), 1068 (C-O), 698 (CHaromat., out of plane).
(1R,2R,3S,4R,5S)-4-[6-(Dibenzylamino)-2-chloro-9H-purin-9-yl]-1-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,3-O-isopropylidenebicyclo[3.1.0]hexane-2,3-diol (25). An amount of 2,6-dichloropurine (2.02 g, 10.47 mmol) was dissolved in isopropanol (100 mL). Dibenzylamine (8.0 mL, 41.6 mmol, 3.9 eq.) was added. The mixture was stirred at 90 °C under reflux for 1.5 h. The precipitated product was filtered off and purified by fc (CH2Cl2:CH3OH = 59:1 ⭢ 29:1 + 0.5% HCOOH, Ø = 8 cm, l = 20 cm, V = 100 mL) to afford the purine derivative as a colorless solid (Rf = 0.37, cyclohexane:ethyl acetate = 1:1), yield 3.27 g (88%). C19H16ClN5 (349.82 g/mol). Melting point: 260.0 °C. Purity (HPLC: method B): > 99% (tR = 16.17 min). Exact mass (APCI): m/z calculated for C19H17ClN5 [M + H]+ 350.1167, found 350.1167. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 13.29 (s, 1H, 9-NH), 8.16 (s, 1H, 8-CH), 7.367.30 (m, 4H, 3, 5-CHbenzyl), 7.30–7.24 (m, 6H, 2, 4, 6-CHbenzyl), 5.53 (s, 2H, CH2 benzyl), 4.81 (s, 2H, CH2 benzyl). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 154.5 (1C, C-6), 152.8 (1C, C-4), 152.4 (1C, C-2), 139.2 (1C, C-8), 137.3 (1C, C1benzyl), 128.5 (4C, C3, 5benzyl), 127.5 (4C, C2, 6benzyl), 127.2 (2C, C4benzyl), 117.6 (1C, C5), 50.8 (1C, CH2 benzyl), 48.9 (1C, CH2 benzyl). FT-IR (neat) ṽ (cm−1) = 3066 (v C-Haromat.), 2978, (C-Haliphat.), 1578 (C = Caromat.), 1076 (C-Cl), 741, 694 (CHaromat., out of plane).
Next, the purine derivative (1.04 g, 2.98 mmol, 1.3 eq.) and triphenylphospane (0.90 g, 3.44 mmol, 1.5 eq.) were dissolved in THF (20 mL) under nitrogen atmosphere. DIAD (0.67 mL, 3.41 mmol, 1.5 eq.) was added dropwise at 0 °C. The mixture was stirred for 15 min at rt. A solution of the alcohol 4 (1.03 g, 2.35 mmol) in THF (20 mL) was added and the solution was stirred overnight. The solvent was evaporated and the residue was purified by fc (cyclohexane:ethyl acetate = 19:1 ⭢ 9:1, Ø = 6 cm, l = 20 cm, V = 65 mL) to afford the product 26 as a colorless solid (Rf = 0.35, cyclohexane:ethyl acetate = 1:1), yield 1.67 g (92%). C45H48ClN5O3Si (770.45 g/mol). Melting point: 84.7 °C. Purity (HPLC: method C): >99% (tR = 19.49 min). Exact mass (LC-MS-ESI): m/z calculated for C45H49ClN5O3Si [M + H]+ 770.3288, found 770.3285. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.29 (s, 1H, 8-CHpurine), 7.60–7.56 (m, 4H, 2, 6-CHPh), 7.42–7.25 (m, 16H, 2, 3, 4, 5, 6-CHbenzyl, 3, 4, 5-CHPh), 5.57 (d, J = 15.7 Hz, 1H, CHHbenzyl), 5.47 (d, J = 15.7 Hz, 1H, CHHbenzyl), 5.21 (dd, J = 7.1, 1.3 Hz, 1H, 2-CH), 4.91 (s, 1H, 4-CH), 4.86 (d, J = 15.4 Hz, 1H, CHHbenzyl), 4.79 (d, J = 15.4 Hz, 1H, CHHbenzyl), 4.75 (dd, J = 7.2, 1.5 Hz, 1H, 3-CH), 4.03 (d, J = 10.7 Hz, 1H, OCHH), 3.87 (d, J = 10.7 Hz, 1H, OCHH), 1.64 (ddd, J = 9.2, 4.5, 1.6 Hz, 1H, 5-CH), 1.46 (s, 3H, C(CH3)2), 1.43 (s, 0.7H, CH2, solvent: cyclohexane), 1.19 (s, 3H, C(CH3)2), 0.98 (s, 10H, 6-CHH, C(CH3)3), 0.93 (ddd, J = 9.1, 5.1, 1.4 Hz, 1H, 6-CHH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 154.5 (1C, C-6purine), 152.5 (1C, C-2purine), 151.1 (1C, C-4purine), 139.3 (1C, C-8purine), 137.4 (1C, C-1benzyl), 136.9 (1C, C-1benzyl), 135.0 (4C, C-2, 6Ph), 132.9 (2C, C-1Ph), 129.8 (2C, C-4Ph), 128.6 (4C, C-2, 6benzyl), 127.8 (4C, C-3, 5Ph), 127.7 (2C, C-2, 6benzyl), 127.5 (1C, C-2, 6benzyl), 127.3 (2C, C-4benzyl), 118.3 (1C, C-5purine), 111,3 (1C, C(CH3)2), 87.9 (1C, C-3), 81.6 (1C, C-2), 64.5 (1C, OCH2), 58.9 (1C, C-4), 50.9 (1C, CH2 benzyl), 49.2 (1C, CH2 benzyl), 38.3 (1C, C-1), 29.6 (1C, C-5), 26.7 (3C, C(CH3)3), 26.3 (0.4C, CH2, solvent: cyclohexane), 25.9 (1C, C(CH3)2), 24.3 (1C, C(CH3)2), 18.8 (1C, C(CH3)3), 12.0 (1C, C-6). FT-IR (neat) ṽ (cm−1) = 2978 (C-Haliphat.), 1574 (C = Caromat.), 1111, 1069, 1042 (C-O), 740, 698 (C-Haromat., out of plane).
(1R,2R,3S,4R,5S)-4-[6-(Dibenzylamino)-2-chloro-9H-purin-9-yl]-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (27). Compound 25 (0.099 g, 0.13 mmol) was dissolved in CH3OH (3.6 mL), trifluoroacetic acid (0.40 mL) and H2O (0.40 mL) were added. The mixture was heated to 70 °C for 2 d. The solvent was evaporated and the residue was purified by fc (CH2Cl2:CH3OH = 96:4, Ø = 2 cm, l = 24 cm, V = 10 mL) but still showed a small impurity by 1H-NMR. The impure product was purified again by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the pure alcohol 27 as a colorless solid (Rf = 0.32, ethyl acetate = 100%), yield 0.052 g (82%). C26H26ClN5O3 (491.98 g/mol). Purity (HPLC: method B): > 99% (tR = 14.14 min). Exact mass (APCI): m/z calculated for C26H27ClN5O3 [M + H]+ 492.1797, found 492.1784. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.51 (s, 1H, 8-CHpurine), 7.36–7.31 (m, 4H, 3, 5-CHbenzyl), 7.30–7.25 (m, 6H, 2, 4, 6-CHbenzyl), 5.61 (d, J = 15.7 Hz, 1H, CHHbenzyl), 5.41 (d, J = 15.8 Hz, 1H, CHHbenzyl), 5.27 (s, 1H, 3-OH), 4.99 (t, J = 5.0 Hz, 1H, CH2OH), 4.88 (d, J = 15.4 Hz, 1H, CHHbenzyl), 4.75 (s, 1H, CHHbenzyl), 4.72 (s, 1H, 4-CH), 4.56 (d, J = 6.5 Hz, 1H, 2-CH), 4.50 (s, 1H, 2-OH), 4.07 (d, J = 11.0 Hz, 1H, OCHH), 3.73 (d, J = 6.2 Hz, 1H, 3-CH), 3.13 (d, J = 11.3 Hz, 1H, OCHH), 1.47 (ddd, J = 8.8, 3.8, 1.5 Hz, 1H, 5-CH), 1.36 (t, J = 4.3 Hz, 1H, 6-CHH), 0.61 (ddd, J = 8.6, 4.7, 1.5 Hz, 1H, 6-CHH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 154.5 (1C, C-6purine), 152.5 (1C, C-2purine), 151.4 (1C, C-4purine), 138.9 (1C, C-8purine), 137.4 (1C, C-1benzyl), 136.9 (1C, C-1benzyl), 128.6 (4C, C-3, 5benzyl), 127.7 (2C, C-2, 6benzyl), 127.4 (2C, C-2, 6benzyl), 127.3 (2C, C-4benzyl), 118.0 (1C, C-5purine), 75.8 (1C, C-3), 70.2 (1C, C-2), 62.2 (1C, OCH2), 61.0 (1C, C-4), 50.8 (1C, CH2 benzyl), 49.1 (1C, CH2 benzyl), 36.4 (1C, C-1), 23.1 (1C, C-5), 11.1 (1C, C-6).
FT-IR (neat) ṽ (cm−1) = 3341 (O-H), 2978 (C-Haliphat.), 1574 (C = Caromat.), 1069 (C-O), 698 (C-Haromat., out of plane).
(1R,2R,3S,4R,5S)-4-[6-(Dibenzylamino)-2-methylthio-9H-purin-9-yl]-1-(hydroxymethyl)-2,3-O-isopropylidenebicyclo[3.1.0]hexane-2,3-diol (28). Compound 25 (0.30 g, 0.39 mmol) was dissolved in DMF (15 mL). NaSCH3 (0.41 g, 5.86 mmol, 15 eq.) was added. The mixture was stirred at 90 °C for 1 h in the microwave at a power of 200 W. The solvent was evaporated and the residue was purified by fc (cyclohexane:ethyl acetate = 6:4, Ø = 5 cm, l = 24 cm, V = 30 mL) to afford the product 28 as a colorless solid (Rf = 0.35, cyclohexane:ethyl acetate = 1:1), yield 0.227 g (89%). C30H33N5O3S (543.69 g/mol). Melting point: 97.8 °C. Purity (HPLC: method B): 93% (tR = 18.60 min). Exact mass (LC-MS-ESI): m/z calculated for C30H34N5O3S [M + H]+ 544.2377, found 544.2378. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 8.24 (s, 1H, 8-CHpurine), 7.36–7.22 (m, 10H, 2, 3, 4, 5, 6-CHbenzyl), 5.11 (dd, J = 15.6, 15.1 Hz, 2H, CH2 benzyl), 5.20 (d, J = 7.1, 1.3 Hz, 1H, 2CH), 4.94 (s, 1H, OH), 4.92 (s, 1H, 4-CH), 4.91–4.74 (m, 2H, CH2 benzyl), 4.61 (dd, J = 7.2, 1.5 Hz, 1H, 3-CH), 3.84 (dd, J = 11.6, 4.0 Hz, 1H, OCHH), 3.35 (d, J = 11.6, 3.9 Hz, 1H, OCHH), 2.41 (s, 3H, SCH3), 1.65 (ddd, J = 9.2, 4.4, 1.5 Hz, 1H, 5CH), 1.45 (s, 3H, C(CH3)2), 1.15 (s, 3H, C(CH3)2), 0.98 (t, J = 4.9 Hz, 1H, 6CHH), 0.88 (ddd, J = 9.1, 5.1, 1.5 Hz, 1H, 6CHH); the 1H-NMR spectrum displayed small impurities in the range of about 5%. 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 163.6 (1C, C-2purine), 153.3 (1C, C-6purine), 150.9 (1C, C-4purine), 137.8 (1C, C-1benzyl), 137.6 (1C, C-8purine), 128.5 (4C, C3, 5benzyl), 127.4 (4C, C2, 6benzyl), 127.1 (2C, C-4benzyl), 116.6 (1C, C-5purine), 111.2 (1C, C(CH3)2), 88.3 (1C, C-3), 80.9 (1C, C-2), 62.6 (1C, OCH2), 58.1 (1C, C4), 50.9 (1C, CH2 benzyl), 48.9 (1C, CH2 benzyl), 38.7 (1C, C-1), 29.6 (1C, C5), 25.8 (1C, C(CH3)2), 24.2 (1C, C(CH3)2), 13.8 (1C, SCH3), 12.6 (1C, C-6); the 13C-NMR spectrum displayed small impurities in the range of about 5%. FT-IR (neat) ṽ (cm−1) = 3372 (O-H), 2982, 2924 (C-Haliphat.), 1562 (C = Caromat.), 1057, 1030 (CO), 748, 733, 698 (CHaromat., out of plane).
(1S,2R,3S,4R,5S)-4-[6-(Dibenzylamino)-2-methylthio-9H-purin-9-yl]-1-(chloromethyl)-2,3-O-isopropylidenebicyclo[3.1.0]hexane-2,3-diol (29). Cyanuric chloride (0.051 g, 0.28 mmol, 1.5 eq.) was stirred with DMF (0.08 mL, 1.04 mmol, 5.8 eq.) for 2 h at rt. Then CH2Cl2 (1 mL) and the alcohol 28 (0.098 g, 0.18 mmol) were added and the mixture was stirred overnight. Water was added and the phases were separated. The organic phase was washed with K2CO3 solution, 0.05 M HCl, and water. The organic phase was dried over anh. Na2SO4, and filtered and concentrated in vacuo. The residue was purified by fc (cyclohexane:ethyl acetate = 7:1, Ø = 2 cm, l = 20 cm, V = 10 mL) to afford the chloride 29 as a colorless solid (Rf = 0.35, cyclohexane:ethyl acetate = 5:1), yield 0.066 g (65%). C30H32ClN5O2S (562.13 g/mol). Melting point: 182.4 °C. Purity (HPLC: method B): > 99% (tR = 21.54 min). Exact mass (LC-MS-ESI): m/z calculated for C30H33ClN5O2S [M + H]+ 562.2038, found 562.2036. 1H-NMR (600 MHz, CDCl3) δ (ppm) = 7.86 (s, 1H, 8-CHpurine), 7.34–7.29 (m, 4H, 3, 5CHbenzyl), 7.29–7.24 (m, 6H, 2, 4, 6-CHbenzyl), 5.50 (s, 2H, CH2 benzyl), 5.38 (dd, J = 7.2, 1.5 Hz, 1H, 2CH), 5.01 (s, 1H, 4-CH), 4.95 (s, 2H, CH2 benzyl), 4.69 (dd, J = 7.2, 1.4 Hz, 1H, 3-CH), 3.94 (d, J = 11.6 Hz, 1H, ClCHH), 3.81 (d, J = 11.6 Hz, 1H, ClCHH), 2.50 (s, 3H, SCH3), 1.75 (ddd, J = 9.4, 4.7, 1.5 Hz, 1H, 5CH), 1.56 (s, 3H, C(CH3)2), 1.43 (s, 0.1H, CH2, solvent: cyclohexane), 1.35 (dd, J = 5.9, 4.8 Hz, 1H, 6CHH), 1.27 (s, 3H, C(CH3)2), 1.08 (ddd, J = 9.4, 5.9, 1.6 Hz, 1H, 6CHH). 13C-NMR (151 MHz, CDCl3) δ (ppm) = 165.0 (1C, C-2purine), 154.2 (1C, C-6purine), 151.6 (1C, C4purine), 137.9 (1C, C-1benzyl), 136.5 (1C, C-8purine), 128.7 (4C, C3, 5benzyl), 128.0 (4C, C2, 6benzyl), 127.5 (2C, C4benzyl), 117.5 (1C, C-5purine), 112.6 (1C, C(CH3)2), 89.4 (1C, C-3), 82.4 (1C, C2), 59.6 (1C, C-4), 51.2 (1C, CH2 benzyl), 49.2 (1C, ClCH2), 49.0 (1C, CH2 benzyl), 39.0 (1C, C-1), 33.0 (1C, C-5), 26.2 (1C, C(CH3)2), 24.4 (1C, C(CH3)2), 16.4 (1C, C-6), 14.8 (1C, SCH3). FT-IR (neat) ṽ (cm−1) = 2978, 2928 (C-Haliphat.), 1589, 1566 (C = Caromat.), 1072, 1049 (CO), 798 (C-Cl), 733, 694 (CHaromat., out of plane).
(1R,2R,3S,4R,5S)-4-[6-(Dibenzylamino)-2-methylthio-9H-purin-9-yl]-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (30). Compound 28 (0.080 g, 0.15 mmol) was dissolved in CH3OH (2.5 mL), trifluoroacetic acid (0.32 mL) and H2O (0.32 mL) were added. The mixture was heated to 70 °C for 2 h. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the alcohol 111 as a colorless solid (Rf = 0.32, ethyl acetate = 100%), yield 0.036 g (48%). C27H29N5O3S (503.62 g/mol). Purity (HPLC: method B): 98% (tR = 14.50 min). Exact mass (APCI): m/z calculated for C27H30N5O3S [M + H]+ 492.2064, found 492.2066. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.35 (s, 1H, 8-CHpurine), 7.95 (s, 0.1H, CH, solvent: DMF), 7.31 (t, J = 7.5 Hz, 4H, 3, 5-CHbenzyl), 7.29–7.23 (m, 6H, 2, 4, 6-CHbenzyl), 5.61 (d, J = 15.8 Hz, 1H, CHHbenzyl), 5.41 (d, J = 15.8 Hz, 1H, CHHbenzyl), 5.19 (s, 1H, 3-OHl), 4.97 (t, J = 5.1 Hz, 1H, CH2OH), 4.92 (d, J = 14.7 Hz, 1H, CHHbenzyl), 4.78 (d, J = 14.7 Hz, 1H, CHHbenzyl), 4.75 (s, 1H, 4-CH), 4.57 (ddd, J = 8.1, 6.5, 1.6 Hz, 1H, 2CH), 4.49 (d, 7.9 Hz, 1H, 2-OH), 4.05 (dd, J = 11.3, 5.3 Hz, 1H, OCHH), 3.72 (ddt, J = 6.4, 4.7, 1.3 Hz, 1H, 3-CH), 3.13 (dd, J = 11.4, 4.8 Hz, 1H, OCHH), 2.89 (s, 0.4H, CH3, solvent: DMF), 2.73 (s, 0.3H, CH3, solvent: DMF), 2.41 (s, 3H, SCH3), 2.07 (s, 0.1H, CH3CN, solvent: acetonitrile), 1.45 (ddd, J = 8.8, 3.9, 1.4 Hz, 1H, 5CH), 1.34 (t, J = 4.3 Hz, 1H, 6CHH), 0.60 (ddd, J = 8.6, 4.7, 1.7 Hz, 1H, 6CHH); the 1HNMR spectrum displayed small impurities in the range of about 5%. 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 163.4 (1C, C-2purine), 162.3 (0.1C, CH, solvent: DMF), 153.3 (1C, C6purine), 151.1 (1C, C-4purine), 137.8 (2C, C-1benzyl), 137.4 (1C, C-8purine), 128.5 (4C, C3, 5benzyl), 127.4 (4C, C-2, 6benzyl), 127.1 (2C, C-4benzyl), 116.5 (1C, C-5purine), 76.0 (1C, C-3), 70.2 (1C, C2), 62.2 (1C, OCH2), 60.6 (1C, C-4), 50.8 (1C, CH2 benzyl), 48.9 (1C, CH2 benzyl), 36.4 (1C, C-1), 35.8 (0.1C, CH3, solvent: DMF), 30.8 (0.1C, CH3, solvent: DMF), 23.2 (1C, C5), 13.8 (1C, SCH3), 11.1 (1C, C6). FT-IR (neat) ṽ (cm−1) = 3368 (O-H), 2978, 2924 (C-Haliphat.), 1562 (C = Caromat.), 1069 (CO), 733, 698 (CHaromat., out of plane).
