
Target Design of Novel Histone Deacetylase 6 Selective Inhibitors with 2-Mercaptoquinazolinone as the Cap Moiety


 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
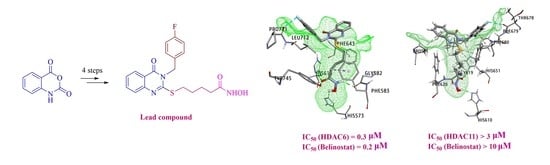
2.1. Structural Design and Synthesis
2.2. Biological Evaluation
2.2.1. Inhibition Screening
2.2.2. In Vitro HDAC Isoform Inhibition Activity of Compound 8
2.2.3. Selective Upregulation in the Acetylation Level of Tubulin
2.3. Molecular Docking Studies
3. Conclusions
4. Experimental Section
4.1. Chemistry
General Information
4.2. Molecular Docking Studies
4.2.1. Ligand and Receptor Preparation
4.2.2. Docking Using AutoDock4Zn
4.3. Biological Methods
4.3.1. Antiproliferative Assay
4.3.2. HDAC Enzyme Inhibition Assays
4.3.3. Western Blotting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, K.; Kakkar, R. Exploring structural requirements of isoform selective histone deacetylase inhibitors: A comparative in silico study. J. Biomol. Struct. Dyn. 2021, 39, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, W. Isoform-selective histone deacetylase inhibitors: The trend and promise of disease treatment. Epigenomics 2015, 39, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Hadjipavlou-Litina, D. Histone deacetylase inhibitors (HDACIs). Structure--activity relationships: History and new QSAR perspectives. Med. Res. Rev. 2012, 32, 1–165. [Google Scholar] [CrossRef]
- Scafuri, B.; Bontempo, P.; Altucci, L.; Masi, L.D.; Facchiano, A. Molecular Docking Simulations on Histone Deacetylases (HDAC)-1 and -2 to Investigate the Flavone Binding. Biomedicines 2020, 8, 568. [Google Scholar] [CrossRef]
- Miceli, M.; Bontempo, P.; Nebbioso, A.; Altucci, L. Natural compounds in epigenetics: A current view. Food Chem. Toxicol. 2014, 73, 71–83. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Pal, D.; Saha, S. Hydroxamic acid-A novel molecule for anticancer therapy. J. Adv. Pharm. Technol. Res. 2012, 3, 92–99. [Google Scholar] [CrossRef]
- Kelly, W.K.; O’Connor, O.A.; Krug, L.M.; Chiao, J.H.; Heaney, M.; Curley, T.; MacGregore-Cortelli, B.; Tong, W.; Secrist, J.P.; Schwartz, L.; et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilidehydroxamic acid, in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 3923–3931. [Google Scholar] [CrossRef]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Reviews. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Liao, D. Profiling technologies for the identification and characterization of small-molecule histone deacetylase inhibitors. Drug Discov. Today. Technol. 2015, 18, 24–28. [Google Scholar] [CrossRef][Green Version]
- Chang, P.; Weykamp, M.; Dennahy, I.S.; Williams, A.M.; Bhatti, U.F.; Liu, B.; Nikolian, V.C.; Li, Y.; Alam, H.B. Histone deacetylase inhibitors: Isoform selectivity improves survival in a hemorrhagic shock model. J. Trauma Acute Care Surg. 2018, 84, 795–801. [Google Scholar] [CrossRef]
- Micelli, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef]
- Zhang, L.; Han, Y.; Jiang, Q.; Wang, C.; Chen, X.; Li, X.; Xu, F.; Jiang, Y.; Wang, Q.; Xu, W. Trend of histone deacetylase inhibitors in cancer therapy: Isoform selectivity or multitargeted strategy. Med. Res. Rev. 2015, 35, 63–84. [Google Scholar] [CrossRef]