(1S,2R,3S,4R,5S)-4-[6-(Dibenzylamino)-2-methylthio-9H-purin-9-yl]-1-(chloromethyl)bicyclo[3.1.0]hexane-2,3-diol (31). Compound 29 (0.055 g, 0.10 mmol) was dissolved in a mixture of CH3OH (1.6 mL) and CH2Cl2 (1.5 mL). Trifluoroacetic acid (0.20 mL) and H2O (0.20 mL) were added. The mixture was heated to 70 °C for 6 h. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the product 31 as a colorless solid (Rf = 0.31, cyclohexane:ethyl acetate = 1:1), yield 0.042 g (82%). C27H28ClN5O2S (522.06 g/mol). Purity (HPLC: method B): 97% (tR = 17.55 min). Exact mass (LC-MS-ESI): m/z calculated for C27H29ClN5O2S [M + H]+ 522.1725, found 522.1713. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.09 (s, 1H, 8-CHpurine), 7.32 (t, J = 7.5 Hz, 4H, 3, 5-CHbenzyl), 7.27 (d, J = 7.4 Hz, 6H, 2, 4, 6-CHbenzyl), 5.54 (d, J = 15.2 Hz, 1H, CHHbenzyl), 5.48 (d, J = 15.4 Hz, 1H, CHHbenzyl), 5.30 (s, 1H, 3-OH), 4.87 (d, J = 14.9 Hz, 1H, CHHbenzyl), 4.82 (d, J = 14.9 Hz, 1H, CHHbenzyl), 4.79 (t, J = 7.5 Hz, 1H, 2-OH), 4.70 (s, 1H, 4-CH), 4.62 (ddd, J = 7.8, 6.7, 1.6 Hz, 1H, 2-CH), 4.16 (d, J = 11.4 Hz, 1H, ClCHH), 4.03 (q, J = 7.1 Hz, 0.2H, CH2, solvent: ethyl acetate), 3.94 (ddt, J = 6.4, 4.7, 1.5 Hz, 1H, 3-CH), 3.74 (d, J = 11.4 Hz, 1H, ClCHH), 2.41 (s, 3H, SCH3), 1.99 (s, 0.3H, OCH3, solvent: ethyl acetate), 1.69 (ddd, J = 9.2, 4.1, 1.2 Hz, 1H, 5-CH), 1.53 (t, J = 4.5 Hz, 1H, 6-CHH), 1.17 (t, J = 7.1 Hz, 0.1H, CH2CH3, solvent: ethyl acetate), 0.88 (ddd, J = 8.7, 4.8, 1.7 Hz, 1H, 6-CHH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 163.5 (1C, C-2purine), 153.4 (1C, C-6purine), 151.2 (1C, C-4purine), 137.9 (1C, C-1benzyl), 137.6 (1C, C-1benzyl), 137.0 (1C, C-8purine), 128.5 (4C, C-3, 5benzyl), 127.4 (4C, C-2, 6benzyl), 127.1 (2C, C-4purine), 116.5 (1C, C-5purine), 76.1 (1C, C-3), 70.9 (1C, C-2), 61.0 (1C, C-4), 59.8 (0.1C, CH2, solvent: ethyl acetate), 50.8 (1C, CH2 benzyl), 49.4 (1C, ClCH2), 48.8 (1C, CH2 benzyl), 35.9 (1C, C-1), 25.5 (1C, C-5), 20.8 (0.1C, OCH3, solvent: ethyl acetate), 14.8 (1C, C-6), 14.1 (0.1C, CH2CH3, solvent: ethyl acetate), 13.8 (1C, SCH3). FT-IR (neat) ṽ (cm−1) = 3341 (O-H), 2978 (C-Haliphat.), 1562 (C = Caromat.), 1072 (C-O), 783 (C-Cl), 733, 694 (C-Haromat., out of plane).
Di-(tert-butyl)-N-[9-((1R,2R,3S,4R,5S)-1-{[(tert-butyldiphenylsilyl)oxy]methyl}-2,3-dihydroxy-2,3-O-isopropylidenebicyclo[3.1.0]hex-4-yl)-2-chloro 9H-purin-6-yl]dicarbamate (35). An amount of 2-chloroadenine (3.02 g, 17.8 mmol) was suspended in THF (88 mL) and di-tert-butyl dicarbonate (15.9 g, 73.1 mmol, 4.1 eq.) and DMAP (0.22 g, 1.82 mmol, 0.1 eq.) were added. The mixture was stirred at rt overnight. The solvent was evaporated and the residue redissolved in ethyl acetate. The organic phase was washed with 1 M HCl and brine. After drying over anh. Na2SO4, the solvent was evaporated. The residue was dissolved in CH3OH (177 mL) and saturated NaHCO3 solution (80 mL) was added. The mixture was stirred for 2.5 h at 50 °C. CH3OH was evaporated and the aqueous residue diluted with H2O. The aqueous phase was extracted four times with CH2Cl2 After drying over anh. Na2SO4, the solvent was evaporated. The residue was purified by fc (cyclohexane:ethyl acetate = 1:4, Ø = 6 cm, l = 10 cm, V = 65 mL), but only a mixture of product and byproducts were obtained. It was purified again by fc (cyclohexane:ethyl acetate = 1:1, Ø = 6 cm, l = 10 cm, V = 65 mL) to afford the pure product as a colorless solid (Rf = 0.17, cyclohexane:ethyl acetate = 1:1), yield 4.85 g (74%). C15H20ClN5O4 (369.81 g/mol). Melting point: 85.9 °C. Purity (HPLC: method B): 99% (tR = 13.25 min). Exact mass (APCI): m/z calculated for C15H21ClN5O4 [M + H]+ 370.1277, found 370.1277. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 13.60 (s, 1H, NH), 8.60 (s, 1H, 8-CHpurine), 1.41 (s, 18H, C(CH3)3). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 150.7 (1C, C-2purine), 149.0 (2C, C = O), 146.5 (1C, C-8purine), 83.5 (2C, C(CH3)3), 27.0 (6C, C(CH3)3); C-1, C-3 and C-5 were not visible. FT-IR (neat) ṽ (cm−1) = 3244 (N-H), 2978 (C-Haliphat.), 1778, 1736 (C = O), 1134, 1107 (C-O).
Next, the purine (1.14 g, 3.08 mmol, 1.1 eq.) and triphenylphospane (1.05 g, 4.00 mmol, 1.5 eq.) were dissolved in THF (25 mL) under nitrogen atmosphere. DIAD (0.78 mL, 3.97 mmol, 1.5 eq.) was added dropwise at 0 °C. The mixture was stirred for 30 min at rt. A solution of the alcohol 4 (1.19 g, 2.70 mmol) in THF (22 mL) was added and the solution was stirred overnight. The solvent was evaporated and the residue was purified by fc (cyclohexane:ethyl acetate = 5:1 + 0.5% triethylamine, Ø = 6 cm, l = 10 cm, V = 65 mL) to afford the product 35 as a colorless solid (Rf = 0.32, cyclohexane:ethyl acetate = 5:1), yield 1.80 g (84%). C41H52ClN5O7Si (790.43 g/mol). Melting point: 88.6 °C. Purity (HPLC: method C): 98% (tR = 18.48 min). Exact mass (APCI): m/z calculated for C31H37ClN5O3Si [M + H+, -2 COOC(CH3)3, +2H]+ 590.2349, found 590.2362. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 8.74 (s, 1H, 8-CHpurine), 7.59 (ddd, J = 7.9, 6.4, 1.5 Hz, 4H, 2, 6-CHPh), 7.47–7.31 (m, 6H, 3, 4, 5-CHPh), 5.23 (dd, J = 7.1, 1.2 Hz, 1H, 2-CHbicyclohexane), 5.04 (s, 1H, 4-CHbicyclohexane), 4.83 (dd, J = 7.2, 1.6 Hz, 1H, 3-CHbicyclohexane), 4.06 (d, J = 10.6 Hz, 1H, OCHH), 3.83 (d, J = 10.6 Hz, 1H, OCHH), 1.72 (ddd, J = 9.2, 4.5, 1.5 Hz, 1H, 5-CHbicyclohexane), 1.46 (s, 3H, C(CH3)2), 1.41 (s, 18H, OC(CH3)3), 1.39 (s, 0.4H, CH2, solvent: cyclohexane), 1.20 (s, 3H, C(CH3)2), 0.99 (s, 11H, 6-CH2 bicyclohexane, SiC(CH3)3). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 153.8 (1C, C-4purine), 151.1 (1C, C-2purine), 149.9 (1C, C-6purine), 149.6 (2C, C = O), 144.8 (1C, C-8purine), 135.0 (4C, C-2, 6Ph), 132.7 (2C, C-1Ph), 129.8 (2C, C-4Ph), 127.8 (4C, C-3, 5Ph), 126.9 (1C, C-5purine), 111.5 (1C, C(CH3)2), 87.6 (1C, C-3bicyclohexane), 84.1 (2C, OC(CH3)3), 81.4 (1C, C-2bicyclohexane), 64.3 (1C, OCH2), 59.4 (1C, C-4bicyclohexane), 38.3 (1C, C-1bicyclohexane), 29.4 (1C, C-5bicyclohexane), 27.2 (6C, OC(CH3)3), 26.7 (3C, SiC(CH3)3), 26.3 (0.1C, CH2, solvent: cyclohexane), 25.8 (1C, C(CH3)2), 24.3 (1C, C(CH3)2), 18.8 (1C, SiC(CH3)3), 11.9 (1C, C-6bicyclohexane). FT-IR (neat) ṽ (cm−1) = 2978, 2932 (C-Haliphat.), 1759 (C = O), 1593, 1574 (C = Caromat.), 1107, 1069, 1038 (C-O), 741, 702 (C-Haromat., out of plane).
Tert-Butyl-N-{9-[(1R,2R,3S,4R,5S)-2,3-dihydroxy-1-(hydroxymethyl)-2,3-O-isopropylidenebicyclo[3.1.0]hex-4-yl]-2-methylthio-9H-purin-6-ylcarbamate (37). Compound 35 (1.00 g, 1.27 mmol) was dissolved in THF (20 mL). Tetrabutylammonium fluoride trihydrate (TBAF x 3H2O, 0.60 g, 1.91 mmol, 1.5 eq.) was added and the mixture was stirred at rt for 1 h. The solvent was evaporated, and the residue was dissolved in DMF (20 mL). NaSCH3 (1.35 g, 19.3 mmol, 15 eq.) was added, and the slurry was stirred overnight. Next, H2O (0.5 mL) was added, and the mixture was heated to 70 °C for 2 h. The reaction was concentrated in vacuo and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 50 mL/min, Biotage®® SNAP C18, 120 g, V = 20 mL) to afford the alcohol 37 as a colorless solid (Rf = 0.24, ethyl acetate = 100%), yield 0.37 g (63%). C21H29N5O5S (463.55 g/mol). Melting point: 110.1 °C. Purity (HPLC: method B): 94% (tR = 12.27 min). Exact mass (LC-MS-ESI): m/z calculated for C21H30N5O5S [M + H]+ 464.1962, found 464.1966. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 10.90 (s, 1H, NH), 8.42 (s, 1H, 8-CHpurine), 5.75 (s, 0.6H, CH2Cl2, solvent: dichloromethane), 5.23 (dd, J = 7.1, 1.3 Hz, 1H, 2CHbicyclohexane), 4.99 (s, 1H, OH), 4.95 (s, 1H, 4-CHbicyclohexane), 4.65 (dd, J = 7.1, 1.5 Hz, 1H, 3-CHbicyclohexane), 3.84 (dd, J = 11.5, 4.0 Hz, 1H, OCHH), 3.37 (dd, J = 11.5, 3.9 Hz, 1H, OCHH), 2.59 (s, 3H, SCH3), 1.64 (ddd, J = 9.2, 4.4, 1.5 Hz, 1H, 5CHbicyclohexane), 1.48 (s, 9H, C(CH3)3), 1.45 (s, 3H, C(CH3)2), 1.18 (s, 3H, C(CH3)2), 0.98 (t, J = 4.8 Hz, 1H, 6CHHbicyclohexane), 0.89 (ddd, J = 9.1, 5.1, 1.5 Hz, 1H, 6CHHbicyclohexane); the 1HNMR spectrum displayed small impurities in the range of about 5%. 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 163.8 (1C, C-2purine), 151.9 (1C, C-4purine), 151.0 (1C, C = O), 149.7 (1C, C-6purine), 140.9 (1C, C-8purine), 120.6 (1C, C-5purine), 111.3 (1C, C(CH3)2), 88.1 (1C, C-3bicyclohexane), 80.9 (1C, C-2bicyclohexane), 80.2 (1C, C(CH3)3), 62.5 (1C, OCH2), 58.4 (1C, C-4bicyclohexane), 54.9 (0.3C, CH2Cl2, solvent: dichloromethane), 38.8 (1C, C-1bicyclohexane), 29.6 (1C, C5bicyclohexane), 27.9 (3C, C(CH3)3), 25.6 (1C, C(CH3)2), 24.2 (1C, C(CH3)2), 14.0 (1C, SCH3), 12.6 (1C, C6bicyclohexane); the 13C-NMR spectrum displayed small impurities in the range of about 5%. FT-IR (neat) ṽ (cm−1) = 3341 (O-H), 2978 (C-Haliphat.), 1759 (C = O), 1609, 1582 (C = Caromat.), 1134, 1061, 1015 (C-O).
Tert-Butyl-N-{9-[(1R,2R,3S,4R,5S)-1-(azidomethyl)-2,3-dihydroxy-2,3-O-isopropylidenebicyclo[3.1.0]hex-4-yl]-2-methylthio-9H-purin-6-yl}carbamate (38). The alcohol 37 (2.19 g, 4.72 mmol) was suspended in CH2Cl2 (110 mL), tosyl chloride (1.81 g, 9.49 mmol, 2 eq.), triethylamine (1.5 mL, 10.8 mmol, 2.3 eq.), and DMAP (0.066 g, 0.54 mmol, 0.1 eq.) were added. The mixture was stirred at rt overnight. Water was added and the mixture was extracted four times with CH2Cl2. The organic phase was dried over anh. Na2SO4 and concentrated in vacuo. The residue was dissolved in DMF (60 mL) and NaN3 (4.60 g, 70.8 mmol, 15 eq.) was added. The mixture was heated to 70 °C for 2 h. Water and brine were added and the reaction mixture was extracted four times with CH2Cl2. The organic phase was dried over anh. Na2SO4 and concentrated in vacuo. The residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 80:20, 50 mL/min, Biotage®® SNAP C18, 120 g, V = 20 mL) to afford the azide 38 as a colorless solid (Rf = 0.27, cyclohexane:ethyl acetate = 1:1), yield 1.26 g (55%). C21H28N8O4S (488.57 g/mol). Melting point: 83.8 °C. Purity (HPLC: method B): 98% (tR = 16.55 min). Exact mass (APCI): m/z calculated for C31H37ClN5O3Si [M + H]+ 489.2027, found 489.2027. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 10.08 (s, 1H, NH), 8.31 (s, 1H, 8-CHpurine), 5.75 (s, 0.2H, CH2Cl2, solvent: dichloromethane), 5.25 (dd, J = 7.1, 1.3 Hz, 1H, 2CHbicyclohexane), 4.97 (s, 1H, 4-CHbicyclohexane), 4.81 (dd, J = 7.1, 1.3 Hz, 1H, 3CHbicyclohexane), 3.74 (d, J = 13.0 Hz, 1H, NCHH), 3.47 (d, J = 13.0 Hz, 1H, NCHH), 2.60 (s, 3H, SCH3), 2.08 (s, 0.1H, CH3CN, solvent: acetonitrile), 1.71 (ddd, J = 9.3, 4.6, 1.5 Hz, 1H, 5CHbicyclohexane), 1.48 (s, 9H, C(CH3)3), 1.47 (s, 3H, C(CH3)2), 1.20 (s, 3H, C(CH3)2), 1.09 (t, J = 5.0 Hz, 1H, 6CHHbicyclohexane), 1.04 (ddd, J = 9.2, 5.3, 1.5 Hz, 1H, 6-CHHbicyclohexane). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 163.7 (1C, C-2purine), 152.0 (1C, C-4purine), 150.8 (1C, C = O), 149.6 (1C, C-6purine), 141.5 (1C, C-8purine), 120.8 (1C, C-5purine), 111.5 (1C, C(CH3)2), 88.1 (1C, C-3bicyclohexane), 82.8 (1C, C-2bicyclohexane), 80.2 (1C, C(CH3)3), 59.0 (1C, C-4bicyclohexane), 54.1 (1C, NCH2), 36.3 (1C, C-1bicyclohexane), 30.0 (1C, C5bicyclohexane), 27.8 (3C, C(CH3)3), 25.8 (1C, C(CH3)2), 24.1 (1C, C(CH3)2), 14.0 (1C, SCH3), 13.9 (1C, C-6bicyclohexane). FT-IR (neat) ṽ (cm−1) = 2982, 2924 (C-Haliphat.), 2099 (N = N=N), 1751, 1712 (C = O), 1605, 1578 (C = Caromat.), 1138, 1053 (C-O).
(1R,2R,3S,4R,5S)-4-(6-Amino-2-methylthio-9H-purin-9-yl)-1-{[4-(hydroxymethyl)-1H-1,2,3-triazol-1-yl]methyl}bicyclo[3.1.0]hexane-2,3-diol (39). The azide 38 (0.030 g, 0.06 mmol) was dissolved in tert-butanol (0.5 mL). Propargyl alcohol (0.015 mL, 0.26 mmol, 4.2 eq.), copper(II) acetylacetonate (0.001 g, 0.004 mmol, 0.06 eq.), sodium ascorbate (0.007 g, 0.04 mmol, 0.6 eq.), and H2O (0.5 mL) were added. The mixture was stirred for 5 h at rt. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the Boc-protected triazole as a colorless solid (Rf = 0.30, CH2Cl2:CH3OH = 95:5), yield 0.020 g (59%). C24H32N8O5S (544.63 g/mol). Purity (HPLC: method B): 97% (tR = 11.30 min). Exact mass (LC-MS-ESI): m/z calculated for C24H32DN8O5S [M + H]+ 546.2352, found 546.2337. Exact mass (APCI): m/z calculated for C19H25N8O3S [M + 2H+, -COOC(CH3)3]+ 445.1765, found 445.1765. 1H-NMR (400 MHz, CD3OD) δ (ppm) = 7.97 (s, 1H, 5-CHtriazole), 5.34 (dd, J = 7.1, 1.4 Hz, 1H, 2-CHbicyclohexane), 5.00 (s, 1H, 4-CHbicyclohexane), 4.95 (d, J = 14.6 Hz, 1H, NCHH), 4.87 (dd, J = 7.3, 1.5 Hz, 1H, 3-CHbicyclohexane), 4.69 (dd, J = 13.2, 2.1 Hz, 2H, OCH2), 4.52 (d, J = 14.6 Hz, 1H, NCHH), 3.35 (s, 0.8H, CH3OH, solvent: methanol), 2.63 (s, 3H, SCH3), 1.89 (ddd, J = 9.4, 4.7, 1.6 Hz, 1H, 5CHbicyclohexane), 1.58 (s, 9H, C(CH3)3), 1.47 (s, 3H, C(CH3)2), 1.26 (t, J = 5.2 Hz, 1H, 6-CHHbicyclohexane), 1.22 (s, 3H, C(CH3)2), 1.15 (ddd, J = 9.4, 5.7, 1.5 Hz, 1H, 6-CHHbicyclohexane); 8-CHpurine was not visible due to occurrence of deuterium exchange at this position. 13C-NMR (101 MHz, CD3OD) δ (ppm) = 167.3 (1C, C-2purine), 153.3 (1C, C-4purine), 152.5 (1C, C = O), 150.9 (1C, C-6purine), 149.2 (1C, C-4triazole), 124.4 (1C, C-5triazole), 120.8 (1C, C-5purine), 113.6 (1C, C(CH3)2), 90.2 (1C, C-3bicyclohexane), 84.6 (1C, C2bicyclohexane), 82.7 (1C, C(CH3)3), 61.7 (1C, C-4bicyclohexane), 56.6 (1C, OCH2), 54.8 (1C, NCH2), 38.2 (1C, C-1bicyclohexane), 33.4 (1C, C5bicyclohexane), 28.5 (3C, C(CH3)3), 26.2 (1C, C(CH3)2), 24.4 (1C, C(CH3)2), 15.2 (1C, C6bicyclohexane), 14.8 (1C, SCH3); C8purine was not visible. FT-IR (neat) ṽ (cm−1) = 3341 (O-H), 3148 (N-H), 2978 (C-Haliphat.), 1751, 1717 (C = O), 1605, 1578 (C = Caromat.), 1142, 1053 (C-O).
The triazole (0.015 g, 0.03 mmol) was dissolved in CH3OH (0.8 mL) and trifluoroacetic acid (0.10 mL) and H2O (0.10 mL) were added. The mixture was heated to 70 °C for 6 h. The solvent was evaporated and the residue was purified by semi-preparative HPLC (method B) to afford the product 39 as a colorless solid (Rf = 0.30, CH2Cl2:CH3OH = 8:2), yield 0.006 g (54%). C16H20N8O3S (404.14 g/mol). Purity (HPLC: method B): 97% (tR = 3.55 min). Exact mass (LC-MS-ESI): m/z calculated for C16H20DN8O3S [M + H]+ 406.1515, found 406.1515 and for C16H21N8O3S [M + H]+ 405.1452, found 405.1454. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.00 (s, 1H, 5-CHtriazole), 7.42 (s, 0.3H, 8CHpurine), 7.32 (s, 1H, NH2), 4.73 (d, J = 14.5 Hz, 1H, NCHH), 4.61 (d, J = 1.8 Hz, 1H, 4-CH), 4.55–4.49 (m, 3H, NCHH, OCH2), 4.52 (dd, J = 6.6, 1.4 Hz, 1H, 2-CH), 4.03 (q, J = 7.1 Hz, 0.1H, CH2, solvent: ethyl acetate),3.81 (dt, J = 6.5, 1.6 Hz, 1H, 3-CH), 2.47 (s, 3H, SCH3), 1.99 (s, 0.1H, OCH3, solvent: ethyl acetate), 1.67 (dd, J = 8.5, 4.0 Hz, 1H, 5CH), 1.42 (t, J = 4.5 Hz, 1H, 6-CHH), 1.17 (t, J = 7.1 Hz, 0.1H, CH2CH3, solvent: ethyl acetate), 0.83 (ddd, J = 8.7, 4.9, 1.6 Hz, 1H, 6-CHH); 8-CHpurine showed a reduced intensity due to occurrence of deuterium exchange at this position. 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 163.8 (1C, C-2purine), 155.4 (1C, C-6Purine), 149.7 (1C, C-4purine), 148.0 (1C, C-4triazole), 137.4 (1C, C-8purine), 123.4 (1C, C-5triazole), 116.4 (1C, C-5purine), 76.5 (1C, C-3), 71.8 (1C, C-2), 61.1 (1C, C-4), 55.1 (1C, OCH2) 52.2 (1C, NCH2), 34.7 (1C, C1), 24.3 (1C, C-5), 13.7 (1C, SCH3), 12.8 (1C, C-6).
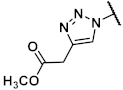
Methyl 1-{[(1R,2R,3S,4R,5S)-4-(6-amino-2-methylthio-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl}-1H-1,2,3-triazole-4-carboxylate (40). The azide 38 (0.054 g, 0.11 mmol) was dissolved in tert-butanol (0.8 mL) and methyl propiolate (0.045 mL, 0.51 mmol, 4.6 eq.), copper(II) acetylacetonate (0.004 g, 0.02 mmol, 0.1 eq.), sodium ascorbate (0.010 g, 0.05 mmol, 0.5 eq.), and H2O (0.8 mL) were added. The mixture was stirred at 80° C for 1.5 h. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the triazole 125 as a colorless solid (Rf = 0.34, ethyl acetate = 100%), yield 0.042 g (66%). C25H32N8O6S (572.64 g/mol). Purity (HPLC: method B): 91% (tR = 14.38 min). Exact mass (APCI): m/z calculated for C20H25N8O4S [M + 2H, -COOC(CH3)3]+ 473.1714, found 473.1709. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 10.11 (s, 1H, NH), 8.77 (s, 1H, 5-CH), 8.18 (s, 1H, 8CHpurine), 5.75 (s, 0.2H, CH2Cl2, solvent: dichloromethane), 5.24 (dd, J = 7.1, 1.4 Hz, 1H, 2CHbicyclohexane), 4.98 (s, 1H, 4-CHbicyclohexane), 4.97 (d, J = 14.5 Hz, 1H, NCHH), 4.83 (dd, J = 7.3, 1.5 Hz, 1H, 3-CHbicyclohexane), 4.49 (d, J = 14.5 Hz, 1H, NCHH), 3.84 (s, 3H, OCH3), 2.57 (s, 3H, SCH3), 2.01 (ddd, J = 9.4, 4.7, 1.6 Hz, 1H, 5CHbicyclohexane), 1.48 (s, 9H, C(CH3)3), 1.39 (s, 3H, C(CH3)2), 1.25 (ddd, J = 9.2, 5.4, 1.6 Hz, 1H, 6CHHbicyclohexane), 1.14 (s, 3H, C(CH3)2), 1.08 (t, J = 5.0 Hz, 1H, 6CHHbicyclohexane); the 1HNMR spectrum displayed small impurities in the range of about 5%. 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 163.8 (1C, C-2purine), 160.7 (1C, C-4carbonyl), 152.0 (1C, C-4purine), 150.8 (1C, C-Ncarbonyl), 149.6 (1C, C-6purine), 141.4 (1C, C-8purine), 138.7 (1C, C4), 129.1 (1C, C-5), 120.7 (1C, C-5purine), 111.5 (1C, C(CH3)2), 88.2 (1C, C-3bicyclohexane), 82.4 (1C, C-2bicyclohexane), 80.2 (1C, C(CH3)3), 59.1 (1C, C-4bicyclohexane), 54.9 (0.1C, CH2Cl2, solvent: dichloromethane), 53.3 (1C, NCH2), 51.8 (1C, OCH3), 36.7 (1C, C-1bicyclohexane), 31.8 (1C, C5bicyclohexane), 27.9 (3C, C(CH3)3), 25.8 (1C, C(CH3)2), 24.1 (1C, C(CH3)2), 14.1 (1C, C6bicyclohexane), 14.0 (1C, SCH3); the 13CNMR spectrum displayed small impurities in the range of about 5%. FT-IR (neat) ṽ (cm−1) = 2978 (C-Haliphat.), 1732 (C = O), 1605, 1578 (C = Caromat.), 1142, 1069, 1053 (C-O).
The triazole (0.038 g, 0.07 mmol) was dissolved in CH3OH (1.6 mL), trifluoroacetic acid (0.20 mL) and H2O (0.20 mL) were added. The mixture was heated to 70 °C overnight. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP C18, 12 g, V = 20 mL) to afford the product 40 as a colorless solid (Rf = 0.32, CH2Cl2:CH3OH = 9:1), yield 0.013 g (44%). C17H20N8O4S (432.46 g/mol). Purity (HPLC: method B): 96% (tR = 4.86 min). Exact mass (LC-MS-ESI): m/z calculated for C17H21N8O4S [M + H]+ 433.1401, found 433.1402. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.76 (s, 1H, 5-CH), 7.55 (s, 1H, 8-CHpurine), 7.31 (s, 2H, NH2), 5.26 (d, J = 4.8 Hz, 1H, 3-OHbicyclohexane), 4.76–4.72 (m, 2H, 2-OHbicyclohexane, NCHH), 4.69 (d, J = 14.5 Hz, 1H, NCHH), 4.63 (s, 1H, 4-CHbicyclohexane), 4.52 (td, J = 7.3, 1.5 Hz, 1H, 2-CHbicyclohexane), 4.09 (q, J = 5.3 Hz, 0.4H, CH3OH, solvent: methanol), 3.90 (ddt, J = 6.5, 4.8, 1.6 Hz, 1H, 3-CHbicyclohexane), 3.83 (s, 3H, OCH3), 3.17 (d, J = 5.2 Hz, 0.8H, CH3OH, solvent: methanol), 2.45 (s, 3H, SCH3), 1.77 (dd, J = 8.5, 4.0 Hz, 1H, 5-CHbicyclohexane), 1.43 (t, J = 4.5 Hz, 1H, 6-CHHbicyclohexane), 0.91 (ddd, J = 8.7, 4.9, 1.6 Hz, 1H, 6-CHHbicyclohexane). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 163.9 (1C, C-2purine), 160.7 (1C, C = O), 155.4 (1C, C-6purine), 149.7 (1C, C-4purine), 138.5 (1C, C-4), 137.6 (1C, C-8purine), 129.4 (1C, C-5), 116.6 (1C, C-5purine), 76.3 (1C, C-3bicyclohexane), 72.2 (1C, C-2bicyclohexane), 61.4 (1C, C-4bicyclohexane), 53.2 (1C, NCH2), 51.7 (1C, OCH3), 48.6 (0.2C, CH3OH, solvent: methanol), 34.4 (1C, C-1bicyclohexane), 24.8 (1C, C-5bicyclohexane), 13.7 (1C, SCH3), 12.9 (1C, C-6bicyclohexane). FT-IR (neat) ṽ (cm−1) = 3341 (O-H), 3148 (N-H), 2978 (C-Haliphat.), 1678 (C = O), 1589 (C = Caromat.), 1130, 1080, 1053 (C-O).
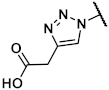
Methyl 2-(1-{[(1R,2R,3S,4R,5S)-4-(6-amino-2-methylthio-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl}-1H-1,2,3-triazol-4-yl)acetate (41). The azide 38 (0.045 g, 0.09 mmol) was dissolved in tert-butanol (0.75 mL) and 3-butynoic acid (0.031 g, 0.37 mmol, 4 eq.), copper(II) acetylacetonate (0.001 g, 0.004 mmol, 0.04 eq.), sodium ascorbate (0.007 g, 0.04 mmol, 0.4 eq.), and H2O (0.75 mL) were added. The mixture was stirred at rt for 6 h. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the triazole as a colorless solid (Rf = 0.18, CH2Cl2:CH3OH = 9:1), yield 0.017 g (33%). C25H32N8O6S (572.64 g/mol). Purity (HPLC: method B): 84% (tR = 12.93 min). Exact mass (LC-MS-ESI): m/z calculated for C25H33N8O6S [M + H]+ 573.2238, found 573.2223. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 10.11 (s, 1H, NH), 8.14 (s, 1H, 8-CHpurine), 8.00 (s, 1H, 5-CHtriazole), 5.19 (s, 1H, 2-CHbicyclohexane), 4.97 (s, 1H, 4-CHbicyclohexane), 4.83 (d, J = 13.7 Hz, 1H, NCHH), 4.79 (d, J = 7.0 Hz, 1H, 3-CHbicyclohexane), 4.50 (d, J = 14.5 Hz, 1H, NCHH), 3.66 (s, 2H, 2-CH2), 3.17 (s, 0.1H, CH3OH, solvent: methanol), 2.58 (s, 3H, SCH3), 1.90 (s, 1H, 5-CHbicyclohexane), 1.49 (s, 9H, C(CH3)3), 1.42 (s, 3H, C(CH3)2), 1.15 (s, 4H, C(CH3)2, 6-CHHbicyclohexane), 1.09 (s, 1H, 6-CHHbicyclohexane). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 171.5 (1C, C-1), 163.8 (1C, C-2purine), 152.0 (1C, C-4purine), 150.8 (1C, C-Ncarbonyl), 149.6 (1C, C-6purine), 141.3 (1C, C-8purine), 140.5 (1C, C-4triazole), 123.7 (1C, C-5triazole), 120.5 (1C, C-5purine), 111.5 (1C, C(CH3)2), 88.3 (1C, C-3bicyclohexane), 82.3 (1C, C-2bicyclohexane), 80.3 (1C, C(CH3)3), 58.9 (1C, C-4bicyclohexane), 52.5 (1C, NCH2), 36.6 (1C, C-1bicyclohexane), 31.7 (1C, C-5bicyclohexane), 31.6 (1C, C-2), 27.9 (3C, C(CH3)3), 25.8 (1C, C(CH3)2), 24.1 (1C, C(CH3)2), 14.0 (2C, C-6bicyclohexane, SCH3). FT-IR (neat) ṽ (cm−1) = 2986 (C-Haliphat.), 1721 (C = O), 1605, 1578 (C = Caromat.), 1142, 1053 (C-O).