- Liu, Y.; Li, L.; Min, J. Corrigendum: Structural biology: HDAC6 finally crystal clear. Nat. Chem. Biol. 2016, 12, 885. [Google Scholar] [CrossRef]
- Wang, X.X.; Wan, R.Z.; Liu, Z.P. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 2018, 143, 1406–1418. [Google Scholar] [CrossRef]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schluter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef]
- Kalin, J.H.; Bergman, J.A. Development and therapeutic implications of selective histone deacetylase 6 inhibitors. J. Med. Chem. 2013, 56, 6297–6313. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, N.; Ge, D.; Chen, Y. Next-generation of selective histone deacetylase inhibitors. RSC Adv. 2019, 9, 19571–19583. [Google Scholar] [CrossRef]
- Juliandi, B.; Abematsu, M.; Sanosaka, T.; Tsujimura, K.; Smith, A.; Nakashima, K. Induction of superficial cortical layer neurons from mouse embryonic stem cells by valproic acid. Neurosci. Res. 2012, 72, 23–31. [Google Scholar] [CrossRef]
- Zhan, P.; Itoh, Y.; Suzuki, T.; Liu, X. Strategies for the Discovery of Target-Specific or Isoform-Selective Modulators. J. Med. Chem. 2015, 58, 7611–7633. [Google Scholar] [CrossRef]
- Osipov, V.N.; Khachatryan, D.S.; Balaev, A.N. Biologically active quinazoline-based hydroxamic acids. Med. Chem. Res. 2020, 29, 831–845. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, T.; Wang, F.; Niu, T.; Liu, Z.; Chen, X.; Long, C.; Tang, M.; Cao, D.; Wang, X.; et al. Discovery of Selective Histone Deacetylase 6 Inhibitors Using the Quinazoline as the Cap for the Treatment of Cancer. J. Med. Chem. 2016, 59, 1455–1470. [Google Scholar] [CrossRef]
- Yu, C.W.; Chang, P.T.; Hsin, L.W.; Chern, J.W. Quinazolin-4-one derivatives as selective histone deacetylase-6 inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2013, 56, 6775–6791. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Abdel-Aziz, A.A.; Ghabbour, H.A.; Al-Gendy, M.A. Synthesis, in vitro antitumour activity, and molecular docking study of novel 2-substituted mercapto-3-(3,4,5-trimethoxybenzyl)-4(3H)-quinazolinone analogues. J. Enzym. Inhib. Med. Chem. 2017, 32, 1229–1239. [Google Scholar] [CrossRef]
- Bui, H.T.B.; Do, K.M.; Nguyen, H.T.D.; Mai, H.V.; Danh, T.L.D.; Tran, D.Q.; Morita, H. Efficient one-pot tandem synthesis and cytotoxicity evaluation of 2,3-disubstituted quinazolin-4(3H)-one derivatives. Tetrahedron 2021, 98, 132426. [Google Scholar] [CrossRef]
- Bui, T.B.H.; Nguyen, H.M.; Mai, V.H.; Danh, L.D.T.; Nguyen, H.S.; Tran, Q.D.; Morita, H. Facile Sodium Metabisulfite Mediated Synthesis of 1,2-Disubstituted Benzimidazoles and Cytotoxicity Evaluation. Heterocycles 2019, 98, 650–665. [Google Scholar]
- Bui, H.T.B.; Ha, Q.T.K.; Oh, W.K.; Vo, D.D.; Chau, Y.N.T.; Tu, C.T.K.; Pham, E.C.; Tran, P.T.; Tran, L.T.; Mai, H.V. Microwave assisted synthesis and cytotoxic activity evaluations of new benzimidazole derivatives. Tetrahedron Lett. 2016, 57, 887–891. [Google Scholar] [CrossRef]