The triazole (0.065 g, 0.11 mmol) was dissolved in CH3OH (1.7 mL) and trifluoroacetic acid (0.20 mL) and H2O (0.10 mL) were added. The mixture was heated to 60 °C for 1 d and then at rt overnight. The solvent was evaporated and the residue was purified by semi-preparative HPLC (method F) to afford the ester 41 as a colorless solid (Rf = 0.24, CH2Cl2:CH3OH = 9:1), yield 0.024 g (48%). C18H22N8O4S (446.15 g/mol). Melting point: 214.6 °C. Purity (HPLC: method B): 98% (tR = 5.15 min). Exact mass (LC-MS-ESI): m/z calculated for C18H23N8O4S [M + H]+ 447.1557, found 447.1558. The compound 41 shows two different rotamers a and b in the NMR spectra in a ratio of approximately 10:1. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 8.58 (s, 0.1H, 5-CHtriazole, rotam. b), 8.04 (s, 1H, 5-CHtriazole, rotam. a), 7.72 (s, 0.1H, 8-CHpurine, rotam. b), 7.42 (s, 1H, 8-CHpurine, rotam. a), 7.30 (s, 2.1H, NH2, rotam. a, b), 5.25 (d, J = 4.3 Hz, 1H, 3a-OHbicyclohexane), 4.90 (d, J = 14.4 Hz, 0.1H, NCHHrotam. b), 4.82 (d, J = 7.3 Hz, 0.1H, 2-CHbicyclohexane, rotam. b), 4.81–4.72 (m, 2H, 2-OHbicyclohexane, rotam. a, NCHHrotam. a), 4.68 (s, 0.1H, 4-CHbicyclohexane, rotam. b), 4.61 (s, 1H, 4-CHbicyclohexane, rotam. a), 4.52 (d, J = 14.5 Hz, 1H, NCHHrotam. a), 4.42 (t, J = 5.7 Hz, 1.1H, 2-CHbicyclohexane, rotam. a, NCHHrotam. b), 3.97 (dd, J = 7.0, 1.7 Hz, 0.1H, 3-CHbicyclohexane, rotam. b), 3.86-3.72 (m, 3H, 3-CHbicyclohexane, rotam. a, 2-CH2, rotam. a), 3.63 (s, 3H, OCH3, rotam. a), 3.58 (s, 0.2H, OCH3, rotam. b), 3.52 (d, J = 16.8 Hz, 0.1H, 2-CHHrotam. b), 2.47 (s, 2.9H, SCH3, rotam. a, b), 1.85 (t, J = 4.7 Hz, 0.1H, 6-CHHbicyclohexane, rotam. b), 1.67 (dd, J = 8.7, 4.0 Hz, 1H, 5-CHbicyclohexane, rotam. a), 1.60 (dd, J = 9.1, 4.6 Hz, 0.1H, 5-CHbicyclohexane, rotam. b), 1.42 (t, J = 4.5 Hz, 1H, 6-CHHbicyclohexane, rotam. a), 0.83 (ddd, J = 8.7, 4.9, 1.5 Hz, 1H, 6-CHHbicyclohexane, rotam. a), 0.67–0.57 (m, 0.1H, 6-CHHbicyclohexane, rotam. b); the signal for 2-CHHrotam. b is located under the H2O signal and therefore only visible in 2D spectra. 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 170.4 (1C, C = O), 163.8 (1C, C-2purine), 155.4 (1C, C-6purine), 149.8 (1C, C-4purine), 139.8 (1C, C-4triazole), 137.4 (1C, C-8purine), 124.2 (1C, C-5triazole), 116.5 (1C, C-5purine), 76.5 (1C, C-3bicyclohexane), 71.8 (1C, C-2bicyclohexane), 61.2 (1C, C-4bicyclohexane), 52.2 (1C, NCH2), 51.9 (1C, OCH3), 34.7 (1C, C-1bicyclohexane), 31.1 (1C, C-2), 24.4 (1C, C-5bicyclohexane), 13.7 (1C, SCH3), 12.9 (1C, C-6bicyclohexane). The resolution was too low to identify the 13C signals for rotamer b, therefore only rotamer a is described here. FT-IR (neat) ṽ (cm−1) = 3352 (O-H), 3244 (N-H), 1751 (C = O), 1624, 1585 (C = Caromat.), 1142, 1119, 1084, 1057 (C-O).
Methyl 3-(1-{[(1R,2R,3S,4R,5S)-4-(6-amino-2-methylthio-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl}-1H-1,2,3-triazol-4-yl)propanoate (42). The azide 38 (0.028 g, 0.06 mmol) was dissolved in tert-butanol (0.5 mL) and 4-pentynoic acid (0.024 g, 0.24 mmol, 4.3 eq.), copper(II) acetylacetonate (0.001 g, 0.004 mmol, 0.07 eq.), sodium ascorbate (0.006 g, 0.03 mmol, 0.5 eq.), and H2O (0.5 mL) were added. The mixture was stirred at rt for 5 h. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the triazole as a colorless solid (Rf = 0.27, CH2Cl2:CH3OH = 9:1), yield 0.018 g (54%). C26H34N8O6S (586.67 g/mol). Purity (HPLC: method B): 96% (tR = 12.64 min). Exact mass (LC-MS-ESI): m/z calculated for C26H34DN8O6S [M + H]+ 588.2458, found 588.2452. 1H-NMR (400 MHz, CD3OD) δ (ppm) = 7.99 (s, 0.1H, 8-CHpurine), 7.82 (s, 1H, 5-CHtriazole), 5.23 (dd, J = 7.1, 1.4 Hz, 1H, 2-CHbicyclohexane), 5.00 (s, 1H, 4-CHbicyclohexane), 4.88–4.82 (m, 1H, 3-CHbicyclohexane, NCHH), 4.52 (d, J = 14.6 Hz, 1H, NCHH), 3.35 (s, 0.2H, CH3OH, solvent: methanol), 2.99 (t, J = 7.4 Hz, 2H, 3-CH2), 2.67 (t, J = 7.4 Hz, 2H, 2-CH2), 2.63 (s, 3H, SCH3), 1.89 (ddd, J = 9.4, 4.6, 1.6 Hz, 1H, 5-CHbicyclohexane), 1.58 (s, 9H, C(CH3)3), 1.48 (s, 3H, C(CH3)2), 1.26 (dd, J = 5.7, 4.7 Hz, 1H, 6-CHHbicyclohexane), 1.22 (s, 3H, C(CH3)2), 1.13 (ddd, J = 9.4, 5.7, 1.5 Hz, 1H, 6-CHHbicyclohexane); 8-CHpurine showed a reduced intensity and no coupling interaction due to occurrence of deuterium exchange at this position. 13C-NMR (101 MHz, CD3OD) δ (ppm) = 176.3 (1C, C-1), 167.4 (1C, C-2purine), 153.3 (1C, C-4purine), 152.5 (1C, C-Ncarbonyl), 150.9 (1C, C-6purine), 147.9 (1C, C-4triazole), 142.6 (1C, C-8purine), 123.9 (1C, C-5triazole), 120.7 (1C, C-5purine), 113.6 (1C, C(CH3)2), 90.0 (1C, C-3bicyclohexane), 84.4 (1C, C-2bicyclohexane), 82.7 (1C, C(CH3)3), 61.6 (1C, C-4bicyclohexane), 54.7 (1C, NCH2), 38.2 (1C, C-1bicyclohexane), 34.5 (1C, C-2), 33.2 (1C, C-5bicyclohexane), 28.5 (3C, C(CH3)3), 26.2 (1C, C(CH3)2), 24.4 (1C, C(CH3)2), 22.0 (1C, C-3), 15.3 (1C, C-6bicyclohexane), 14.8 (1C, SCH3). FT-IR (neat) ṽ (cm−1) = 2978 (C-Haliphat.), 1717 (C = O), 1605, 1578 (C = Caromat.), 1142, 1053 (C-O).
The triazole acid (0.015 g, 0.03 mmol) was dissolved in CH3OH (0.4 mL) and trifluoroacetic acid (0.05 mL) and H2O (0.05 mL) were added. The mixture was stirred at rt for 3 d. The solvent was evaporated and the residue was purified by semi-preparative HPLC (method D) to afford the ester 42 as a colorless solid (Rf = 0.24, CH2Cl2:CH3OH = 9:1), yield 0.004 g (34%). C19H24N8O4S (460.16 g/mol). Purity (HPLC: method B): 98% (tR = 5.67 min). Exact mass (LC-MS-ESI): m/z calculated for C19H24DN8O4S [M + H]+ 462.1777, found 462.1775 and for C19H25N8O4S [M + H]+ 461.1714, found 461.1709. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 7.88 (s, 1H, 5-CHtriazole), 7.30 (s, 0.4H, 8CHpurine), 7.29 (s, 2H, NH2), 5.24 (d, J = 4.7 Hz, 1H, 3-OHbicyclohexane), 4.76 (d, J = 7.3 Hz, 1H, 2OHbicyclohexane), 4.73 (d, J = 14.5 Hz, 1H, NCHH), 4.61 (d, J = 1.7 Hz, 1H, 4-CHbicyclohexane), 4.44 (d, J = 14.5 Hz, 1H, NCHH), 4.40 (td, J = 7.0, 1.7 Hz, 1H, 2CHbicyclohexane), 4.03 (q, J = 7.1 Hz, 0.1H, CH2, solvent: ethyl acetate), 3.81 (tt, J = 6.3, 1.6 Hz, 1H, 3-CHbicyclohexane), 3.58 (s, 3H, OCH3), 2.88 (t, J = 7.5 Hz, 2H, 3-CH2), 2.67 (t, J = 7.5 Hz, 2H, 2-CH2), 2.46 (s, 3H, SCH3), 1.99 (s, 0.1H, OCH3, solvent: ethyl acetate), 1.65 (dd, J = 8.3, 4.1 Hz, 1H, 5CHbicyclohexane), 1.42 (t, J = 4.5 Hz, 1H, 6CHHbicyclohexane), 1.17 (t, J = 7.1 Hz, 0.1H, CH2CH3, solvent: ethyl acetate), 0.81 (ddd, J = 8.7, 4.9, 1.6 Hz, 1H, 6-CHHbicyclohexane); 8-CHpurine showed a reduced intensity due to occurrence of deuterium exchange at this position. 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 172.6 (1C, C-1), 163.8 (1C, C-2purine), 155.4 (1C, C6purine), 149.7 (1C, C-4purine), 145.4 (1C, C-4triazole), 137.3 (1C, C-8purine), 122.7 (1C, C-5triazole), 116.4 (1C, C-5purine), 76.3 (1C, C-3bicyclohexane), 71.7 (1C, C-2bicyclohexane), 61.1 (1C, C-4bicyclohexane), 52.0 (1C, NCH2), 51.4 (1C, OCH3), 34.8 (1C, C-1bicyclohexane), 32.8 (1C, C-2), 24.2 (1C, C-5bicyclohexane), 20.7 (1C, C-3), 13.6 (1C, SCH3), 12.9 (1C, C6bicyclohexane).
(1R,2R,3S,4R,5S)-4-(6-Amino-2-methylthio-9H-purin-9-yl)-1-{[4-(aminomethyl)-1H-1,2,3-triazol-1-yl]methyl}bicyclo[3.1.0]hexane-2,3-diol (43). The azide 38 (0.029 g, 0.06 mmol) was dissolved in tert-butanol (0.5 mL) and propargylamine (0.016 mL, 0.25 mmol, 4.2 eq.), copper(II) acetylacetonate (0.001 g, 0.004 mmol, 0.06 eq.), sodium ascorbate (0.007 g, 0.04 mmol, 0.6 eq.), and H2O (0.5 mL) were added. The mixture was stirred for 5 h at rt. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0 + 0.1% trifluoroacetic acid, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL). The intermediate already partly decomposed (deprotection) during this purification and was therefore dissolved in CH3OH (0.5 mL). Next, triflouroacetic acid (0.05 mL) and H2O (0.10 mL) were added and the mixture was heated to 70 °C for 1 d. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0 + 0.1% trifluoroacetic acid, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL), but the product remained impure. The impure product was purified by semi-preparative HPLC (method D) to afford the pure product 43 as a colorless solid (Rf = 0.15, CH3OH = 100% + 1% triethylamine), yield 0.002 g (8%). C16H21N9O2S (403.47 g/mol). Purity (HPLC: method D): 95% (tR = 8.88 min). Exact mass (LC-MS-ESI): m/z calculated for C16H22N9O2S [M + H]+ 404.1612, found 404.1624. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 7.94 (s, 1H, 5-CHtriazole), 7.30 (s, 2H, NH2), 7.29 (s, 1H, 8-CHpurine), 4.76 (d, J = 14.5 Hz, 1H, NCHH), 4.62 (s, 1H, 4-CH), 4.46 (d, J = 14.4 Hz, 1H, NCHH), 4.41 (dd, J = 6.6, 1.5 Hz, 1H, 2-CH), 3.81 (dt, J = 6.6, 1.6 Hz, 1H, 3-CH), 3.77 (s, 2H, NH2CH2), 2.46 (s, 3H, SCH3), 1.66 (dd, J = 8.7, 4.0 Hz, 1H, 5CH), 1.43 (t, J = 4.5 Hz, 1H, 6-CHH), 0.83 (ddd, J = 8.7, 4.9, 1.6 Hz, 1H, 6-CHH). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 163.8 (1C, C-2purine), 155.4 (1C, C-6purine), 149.7 (1C, C-4purine), 149.1 (1C, C-4triazole), 137.3 (1C, C-8purine), 122.7 (1C, C-5triazole), 116.5 (1C, C-5purine), 76.3 (1C, C-3), 71.7 (1C, C-2), 61.1 (1C, C-4), 52.0 (1C, NCH2), 37.1 (1C, NH2CH2), 34.9 (1C, C-1), 24.3 (1C, C-5), 13.6 (1C, SCH3), 12.9 (1C, C-6).
1-{[(1R,2R,3S,4R,5S)-4-(6-Amino-2-methylthio-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl}-1H-1,2,3-triazole-4-carboxylic acid (44). The ester 40 (0.082 g, 0.19 mmol) was suspended in CH3CN (0.7 mL) and H2O (2 mL), and NaOH solution-(2 M, 0.30 mL) was added. The mixture was heated to 60 °C overnight. The solvent was evaporated and the residue was purified by semi preparative HPLC (method A) to afford the carboxylic acid 44 as a colorless solid (Rf = 0.44, CH2Cl2:CH3OH = 1:1), yield 0.036 g (45%). C16H18N8O4S (418.43 g/mol). Melting point: 139.3 °C. Purity (HPLC: method B): > 99% (tR = 7.05 min). Exact mass (LC-MS-ESI): m/z calculated for C16H19N8O4S [M + H]+ 419.1244, found 419.1241. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.27 (s, 1H, 5-CH), 7.35 (s, 2H, NH2), 7.24 (s, 1H, 8-CHpurine), 5.50 (s, 2H, 2-OHbicyclohexane, 3-OHbicyclohexane), 4.71 (d, J = 14.6 Hz, 1H, NCHH), 4.63 (d, J = 2.0 Hz, 1H, 4-CHbicyclohexane), 4.53 (d, J = 14.5 Hz, 1H, NCHH), 4.44 (dd, J = 6.5, 1.5 Hz, 1H, 2-CHbicyclohexane), 3.76 (dt, J = 6.6, 1.5 Hz, 1H, 3-CHbicyclohexane), 2.47 (s, 3H, SCH3), 2.07 (s, 0.1H, CH3CN, solvent: acetonitrile), 1.67 (dd, J = 8.4, 3.7 Hz, 1H, 5-CHbicyclohexane), 1.46 (t, J = 4.4 Hz, 1H, 6-CHHbicyclohexane), 0.85 (ddd, J = 8.7, 4.8, 1.6 Hz, 1H, 6-CHHbicyclohexane). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 164.2 (1C, C = O), 163.9 (1C, C-2purine), 155.4 (1C, C-6purine), 149.7 (1C, C-4purine), 148.3 (1C, C-4), 137.1 (1C, C-8purine), 126.5 (1C, C-5), 116.4 (1C, C-5purine), 76.8 (1C, C-3bicyclohexane), 71.7 (1C, C-2bicyclohexane), 60.8 (1C, C-4bicyclohexane), 52.2 (1C, NCH2), 34.7 (1C, C-1bicyclohexane), 24.1 (1C, C-5bicyclohexane), 13.7 (1C, SCH3), 13.0 (1C, C-6bicyclohexane). FT-IR (neat) ṽ (cm−1) = 3341 (O-H), 3198 (N-H), 1585 (C = O), 1539 (C = Caromat.), 1057 (C-O).
2-(1-{[(1R,2R,3S,4R,5S)-4-(6-Amino-2-methylthio-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl}-1H-1,2,3-triazol-4-yl)acetic acid (45). The azide 38 (0.045 g, 0.09 mmol) was dissolved in tert-butanol (0.75 mL) and 3-butynoic acid (0.031 g, 0.37 mmol, 4 eq.), copper(II) acetylacetonate (0.001 g, 0.004 mmol, 0.04 eq.), sodium ascorbate (0.007 g, 0.04 mmol, 0.4 eq.), and H2O (0.75 mL) were added. The mixture was stirred at rt for 6 h. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the triazole as a colorless solid (Rf = 0.18, CH2Cl2:CH3OH = 9:1), yield 0.017 g (33%). C25H32N8O6S (572.64 g/mol). Purity (HPLC: method B): 84% (tR = 12.93 min). Exact mass (LC-MS-ESI): m/z calculated for C25H33N8O6S [M + H]+ 573.2238, found 573.2223. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 10.11 (s, 1H, NH), 8.14 (s, 1H, 8-CHpurine), 8.00 (s, 1H, 5-CHtriazole), 5.19 (s, 1H, 2-CHbicyclohexane), 4.97 (s, 1H, 4CHbicyclohexane), 4.83 (d, J = 13.7 Hz, 1H, NCHH), 4.79 (d, J = 7.0 Hz, 1H, 3-CHbicyclohexane), 4.50 (d, J = 14.5 Hz, 1H, NCHH), 3.66 (s, 2H, 2-CH2), 3.17 (s, 0.1H, CH3OH, solvent: methanol), 2.58 (s, 3H, SCH3), 1.90 (s, 1H, 5CHbicyclohexane), 1.49 (s, 9H, C(CH3)3), 1.42 (s, 3H, C(CH3)2), 1.15 (s, 4H, C(CH3)2, 6CHHbicyclohexane), 1.09 (s, 1H, 6CHHbicyclohexane). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 171.5 (1C, C-1), 163.8 (1C, C-2purine), 152.0 (1C, C4purine), 150.8 (1C, C-Ncarbonyl), 149.6 (1C, C-6purine), 141.3 (1C, C-8purine), 140.5 (1C, C-4triazole), 123.7 (1C, C-5triazole), 120.5 (1C, C-5purine), 111.5 (1C, C(CH3)2), 88.3 (1C, C-3bicyclohexane), 82.3 (1C, C-2bicyclohexane), 80.3 (1C, C(CH3)3), 58.9 (1C, C4bicyclohexane), 52.5 (1C, NCH2), 36.6 (1C, C1bicyclohexane), 31.7 (1C, C-5bicyclohexane), 31.6 (1C, C-2), 27.9 (3C, C(CH3)3), 25.8 (1C, C(CH3)2), 24.1 (1C, C(CH3)2), 14.0 (2C, C6bicyclohexane, SCH3). FT-IR (neat) ṽ (cm−1) = 2986 (C-Haliphat.), 1721 (C = O), 1605, 1578 (C = Caromat.), 1142, 1053 (CO).
The triazole (0.048 g, 0.08 mmol) was dissolved in CH3CN (0.4 mL) and H2O (1.4 mL), and trifluoroacetic acid (0.20 mL) was added. The mixture was heated to 60 °C overnight. The solvent was evaporated and the was purified by semi-preparative HPLC (method E) to afford the product 45 as a colorless solid, yield 0.019 g (51%). C17H20N8O4S (432.46 g/mol). Melting point: 179.2 °C. Purity (HPLC: D): 99% (tR = 9.71 min). Exact mass (LC-MS-ESI): m/z calculated for C17H21N8O4S [M + H]+ 433.1401, found 433.1411. The compound 45 shows two different rotamers a and b in the NMR spectra in a ratio of approximately 7:1. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 8.54 (s, 0.2H, 5-CHtriazole, rotam. b), 8.02 (s, 1H, 5-CHtriazole, rotam. a), 7.70 (s, 0.1H, 8-CHpurine, rotam. b), 7.43 (s, 1H, 8-CHpurine, rotam. a), 7.30 (s, 2.4H, NH2, rotam. a, b), 4.86 (d, J = 14.4 Hz, 0.2H, NCHHrotam. b), 4.81 (d, J = 6.5 Hz, 0.2H, 2-CHbicyclohexane, rotam. b), 4.75 (d, 1H, J = 14.5 Hz, NCHHrotam. a), 4.68 (s, 0.2H, 4-CHbicyclohexane, rotam. b), 4.61 (s, 1H, 4-CHbicyclohexane, rotam. a), 4.52 (d, J = 14.5 Hz, 1H, NCHHrotam. a), 4.47 (s, 0.2H, NCHHrotam. b), 4.42 (d, J = 6.6 Hz, 1H, 2-CHbicyclohexane, rotam. a), 3.95 (d, J = 6.7 Hz, 0.2H, 3-CHbicyclohexane, rotam. b), 3.81 (d, 1H, J = 6.4 Hz, 3-CHbicyclohexane, rotam. a) 3.66 (t, J = 19.2 Hz, 2H, 2-CH2, rotam. a), 3.43 (d, J = 16.7 Hz, 0.1H, 2-CHHrotam. b), 3.18 (d, J = 16.9 Hz, 0.1H, 2-CHHrotam. b), 2.47 (s, 3.5H, SCH3, rotam. a, b), 1.85 (s, 0.2H, 6-CHHbicyclohexane, rotam. b), 1.66 (dd, J = 8.9, 3.9 Hz, 1H, 5-CHbicyclohexane, rotam. a), 1.59 (s, 0.2H, 5-CHbicyclohexane, rotam. b), 1.41 (t, J = 4.5 Hz, 1H, 6-CHHbicyclohexane, rotam. a), 0.82 (dd, J = 9.0, 4.9 Hz, 1H, 6-CHHbicyclohexane, rotam. a), 0.63 (s, 0.2H, 6-CHHbicyclohexane, rotam. b). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 171.6 (1C, C = O), 163.8 (1C, C-2purine), 155.4 (1C, C-6purine), 149.8 (1C, C-4purine), 140.6 (1C, C-4triazole), 137.4 (1C, C-8purine), 124.1 (1C, C-5triazole), 116.5 (1C, C-5purine), 76.6 (1C, C-3bicyclohexane), 71.8 (1C, C-2bicyclohexane), 61.1 (1C, C-4bicyclohexane), 52.2 (1C, NCH2), 34.7 (1C, C-1bicyclohexane), 31.7 (1C, C-2), 24.4 (1C, C-5bicyclohexane), 13.7 (1C, SCH3), 12.9 (1C, C-6bicyclohexane); the resolution was too low to identify the 13C signals for rotamer b, therefore only rotamer a is described here. FT-IR (neat) ṽ (cm−1) = 3518, 3329, 3198 (O-H), 2920 (C-Haliphat.), 1651 (C = O), 1586 (C = Caromat.), 1123, 1092 (C-O).
3-[1-({(1R,2R,3S,4R,5S)-4-[6-(tert-Butoxycarbonyl)amino-2-methylthio-9H-purin-9-yl]-2,3-dihydroxy-2,3-O-isopropylidenebicyclo[3.1.0]hex-1-yl}methyl)-1H-1,2,3-triazol-4-yl]propanoic acid (46). The azide 38 (0.028 g, 0.06 mmol) was dissolved in tert-butanol (0.5 mL) and 4p-entynoic acid (0.024 g, 0.24 mmol, 4.3 eq.), copper(II) acetylacetonate (0.001 g, 0.004 mmol, 0.07 eq.), sodium ascorbate (0.006 g, 0.03 mmol, 0.5 eq.), and H2O (0.5 mL) were added. The mixture was stirred at rt for 5 h. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the triazole as a colorless solid (Rf = 0.27, CH2Cl2:CH3OH = 9:1), yield 0.018 g (54%). C26H34N8O6S (586.67 g/mol). Purity (HPLC: method B): 96% (tR = 12.64 min). Exact mass (LC-MS-ESI): m/z calculated for C26H34DN8O6S [M + H]+ 588.2458, found 588.2452. 1H-NMR (400 MHz, CD3OD) δ (ppm) = 7.99 (s, 0.1H, 8-CHpurine), 7.82 (s, 1H, 5-CHtriazole), 5.23 (dd, J = 7.1, 1.4 Hz, 1H, 2-CHbicyclohexane), 5.00 (s, 1H, 4-CHbicyclohexane), 4.88–4.82 (m, 1H, 3-CHbicyclohexane, NCHH), 4.52 (d, J = 14.6 Hz, 1H, NCHH), 3.35 (s, 0.2H, CH3OH, solvent: methanol), 2.99 (t, J = 7.4 Hz, 2H, 3-CH2), 2.67 (t, J = 7.4 Hz, 2H, 2-CH2), 2.63 (s, 3H, SCH3), 1.89 (ddd, J = 9.4, 4.6, 1.6 Hz, 1H, 5-CHbicyclohexane), 1.58 (s, 9H, C(CH3)3), 1.48 (s, 3H, C(CH3)2), 1.26 (dd, J = 5.7, 4.7 Hz, 1H, 6-CHHbicyclohexane), 1.22 (s, 3H, C(CH3)2), 1.13 (ddd, J = 9.4, 5.7, 1.5 Hz, 1H, 6-CHHbicyclohexane); 8-CHpurine showed a reduced intensity and no coupling interaction due to occurrence of deuterium exchange at this position. 13C-NMR (101 MHz, CD3OD) δ (ppm) = 176.3 (1C, C-1), 167.4 (1C, C-2purine), 153.3 (1C, C-4purine), 152.5 (1C, C-Ncarbonyl), 150.9 (1C, C-6purine), 147.9 (1C, C-4triazole), 142.6 (1C, C-8purine), 123.9 (1C, C-5triazole), 120.7 (1C, C-5purine), 113.6 (1C, C(CH3)2), 90.0 (1C, C-3bicyclohexane), 84.4 (1C, C-2bicyclohexane), 82.7 (1C, C(CH3)3), 61.6 (1C, C-4bicyclohexane), 54.7 (1C, NCH2), 38.2 (1C, C-1bicyclohexane), 34.5 (1C, C-2), 33.2 (1C, C-5bicyclohexane), 28.5 (3C, C(CH3)3), 26.2 (1C, C(CH3)2), 24.4 (1C, C(CH3)2), 22.0 (1C, C-3), 15.3 (1C, C-6bicyclohexane), 14.8 (1C, SCH3). FT-IR (neat) ṽ (cm−1) = 2978 (C-Haliphat.), 1717 (C = O), 1605, 1578 (C = Caromat.), 1142, 1053 (C-O).
The triazole (0.070 g, 0.12 mmol) was dissolved in CH3CN (0.7 mL) and H2O (2 mL), and trifluoroacetic acid (0.30 mL) was added. The mixture was stirred at rt for 1 d. The solvent was evaporated and the residue was purified by semi-preparative HPLC (method E) to afford the carboxylic acid 46 as a colorless solid, yield 0.010 g (19%). C18H22N8O4S (446.49 g/mol). Melting point: 133.3 °C Purity (HPLC: D): 99% (tR = 10.32 min). Exact mass (LC-MS-ESI): m/z calculated for C18H23N8O4S [M + H]+ 447.1557, found 447.1557. The compound 46 shows three different rotamers a, b and c in the NMR spectra in a ratio of approximately 10:6:3. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.40 (s, 0.6H, 5-CHtriazole, rotam. b), 7.97 (s, 0.3H, 5-CHtriazole, rotam. c), 7.89 (s, 1H, 5-CHtriazole, rotam. a), 7.58 (s, 0.6H, 8-CHpurine, rotam. b), 7.36 (s, 0.3H, 8-CHpurine, rotam. c), 7.30 (s, 2.5H, 8-CHpurine, rotam. a, NH2, rotam. b, c), 7.26 (s, 2H, NH2, rotam. a), 5.24 (s, 0.6H, 3-OHbicyclohexane), 4.80-4.75 (m, 1.3H, 2-CHbicyclohexane, rotam. b, NCHHrotam. b), 4.73 (d, J = 14.5 Hz, 1H, NCHHrotam. a), 4.69 (s, 0.3H, 4-CHbicyclohexane, rotam. c), 4.67 (s, 0.6H, 4-CHbicyclohexane, rotam. b), 4.64 (d, J = 7.1 Hz, 0.3H, 2-CHbicyclohexane, rotam. c), 4.62 (d, J = 1.7 Hz, 1H, 4-CHbicyclohexane, rotam. a), 4.57 (d, J = 14.5 Hz, 0.3H, NCHHrotam. c), 4.49–4.42 (m, 1.8H, NCHHrotam. a, b, c), 4.41 (dd, J = 6.7, 1.5 Hz, 1H, 2-CHbicyclohexane, rotam. a), 3.98 (d, J = 7.0 Hz, 0.2H, 3-CHbicyclohexane, rotam. c), 3.92 (dd, J = 6.9, 1.7 Hz, 0.6H, 3-CHbicyclohexane, rotam. b), 3.79 (dt, J = 6.6, 1.6 Hz, 1H, 3-CHbicyclohexane, rotam. a), 2.85 (t, J = 7.6 Hz, 2H, 3-CH2, rotam. a), 2.80 (t, J = 7.6 Hz, 0.6H, 3-CH2, rotam. c), 2.66 (t, J = 7.6 Hz, 1.2H, 3-CH2, rotam. b), 2.58 (dd, J = 8.3, 6.9 Hz, 2H, 2-CH2, rotam. a), 2.53 (t, J = 7.0 Hz, 0.6H, 2-CH2, rotam. c), 2.48 (s, 1.2H, 2-CH2, rotam. b), 2.46 (s, 4.5H, SCH3, rotam. a, b, c), 2.07 (s, 0.1H, CH3CN, solvent: acetonitrile), 1.85 (t, J = 4.6 Hz, 0.6H, 6-CHHbicyclohexane, rotam. b), 1.68–1.62 (m, 1.3H, 5-CHbicyclohexane, rotam. a, 6-CHHbicyclohexane, rotam. c), 1.58 (ddd, J = 9.1, 4.5, 1.7 Hz, 0.6H, 5-CHbicyclohexane, rotam. b), 1.44 (dd, J = 4.6, 3.6 Hz, 0.3H, 5-CHbicyclohexane, rotam. c), 1.42 (t, J = 4.5 Hz, 1H, 6-CHHbicyclohexane, rotam. a), 0.81 (ddd, J = 8.7, 4.9, 1.6 Hz, 1H, 6-CHHbicyclohexane, rotam. a), 0.68 (ddd, J = 8.5, 3.0, 1.6 Hz, 0.6H, 6-CHHbicyclohexane, rotam. b), 0.60 (dd, J = 9.4, 4.7 Hz, 0.3H, 6-CHHbicyclohexane, rotam. c); 3-OHbicyclohexane could not be clearly assigned to one of the three rotamers. 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 173.7 (0.6C, C-1rotam. a, c), 173.7 (0.3C, C-1rotam. b), 163.8 (1C, C-2purine, rotam. a), 163.7 (0.3C, C-2purine, rotam. b), 163.6 (0.2C, C-2purine, rotam. c), 155.4 (1C, C-6purine, rotam. a), 155.4 (0.5C, C-6purine, rotam. b, c), 149.7 (1C, C-4purine, rotam. a), 149.6 (0.5C, C-4purine, rotam. b c), 145.8 (0.6C, C-4triazole, rotam. a), 145.4 (0.1C, C-4triazole, rotam. c), 145.1 (0.4C, C-4triazole, rotam. b), 137.8 (0.3C, C-8purine, rotam. b), 137.6 (0.1C, C-8purine, rotam. c), 137.3 (1C, C-8purine, rotam. a), 122.9 (0.4C, C-5triazole, rotam. b), 122.8 (0.2C, C-5triazole, rotam. c), 122.7 (1C, C-5triazole, rotam. a), 116.5 (1C, C-5purine, rotam. a), 116.4 (0.4C, C-5purine, rotam. b), 116.4 (0.1C, C-5purine, rotam. c), 86.4 (0.4C, C-3bicyclohexane, rotam. b), 86.1 (0.1C, C-3bicyclohexane, rotam. c), 79.7 (0.5C, C-2bicyclohexane, rotam. b), 78.6 (0.1C, C-2bicyclohexane, rotam. c), 76.3 (1C, C-3bicyclohexane, rotam. a), 71.7 (1C, C-2bicyclohexane, rotam. a), 61.1 (1C, C-4bicyclohexane, rotam. a), 61.0 (0.2C, C-4bicyclohexane, rotam. c), 60.7 (0.5C, C-4bicyclohexane, rotam. b), 54.4 (0.4C, NCH2,rotam. b), 53.2 (0.2C, NCH2, rotam. c), 52.0 (1C, NCH2, rotam. a), 37.4 (0.1C, C-1bicyclohexane, rotam. c), 36.4 (0.4C, C-1bicyclohexane, rotam. b), 34.8 (1C, C-1bicyclohexane, rotam. a), 33.4 (0.9C, C-2rotam. b, c), 33.3 (1C, C-2rotam. a), 30.1 (0.5C, b-C-5bicyclohexane, rotam. b), 29.1 (0.2C, C-5bicyclohexane, rotam. c), 24.2 (1C, C-5bicyclohexane, rotam. a), 20.8 (0.3C, C-3rotam. c), 20.8 (1C, C-3rotam. a), 20.7 (0.6C, C-3rotam. b), 13.7 (1C, SCH3, rotam. a, c), 13.6 (0.4C, SCH3,rotam. b), 12.9 (1C, C-6bicyclohexane, rotam. a), 12.7 (0.2C, C-6bicyclohexane, rotam. c), 12.5 (1C, C-6bicyclohexane, rotam. b). FT-IR (neat) ṽ (cm−1) = 3098 (N-H), 2943 (C-Haliphat.), 1713, 1670 (C = O), 1539 (C = Caromat.), 1192, 1142 (C-O).
Triethylammonium 2-{[(1R,2R,3S,4R,5S)-4-(6-Amino-2-methylthio-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl}amino-3,4-dioxocyclobut-1-en-1-olate (47). The azide 38 (0.032 g, 0.07 mmol) was dissolved in CH3OH (2 mL) and Pd/C (10 wt%, 0.003 g, 0.003 mmol, 0.004 eq.) was added; the mixture was flushed several times with H2 gas. The reaction was stirred overnight at 5 bar H2 atmosphere. The solvent was evaporated and the residue was purified by fc (CH2Cl2:CH3OH = 95:5 + 1% triethylamine, Ø = 2 cm, l = 20 cm, V = 10 mL) to afford the amine as a colorless solid (Rf = 0.38, CH2Cl2:CH3OH = 95:5 + 1% triethylamine), yield 0.018 g (60%). C21H30N6O4S (462.57 g/mol). Melting point: 111.4 °C. Purity (HPLC: method D): 90% (tR = 15.74 min). Exact mass (LC-MS-ESI): m/z calculated for C21H31N6O4S [M + H]+ 463.2122, found 463.2125. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.45 (s, 1H, 8-CHpurine), 5.26 (d, J = 7.1 Hz, 1H, 2CHbicyclohexane), 4.93 (s, 1H, 4-CHbicyclohexane), 4.73 (dd, J = 7.3, 1.5 Hz, 1H, 3CHbicyclohexane), 3.17 (s, 0.3H, CH3OH, solvent: methanol), 2.95 (d, J = 13.3 Hz, 1H, NCHH), 2.82 (d, J = 13.3 Hz, 1H, NCHH), 2.59 (s, 3H, SCH3), 1.68 (ddd, J = 8.3, 5.2, 1.5 Hz, 1H, 5CHbicyclohexane), 1.48 (s, 9H, C(CH3)3), 1.45 (s, 3H, C(CH3)2), 1.18 (s, 3H, C(CH3)2), 0.94–0.90 (m, 2H, 6CH2 bicyclohexane). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 163.7 (1C, C-2purine), 152.0 (1C, C-4purine), 150.9 (1C, C = O), 149.6 (1C, C-6purine), 141.5 (1C, C-8purine), 120.7 (1C, C-5purine), 111.3 (1C, C(CH3)2), 88.3 (1C, C-3bicyclohexane), 82.1 (1C, C-2bicyclohexane), 80.2 (1C, C(CH3)3), 59.2 (1C, C-4bicyclohexane), 43.6 (1C, NCH2), 38.4 (1C, C-1bicyclohexane), 29.7 (1C, C5bicyclohexane), 27.9 (3C, C(CH3)3), 25.9 (1C, C(CH3)2), 24.2 (1C, C(CH3)2), 14.0 (1C, SCH3), 13.3 (1C, C-6bicyclohexane). FT-IR (neat) ṽ (cm−1) = 2978, 2928 (C-Haliphat.), 1751, 1721 (C = O), 1605, 1574 (C = Caromat.), 1142, 1053 (C-O).
The amine (0.105 g, 0.23 mmol) was dissolved in CH2Cl2 (2 mL). Dimethyl squarate (0.104 g, 0.73 mmol, 3.2 eq.) and triethylamine (0.20 mL, 1.44 mmol, 6.4 eq.) were added and the mixture was stirred at rt overnight. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 100:0, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the squaramide as a colorless solid (Rf = 0.33, ethyl acetate = 100%), yield 0.087 g (67%). C26H32N6O7S (572.64 g/mol). Melting point: 148.4 °C. Purity (HPLC: method B): 90% (tR = 13.02 min). Exact mass (LC-MS-ESI): m/z calculated for C26H33N6O7S [M + H]+ 573.2126, found 573.2130. The compound shows two different rotamers a and b in the NMR spectra in a ratio of approximately 5:4. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 10.09 (s, 1H, NHpurine, rotam. a/b), 8.94 (s, 0.5H, NHsquaramide, rotam. a), 8.74 (s, 0.4H, NHsquaramide, rotam. b), 8.26 (s, 1H, 8-CHpurine, rotam. a/b), 5.25 (t, J = 6.6 Hz, 1H, 2-CHbicyclohexane, rotam. a/b), 4.93 (s, 1H, 4-CHbicyclohexane, rotam. a/b), 4.88 (d, J = 7.1 Hz, 0.4H, 3-CHbicyclohexane, rotam. b), 4.83 (d, J = 7.2 Hz, 0.5H, 3-CHbicyclohexane, rotam. a), 4.29 (s, 1.2H, OCH3, rotam. b), 4.24 (s, 1.5H, OCH3, rotam. a), 3.77 (q, J = 14.2 Hz, 0.9H, NCH2, rotam. b), 3.69 (d, J = 13.2 Hz, 1H, NCHHrotam. a), 3.50 (d, J = 13.2 Hz, 1H, NCHHrotam. a), 3.17 (s, 0.2H, CH3OH, solvent: methanol), 2.58 (d, J = 4.0 Hz, 3H, SCH3, rotam. a/b), 1.77 (dt, J = 13.0, 6.7 Hz, 1H, 5-CHbicyclohexane, rotam. a/b), 1.48 (s, 9H, C(CH3)3, rotam. a/b), 1.45 (s, 3H, C(CH3)2,rotam. a/b), 1.18 (s, 3H, C(CH3)2,rotam. a/b), 0.98 (d, J = 7.0 Hz, 1H, 6-CH2 bicyclohexane, rotam. a/b). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 189.3 (1C, C-1squaramide, rotam. a/b), 182.5 (0.3C, C-2squaramide, rotam. a), 182.1 (0.3C, C-2squaramide, rotam. b), 177.7 (0.3C, C-4squaramide, rotam. b), 176.7 (0.4C, C-4squaramide, rotam. a), 172.4 (0.4C, C-3squaramide, rotam. a), 171.9 (0.3C, C-3squaramide, rotam. a), 163.8 (1C, C-2purine, rotam. a/b), 152.0 (1C, C-4purine, rotam. a/b), 150.8 (1C, C-Ncarbonyl, rotam. a/b), 149.6 (1C, C-6purine, rotam. a/b), 141.4 (1C, C-8purine, rotam. a/b), 120.8 (0.2C, C-5purine, rotam. b), 120.7 (0.3C, C-5purine, rotam. a), 111.4 (1C, C(CH3)2,rotam. a/b), 88.2 (0.6C, C-3bicyclohexane, rotam. a), 88.0 (0.5C, C-3bicyclohexane, rotam. b), 82.4 (0.8C, C-2bicyclohexane, rotam. a), 82.1 (0.6C, C-2bicyclohexane, rotam. b), 80.2 (1C, C(CH3)3,rotam. a/b), 60.1 (0.6C, OCH3,rotam. a), 60.0 (0.5C, OCH3,rotam. b), 59.3 (1C, C-4bicyclohexane, rotam. a/b), 46.7 (0.7C, NCH2,rotam. a), 46.1 (0.5C, NCH2,rotam. b), 37.5 (0.4C, C-1bicyclohexane, rotam. b), 37.2 (0.5C, C-1bicyclohexane, rotam. a), 30.3 (1C, C-5bicyclohexane, rotam. a/b), 27.9 (3C, C(CH3)3, rotam. a/b), 25.9 (1C, C(CH3)2,rotam. a/b), 24.1 (1C, C(CH3)2,rotam. a/b), 14.0 (1C, SCH3,rotam. a/b), 13.2 (0.5C, C-6bicyclohexane, rotam. b), 13.0 (0.6C, C-6bicyclohexane, rotam. a). FT-IR (neat) ṽ (cm−1) = 3202 (N-H), 2986, 2936 (C-Haliphat.), 1802, 1755, 1709 (C = O), 1601 (C = Caromat.), 1138, 1049 (C-O).
The squaramide (0.085 g, 0.15 mmol) was dissolved in CH3OH (3.2 mL) and trifluoroacetic acid (0.40 mL) and H2O (0.40 mL) were added. The mixture was stirred at rt for 5 d and was then heated to 70° C for 1 d. The solvent was evaporated and the residue was purified by semi-preparative HPLC (method G) to afford the product 47 as a colorless solid, yield 0.021 g (27%). C23H33N7O5S (519.62 g/mol). Purity (HPLC: method D): 99% (tR = 6.26 min). Exact mass (LC-MS-ESI): m/z calculated for C17H17N6O5S [M-C6H16N]− 417.0987, found 417.0984. The compound 47 shows three different rotamers a, b and c in the NMR spectra in a ratio of approximately 10:2:1. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 9.36 (s, 0.9H, NHtriethylammonium), 7.94 (s, 1H, 8-CHpurine, rotam. a), 7.90 (s, 0.2H, 8-CHpurine, rotam. b), 7.78 (s, 0.1H, 8-CHpurine, rotam. c), 7.40 (t, J = 6.6 Hz, 1H, NHsquaramide, rotam. a), 7.36 (s, 0.4H, NHsquaramide, rotam. b, c), 5.34 (d, J = 7.7 Hz, 1H, 2-CHbicyclohexane, rotam. b), 5.16 (s, 0.4H, 3-OHbicyclohexane, rotam. b, c), 5.02 (s, 1H, 3-OHbicyclohexane, rotam. a), 4.85 (s, 0.2H, 4-CHbicyclohexane, rotam. b), 4.72 (d, J = 7.1 Hz, 0.1H, 2-CHbicyclohexane, rotam. c), 4.67 (s, 0.1H, 4-CHbicyclohexane, rotam. c), 4.63 (d, J = 8.1 Hz, 0.2H, 3-CHbicyclohexane, rotam. b), 4.61 (d, J = 1.6 Hz, 1H, 4-CHbicyclohexane, rotam. a), 4.44 (d, J = 6.3 Hz, 1H, 2-CHbicyclohexane, rotam. a), 3.99 (dd, J = 14.2, 6.8 Hz, 1H, NCHHrotam. a), 3.89–3.80 (m, 0.3H, 3-CHbicyclohexane, rotam. c, NCHHrotam. c), 3.80–3.72 (m, 1.3H, NCHHrotam. b, 3-CHbicyclohexane, rotam. a), 3.69 (dd, J = 14.1, 6.1 Hz, 1H, NCHHrotam. b), 3.513.47 (m, 0.2H, -NCHHrotam. c), 3.30 (dd, J = 14.3, 6.2 Hz, 1H, NCHHrotam. a), 3.09 (q, J = 7.3 Hz, 6.6H, CH2, triethylammonium), 2.49 (s, 0.6H, SCH3, rotam. b), 2.48 (s, 0.2H, SCH3, rotam. c), 2.47 (s, 3H, SCH3, rotam. a), 1.84 (ddd, J = 9.0, 4.2, 1.5 Hz, 0.2H, 5-CHbicyclohexane, rotam. b), 1.58 (dd, J = 8.3, 3.8 Hz, 1H, 5-CHbicyclohexane, rotam. a), 1.52 (s, 0.1H, 6-CHHbicyclohexane, rotam. c), 1.48 (dd, J = 8.4, 4.3 Hz, 0.1H, 5-CHbicyclohexane, rotam. c), 1.34 (t, J = 4.3 Hz, 1H, 6-CHHbicyclohexane, rotam. a), 1.16 (t, J = 7.3 Hz, 9.9H, 3-CH3, triethylammonium), 1.09–1.06 (m, 0.3H, 6-CHHbicyclohexane, rotam. c), 0.66 (ddd, J = 8.5, 4.6, 1.5 Hz, 1H, 6-CHHbicyclohexane, rotam. a), 0.65–0.62 (m, 0.2H, 6-CHHbicyclohexane, rotam. b), 0.57 (dd, J = 9.0, 4.5 Hz, 0.1H, 6-CHHbicyclohexane, rotam. c). 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 198.1 (0.5C, C-4squaramide, rotam. a, b), 188.7 (2C, C-1squaramide, rotam. a, C-3squaramide, rotam. a), 188.4 (0.4C, C-1squaramide, rotam. b, C-3squaramide, rotam. b), 181.5 (1C, C-2squaramide, rotam. a), 181.4 (0.3C, C-2squaramide, rotam. b), 164.1 (0.1C, C-2purine, rotam. b), 163.7 (1C, C-2purine, rotam. a), 155.4 (1C, C-6purine, rotam. a), 155.4 (0.2C, C-6purine, rotam. b), 149.8 (1C, C-4purine, rotam. a), 149.5 (0.2C, C-4purine, rotam. b), 137.5 (0.1C, C-8purine, rotam. b), 137.3 (1C, C-8purine, rotam. a), 116.5 (0.1C, C-5purine, rotam. b), 116.5 (1C, C-5purine, rotam. a), 87.7 (0.2C, C-3bicyclohexane, rotam. b), 80.8 (0.2C, C-2bicyclohexane, rotam. b), 76.7 (1C, C-3bicyclohexane, rotam. a), 71.0 (1C, C-2bicyclohexane, rotam. a), 60.6 (1C, C-4bicyclohexane, rotam. a), 60.6 (0.3C, C-4bicyclohexane, rotam. b), 45.7 (5.0C, CH2, triethylammonium), 44.7 (1C, NCH2, rotam. a), 44.6 (0.2C, NCH2, rotam. b), 35.8 (1C, C-1bicyclohexane, rotam. a), 27.5 (0.2C, C-5bicyclohexane, rotam. b), 22.5 (C, C-5bicyclohexane, rotam. a), 13.7 (0.2C, SCH3, rotam. b), 13.6 (1C, SCH3, rotam. a), 11.8 (0.2C, C-6bicyclohexane, rotam. b), 11.7 (1C, C-6bicyclohexane, rotam. a), 8.6 (4.6C, CH3, triethylammonium); the signal for C-1bicyclohexane, rotam. b is located under the DMSO-d6 signal and therefore only visible in 2D spectra. The resolution was too low to identify the 13C signals for rotamer c, therefore only rotamer a and b are described here. FT-IR (neat) ṽ (cm−1) = 3321 (O-H), 3190 (N-H), 2924 (C-Haliphat.), 1786 (C = O), 1528 (C = Caromat.), 1065 (C-O).
Triethylammonium 2-(N-{[(1R,2R,3S,4R,5S)-4-(6-amino-2-methylthio-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl}sulfamoyl)acetate (48). The azide 38 (0.032 g, 0.07 mmol) was dissolved in CH3OH (2 mL) and Pd/C (10 wt%, 0.003 g, 0.003 mmol, 0.004 eq.) was added; the mixture was flushed several times with H2 gas. The reaction was stirred overnight at 5 bar H2 atmosphere. The solvent was evaporated and the residue was purified by fc (CH2Cl2:CH3OH = 95:5 + 1% triethylamine, Ø = 2 cm, l = 20 cm, V = 10 mL) to afford the amine as a colorless solid (Rf = 0.38, CH2Cl2:CH3OH = 95:5 + 1% triethylamine), yield 0.018 g (60%).
The amine (0.16 g, 0.35 mmol) was dissolved in CH2Cl2 (8 mL) and methyl 2-(chlorosulfonyl)acetate (0.068 g, 0.39 mmol, 1.1 eq.), triethylamine (0.10 mL, 0.72 mmol, 2 eq.), and a catalytic amount of DMAP (~5 mol%) were added. The mixture was stirred at rt overnight. The solvent was evaporated and the residue was purified by fc (CH3CN:H2O = 5:95 ⭢ 65:35, 12 mL/min, Biotage®® SNAP Ultra C18, 12 g, V = 20 mL) to afford the sulfonamide as a colorless solid (Rf = 0.49, ethyl acetate = 100%), yield 0.103 g (50%). C24H34N6O8S2 (598.69 g/mol). Purity (HPLC: method B): 90% (tR = 14.49 min). Exact mass (LC-MS-ESI): m/z calculated for C24H35N6O8S [M + H]+ 599.1952, found 599.1952. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 10.09 (s, 1H, NHpurine), 8.33 (s, 1H, 8CHpurine), 7.76 (t, J = 6.4 Hz, 1H, NHsulfonamide), 5.75 (s, 0.2H, CH2Cl2, solvent: dichloromethane), 5.20 (dd, J = 7.2, 1.2 Hz, 1H, 2-CHbicyclohexane), 4.93 (s, 1H, 4CHbicyclohexane), 4.75 (dd, J = 7.3, 1.3 Hz, 1H, 3CHbicyclohexane), 4.23 (dd, J = 14.2, 1.6 Hz, 2H, 2-CH2), 3.69 (s, 1H, OCH3), 3.34–3.28 (m, 2H, NCH2), 2.59 (s, 3H, SCH3), 1.71 (ddd, J = 9.0, 4.7, 1.5 Hz, 1H, 5CHbicyclohexane), 1.48 (s, 9H, C(CH3)3), 1.46 (s, 3H, C(CH3)2), 1.21–1.15 (m, 3H, C(CH3)2), 1.03–0.95 (m, 2H, 6-CH2 bicyclohexane). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 164.0 (1C, C-1), 163.8 (1C, C-2purine), 152.0 (1C, C4purine), 150.8 (1C, C-Ncarbonyl), 149.6 (1C, C-6purine), 141.1 (1C, C-8purine), 120.7 (1C, C-5purine), 111.4 (1C, C(CH3)2), 88.2 (1C, C-3bicyclohexane), 81.9 (1C, C-2bicyclohexane), 80.2 (1C, C(CH3)3), 58.9 (1C, C-4bicyclohexane), 56.1 (1C, C-2), 52.5 (1C, OCH3), 45.5 (1C, NCH2), 36.8 (1C, C-1bicyclohexane), 29.9 (1C, C5bicyclohexane), 27.8 (3C, C(CH3)3), 25.8 (1C, C(CH3)2), 24.2 (1C, C(CH3)2), 14.0 (1C, SCH3), 13.4 (1C, C-6bicyclohexane). FT-IR (neat) ṽ (cm−1) = 3283 (N-H), 2982, 2932 (C-Haliphat.), 1744 (C = O), 1605, 1578 (C = Caromat.), 1327, 1207 (S = O), 1138, 1053 (C-O).
The sulfonamide (0.090 g, 0.15 mmol) was dissolved in CH3OH (3.2 mL) and trifluoroacetic acid (0.40 mL) and H2O (0.40 mL) were added. The mixture was stirred at 70 °C overnight. The solvent was evaporated, and the residue was dissolved in CH3CN (3.2 mL), 2 M NaOH-solution and H2O (0.40 mL) were added. The mixture was stirred at 70° C overnight. The solvent was evaporated, and the residue was purified by semi-preparative HPLC (method E) to afford the product 48 as a colorless solid, yield 0.056 g (69%). C21H35N7O6S2 (545.21 g/mol). Purity (HPLC: method D): 99% (tR = 9.86 min). Exact mass (LC-MS-ESI): m/z calculated for C15H19N6O6S2 [M-C6H16N]− 443.0813, found 443.0826. The compound 48 shows two different rotamers a and b in the NMR spectra in a ratio of approximately 2:1. 1H-NMR (600 MHz, DMSO-d6) δ (ppm) = 8.10 (s, 1H, 8-CHpurine, rotam. a), 7.97 (s, 0.5H, 8CHpurine, rotam. b), 7.29 (s, 3H, NH2, rotam. a, NHrotam. a), 7.25 (s, 1.5H, NH2, rotam. b, NHrotam. b), 5.12 (s, 1H, 3OHbicyclohexane, rotam. a), 4.78 (s, 0.3H, 2CHbicyclohexane, rotam. b), 4.70 (s, 0.5H, 4CHbicyclohexane, rotam. b), 4.61 (d, J = 1.9 Hz, 1H, 4-CHbicyclohexane, rotam. a), 4.51 (dd, J = 6.5, 1.4 Hz, 1H, 2CHbicyclohexane, rotam. a), 4.01 (s, 0.3H, 3CHbicyclohexane, rotam. b), 3.91 (s, 0.5H, 2-CHHrotam. b), 3.84 (dt, J = 6.7, 1.6 Hz, 1H, 3CHbicyclohexane, rotam a), 3.73 (dd, J = 14.6, 5.1 Hz, 2H, 2-CH2 rotam. a, 2CHHrotam. b), 3.39 (d, J = 13.5 Hz, 1H, NCHHrotam a), 3.25 (s, 0.5H, NCHHrotam b), 3.13 (s, 0.5H, NCHHrotam b), 3.07 (d, J = 13.5 Hz, 1H, NCHHrotam a), 2.93 (q, J = 7.5 Hz, 7.8H, CH2, triethylammonium), 2.48 (s, 3H, SCH3, rotam. a), 1.54 (t, J = 4.6 Hz, 0.5H, 6CHHbicyclohexane, rotam. b), 1.48 (dd, J = 8.5, 3.8 Hz, 1H, 5CHbicyclohexane, rotam. a), 1.33 (dd, J = 8.7, 4.8 Hz, 0.5H, 5CHbicyclohexane, rotam. b), 1.30 (t, J = 4.4 Hz, 1H, 6CHHbicyclohexane, rotam. a), 1.11 (t, J = 7.3 Hz, 12.3H, CH3, triethylammonium), 0.72 (ddd, J = 8.6, 4.7, 1.5 Hz, 1H, 6CHHbicyclohexane, rotam. a), 0.62 (dd, J = 9.6, 4.8 Hz, 0.5H, 6CHHbicyclohexane, rotam. b); the signal for SCH3, rotam. b is located under the DMSO-d6 signal and therefore only visible in 2D spectra. 13C-NMR (151 MHz, DMSO-d6) δ (ppm) = 165.0 (0.7C, C-1rotam. a, b), 163.7 (1C, C2purine, rotam. a), 163.6 (0.4C, C-2purine, rotam. b), 155.4 (1C, C-6purine, rotam. a), 155.4 (0.4C, C6purine, rotam. b), 149.8 (1C, C4purine, rotam. a), 149.6 (0.4C, C4purine, rotam. b), 138.1 (0.2C, C8purine, rotam. b), 137.9 (1C, C8purine, rotam. a), 116.5 (0.8C, C-5purine, rotam. a, b), 76.9 (1C, C3bicyclohexane, rotam. a), 71.6 (1C, C2bicyclohexane, rotam. a), 61.0 (1C, C-4bicyclohexane, rotam. a), 57.4 (1C, C-2rotam. a), 45.7 (1C, NCH2, rotam. a), 45.3 (6.3C, CH2, triethylammonium), 34.5 (1C, C-1bicyclohexane, rotam. a), 27.6 (0.4C, C5bicyclohexane, rotam. b), 23.2 (1C, C5bicyclohexane, rotam. a), 13.7 (1.3C, SCH3, rotam. a, b), 12.6 (0.4C, C-6bicyclohexane, rotam. b), 12.5 (1C, C-6bicyclohexane, rotam. a), 9.0 (5.6C, CH3, triethylammonium); the resolution was too low to identify the 13C signals for rotamer b, therefore only those signals of rotamer b that are visible in the 13C-NMR spectrum are described here. FT-IR (neat) ṽ (cm−1) = 3306 (O-H), 3132 (N-H), 2982, 2928 (C-Haliphat.), 1612 (C = O), 1582 (C = Caromat.), 1300 (S = O), 1150, 1119 (C-O), 1065 (S = O).


 ,
, 
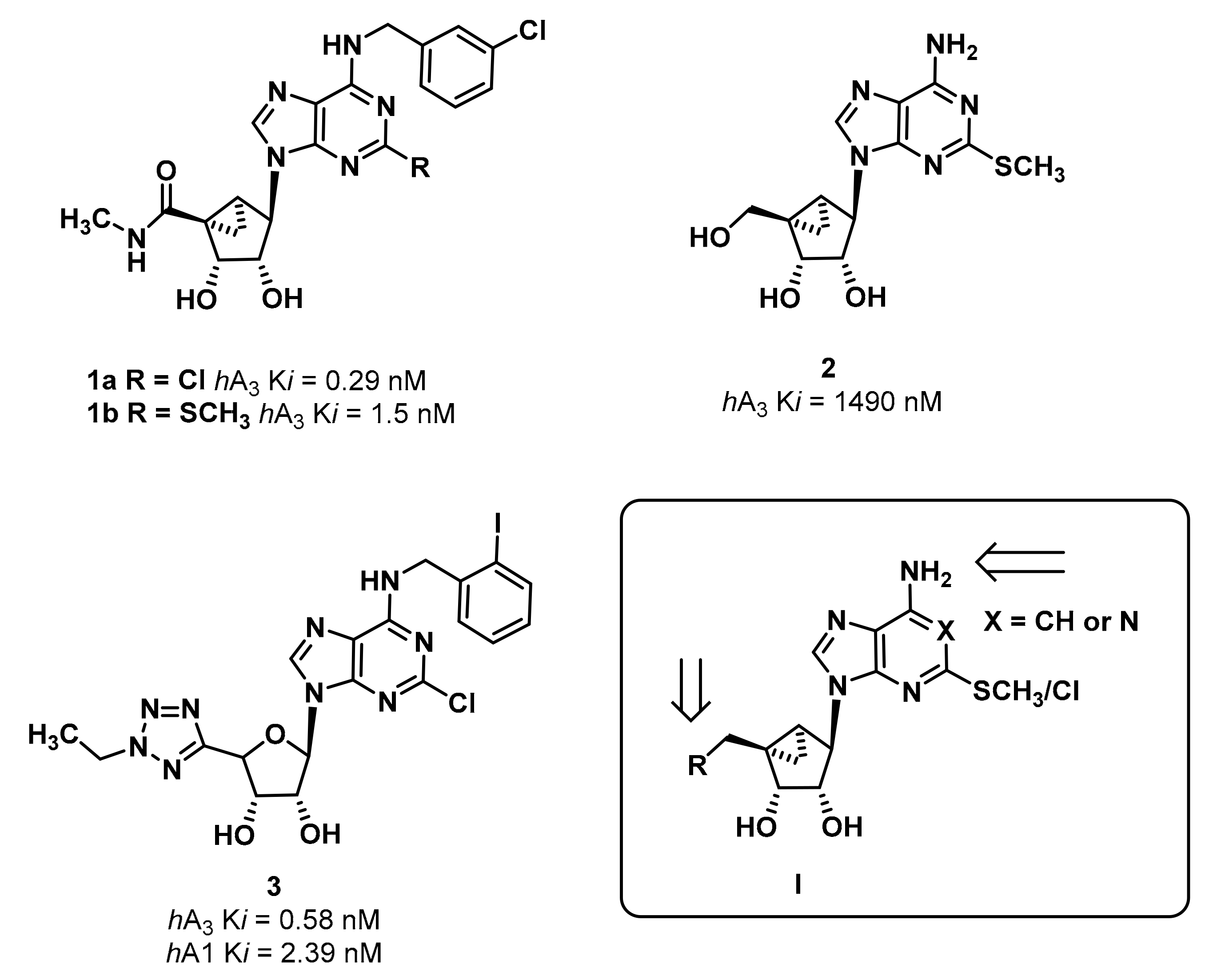
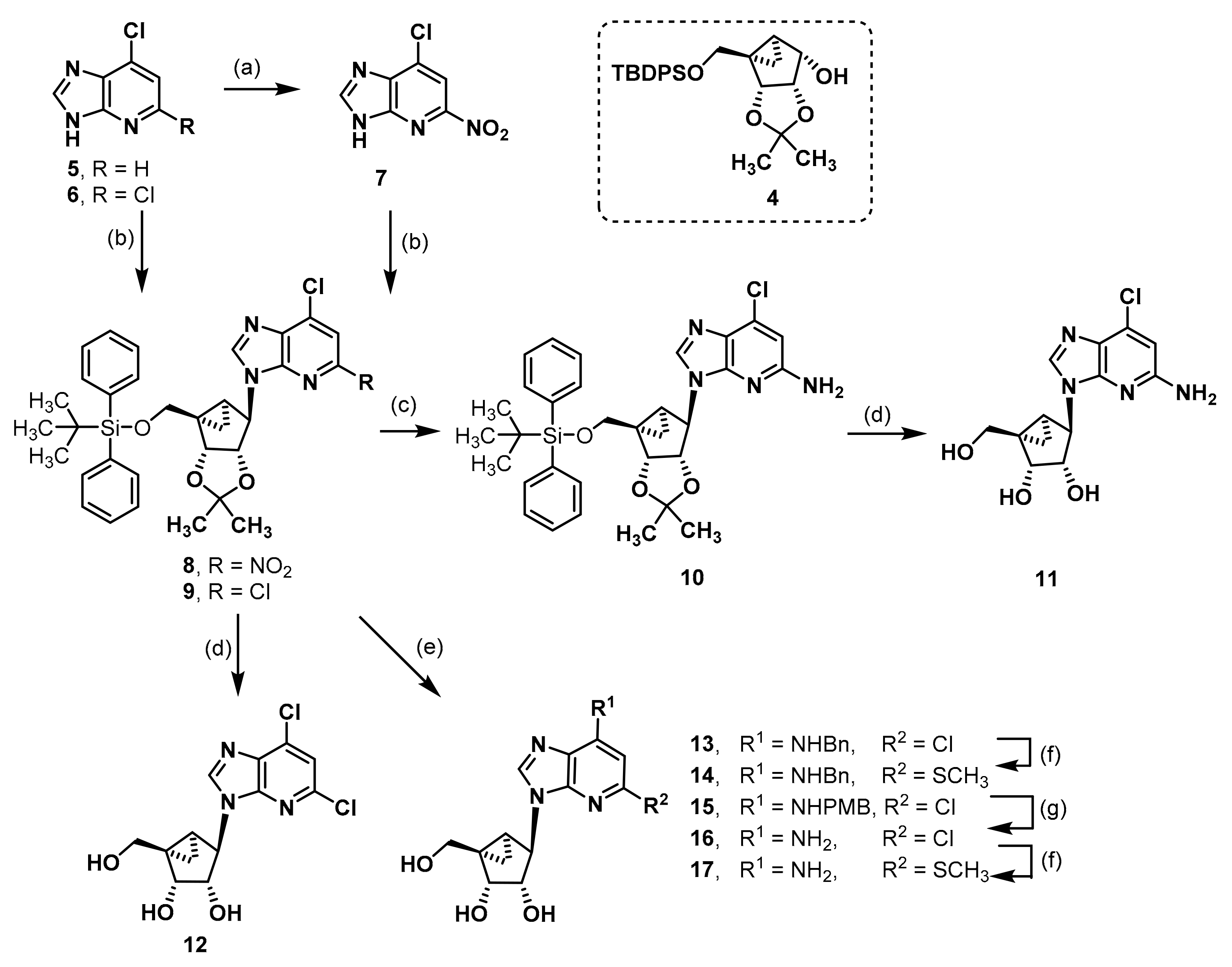
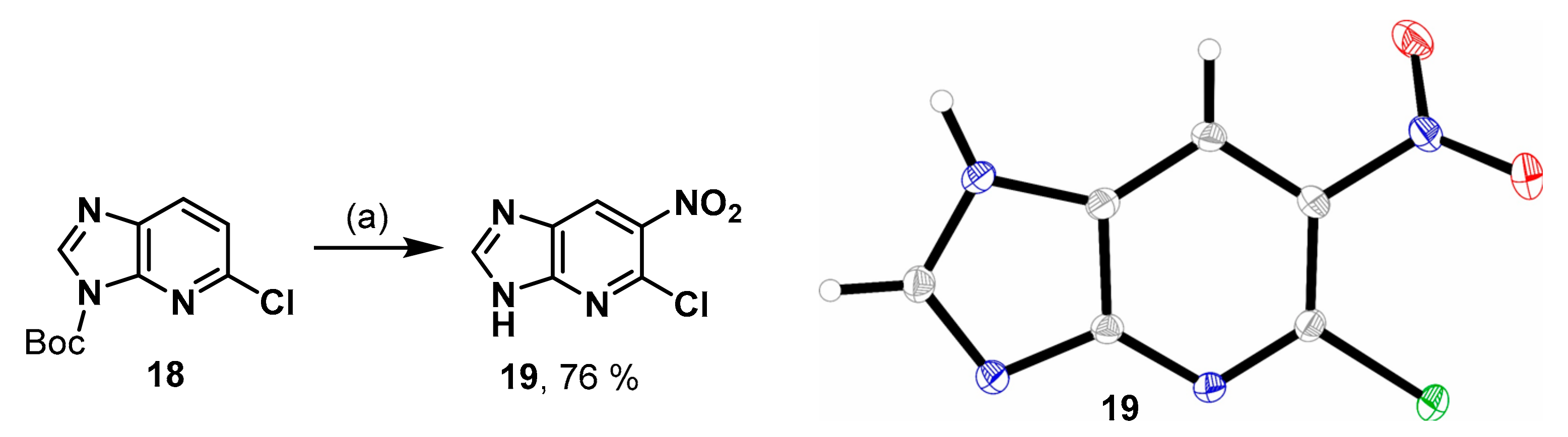
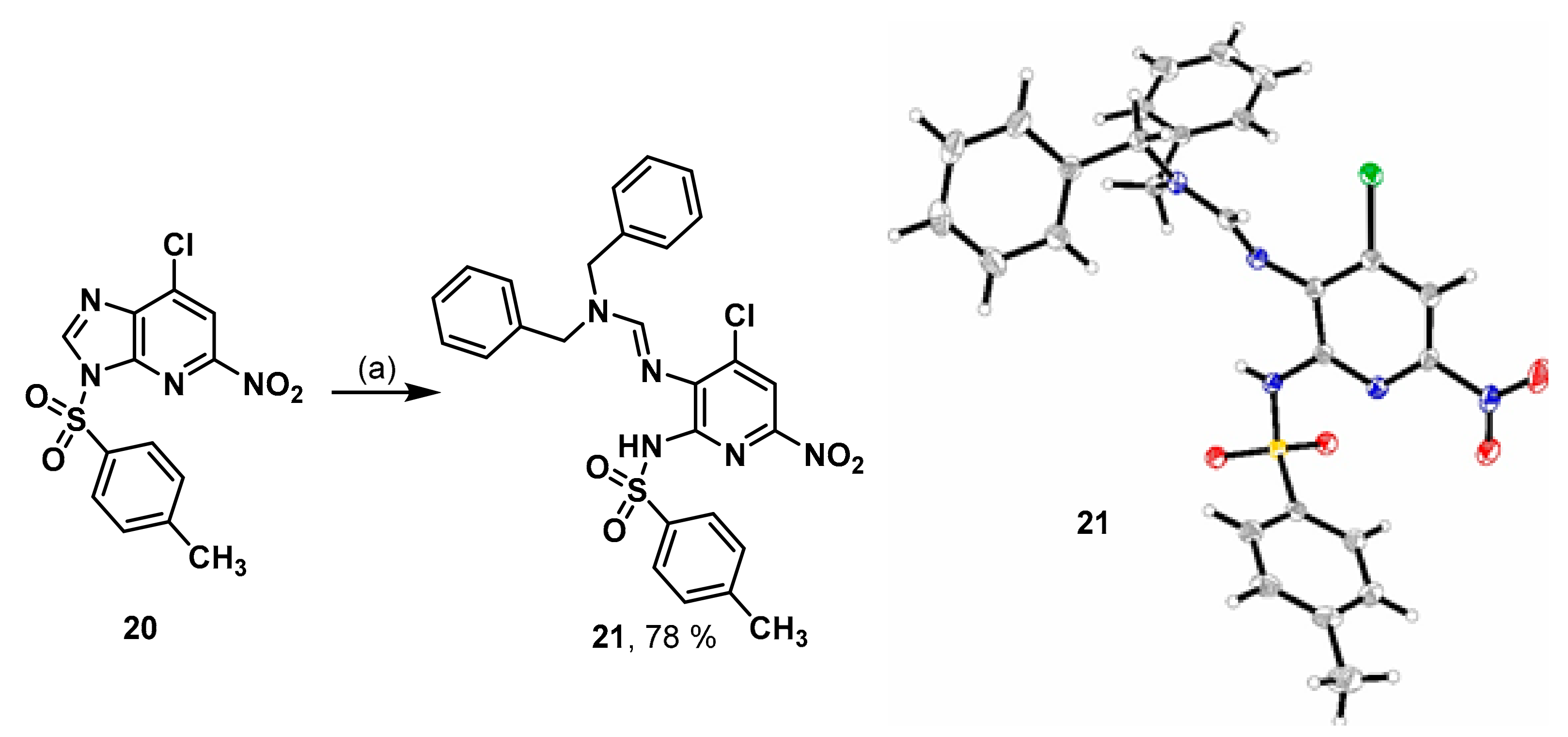
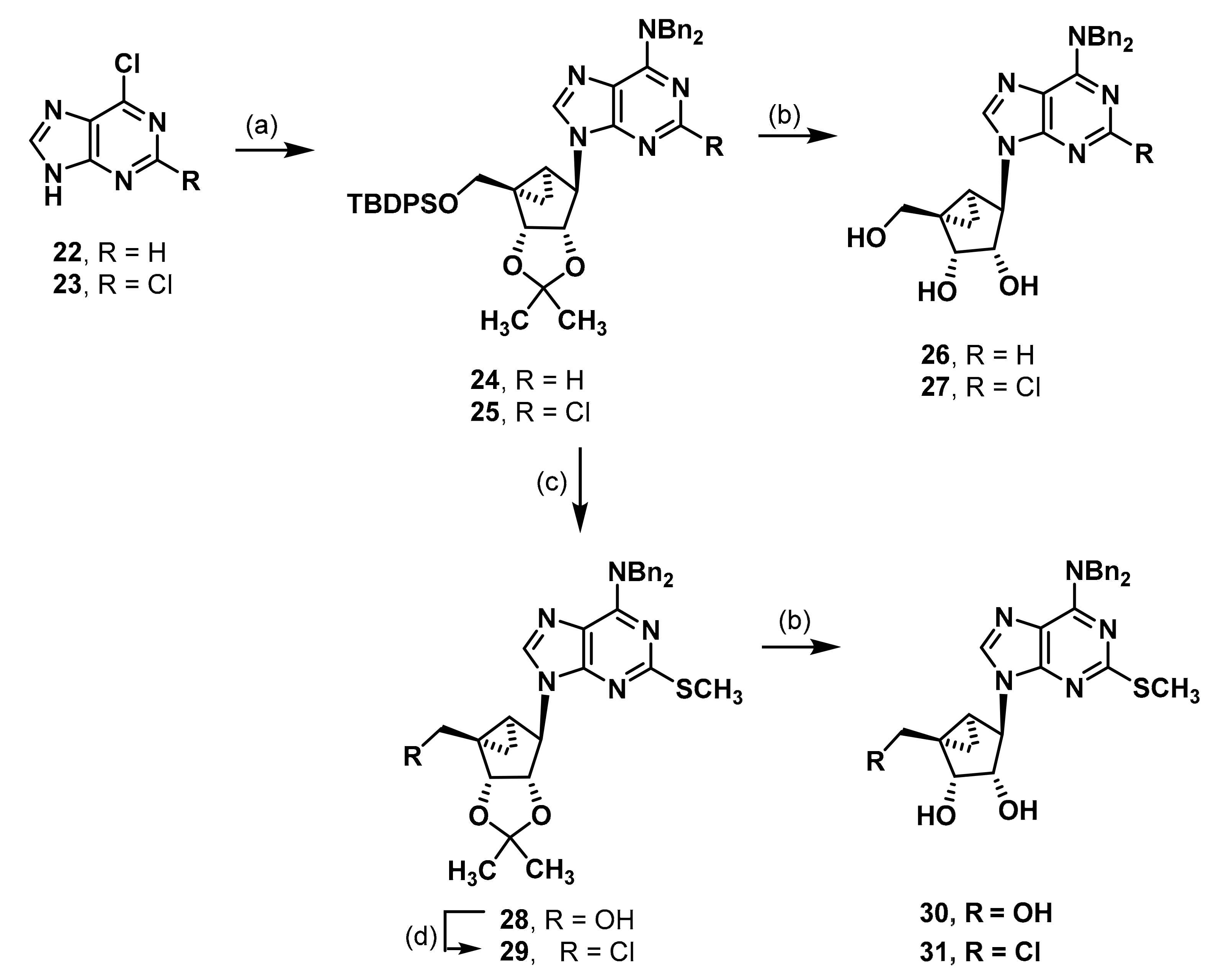
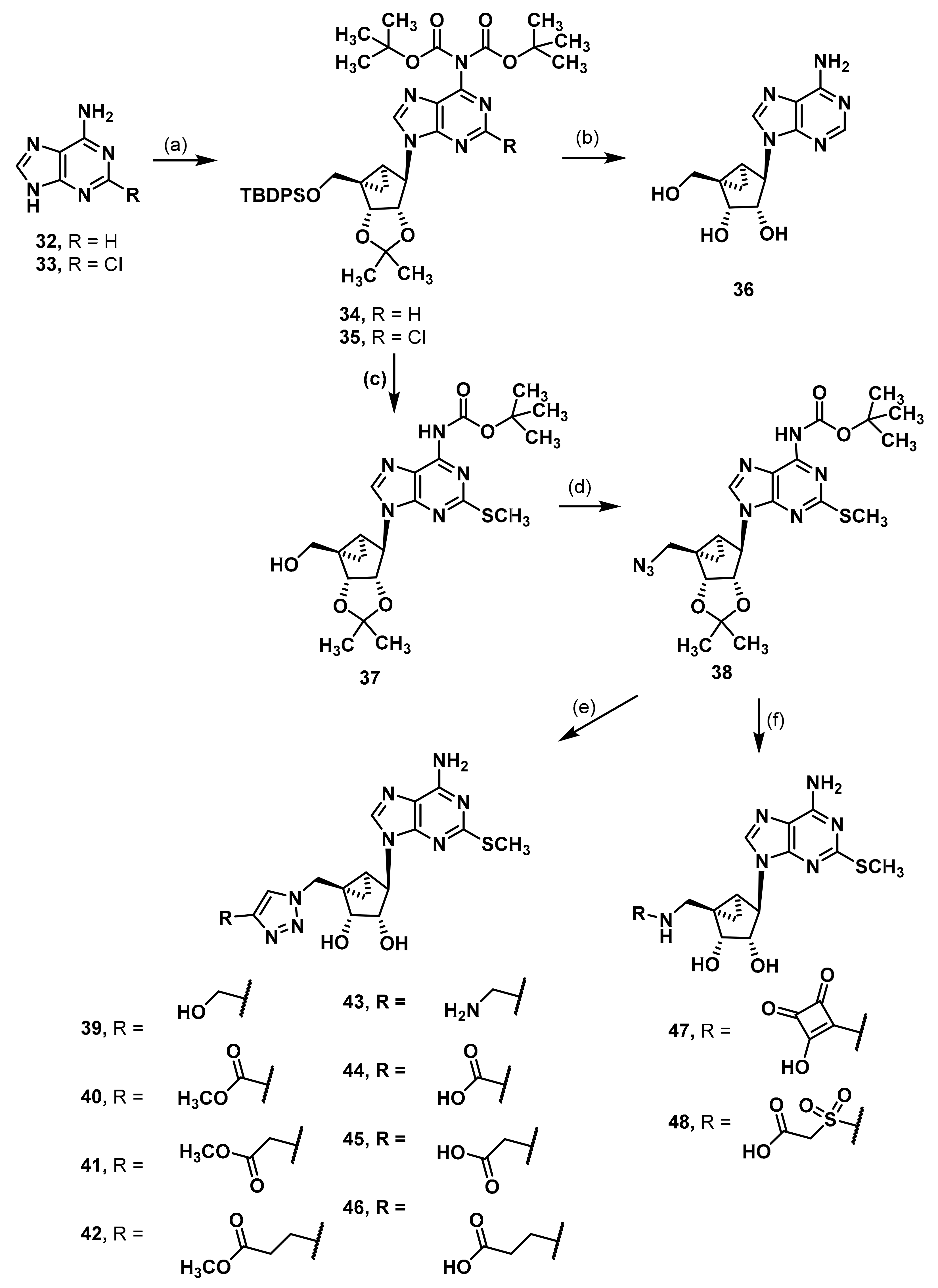

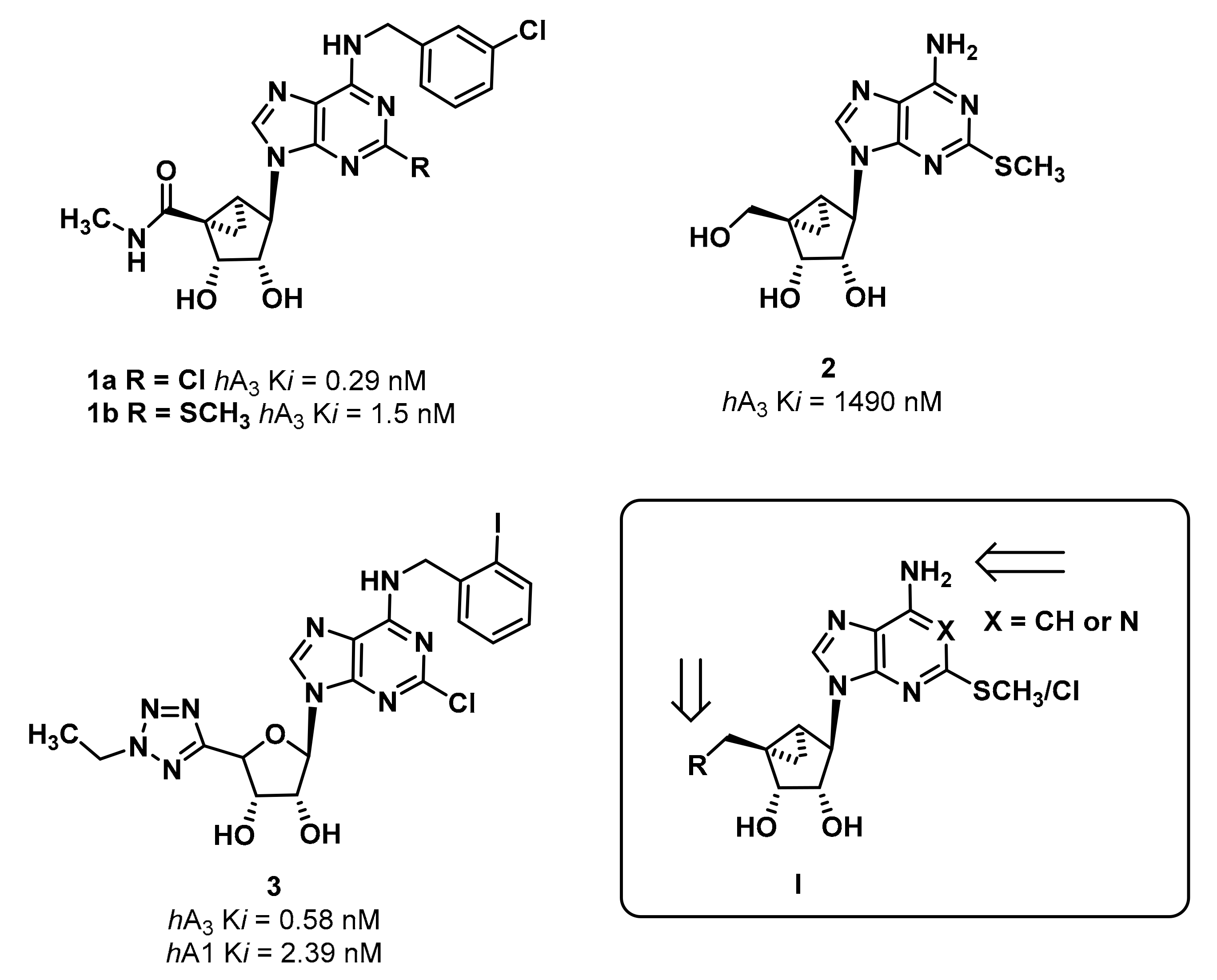{kind=link}
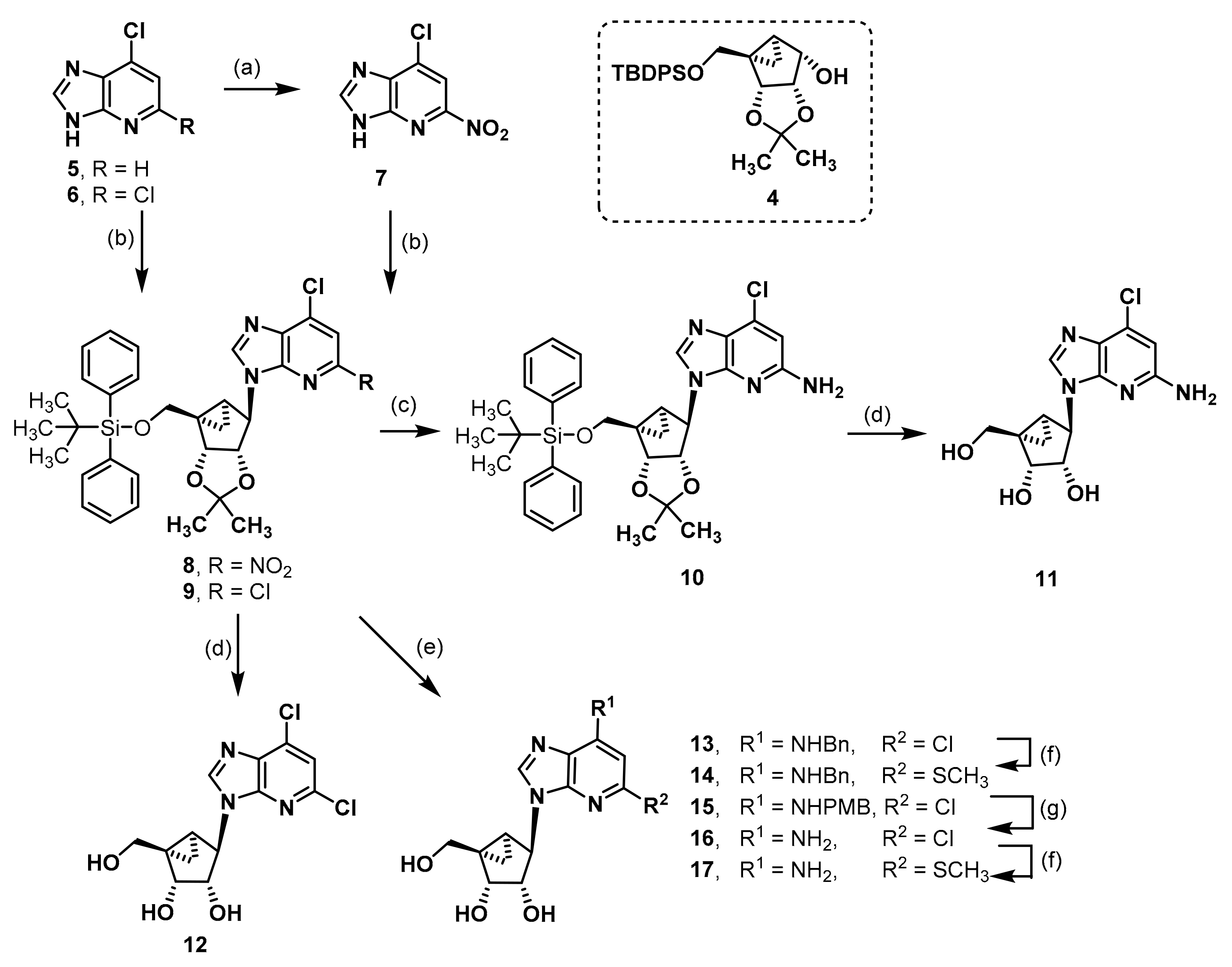{kind=link}
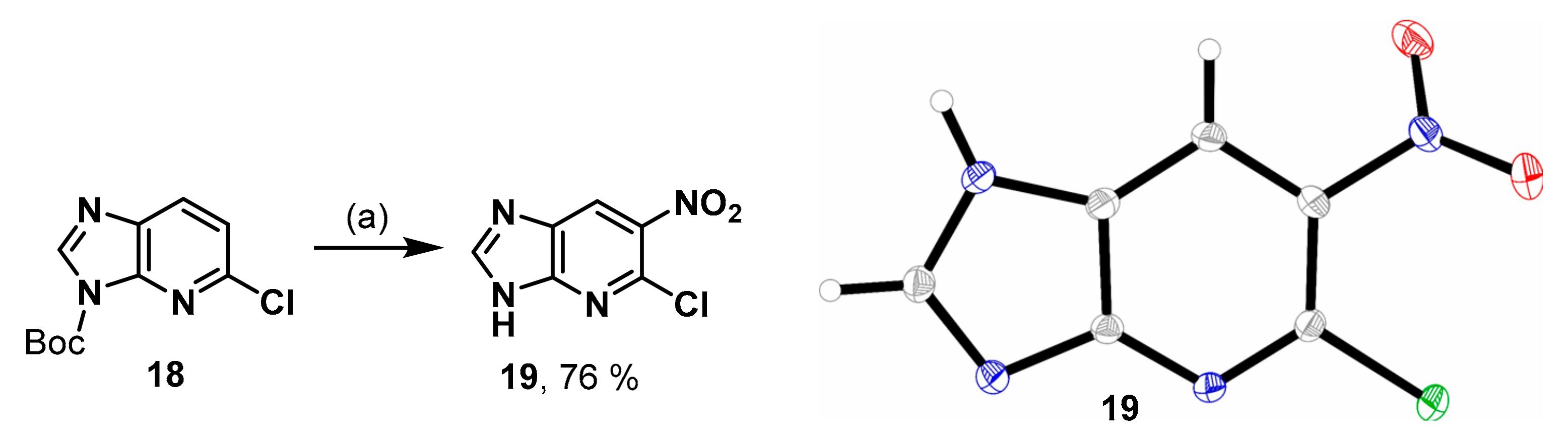{kind=link}
{kind=link}
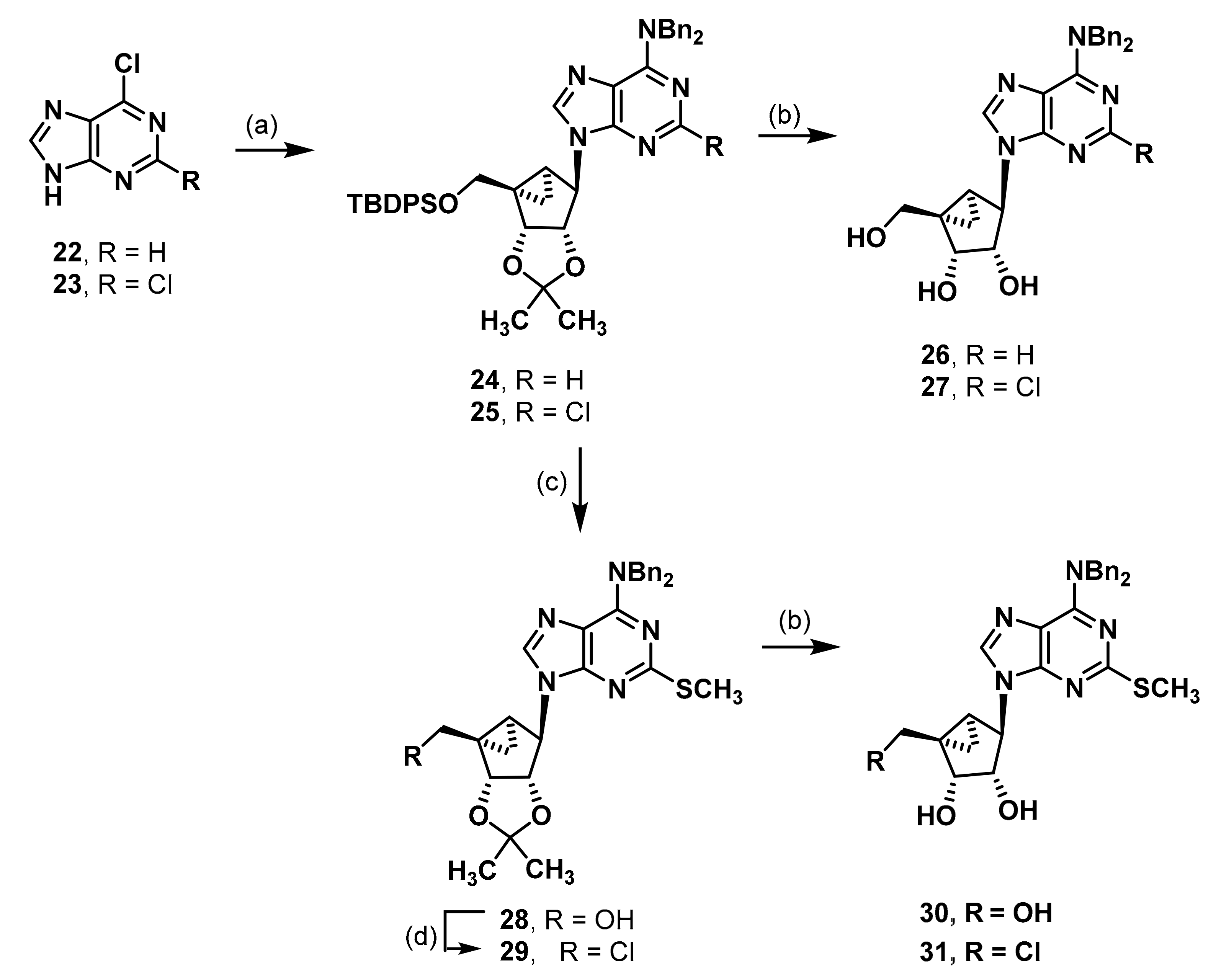{kind=link}
{kind=link}