- Mahdavi, M.; Pedrood, K.; Safavi, M.; Saeedi, M.; Pordeli, M.; Ardestani, S.K.; Emami, S.; Adib, M.; Foroumadi, A.; Shafiee, A. Synthesis and anticancer activity of N-substituted 2-arylquinazolinones bearing trans-stilbene scaffold. Eur. J. Med. Chem. 2015, 95, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Fodor, M.; Price, E.; Wang, P.; Lu, H.; Argintaru, A.; Chen, Z.; Glick, M.; Hao, H.X.; Kato, M.; Koenig, R.; et al. Dual Allosteric Inhibition of SHP2 Phosphatase. ACS Chem. Biol. 2018, 13, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-W.; Miao, C.-B.; Kang, H. Benign and Efficient Synthesis of 2-Substituted 4(3H)-Quinazolinones Mediated by Iron(III) Chloride Hexahydrate in Refluxing Water. Bull. Chem. Soc. Jpn. 2006, 79, 1426–1430. [Google Scholar] [CrossRef]
- Yang, L.; Xue, X.; Zhang, Y. Simple and Efficient Synthesis of Belinostat. Synth. Commun. 2010, 40, 2520–2524. [Google Scholar] [CrossRef]
- Kim, N.Y.; Cheon, C.-H. Synthesis of quinazolinones from anthranilamides and aldehydes via metal-free aerobic oxidation in DMSO. Tetrahedron Lett. 2014, 55, 2340–2344. [Google Scholar] [CrossRef]
- Laskowski, R.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef]
- Dan, N.T.; Quang, H.D.; Van Truong, V.; Huu Nghi, D.; Cuong, N.M.; Cuong, T.D.; Toan, T.Q.; Bach, L.G.; Anh, N.H.T.; Mai, N.T.; et al. Design, synthesis, structure, in vitro cytotoxic activity evaluation and docking studies on target enzyme GSK-3beta of new indirubin-3’-oxime derivatives. Sci. Rep. 2020, 10, 11429–11441. [Google Scholar] [CrossRef]
- Ngo, S.T.; Tam, N.M.; Pham, M.Q.; Nguyen, T.H. Benchmark of Popular Free Energy Approaches Revealing the Inhibitors Binding to SARS-CoV-2 Mpro. J. Chem. Inf. Modeling 2021, 61, 2302–2312. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Nguyen, T.H.; Pham, T.N.H.; Huy, N.T.; Bay, M.V.; Pham, M.Q.; Nam, P.C.; Vu, V.V.; Ngo, S.T. Autodock Vina Adopts More Accurate Binding Poses but Autodock4 Forms Better Binding Affinity. J. Chem. Inf. Modeling 2020, 60, 204–211. [Google Scholar] [CrossRef]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4(Zn): An improved AutoDock force field for small-molecule docking to zinc metalloproteins. J. Chem. Inf. Modeling 2014, 54, 2371–2379. [Google Scholar] [CrossRef]
- Ngo, Q.A.; Thi, T.H.N.; Pham, M.Q.; Delfino, D.; Do, T.T. Antiproliferative and antiinflammatory coxib-combretastatin hybrids suppress cell cycle progression and induce apoptosis of MCF7 breast cancer cells. Mol. Divers. 2021, 25, 2307–2319. [Google Scholar] [CrossRef]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural Snapshots of Human HDAC8 ProvideInsights into the Class I Histone Deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef]
- Ortore, G.; Colo, F.D.; Martinelli, A. Docking of hydroxamic acids into HDAC1 and HDAC8: A rationalization of activity trends and selectivities. J. Chem. Inf. Model. 2009, 49, 2774–2785. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System, version 1.3r1; Schrödinger, Inc.: New York, NY, USA, 2010.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R. Gaussian 09 Rev. d.01, Gaussian Inc.: Wallingford, CT, USA, 2009.
- Buckley, M.T.; Yoon, J.; Yee, H.; Chiriboga, L.; Liebes, L.; Ara, G.; Qian, X.; Bajorin, D.F.; Sun, T.; Osman, I. The histone deacetylase inhibitor belinostat (PXD101) suppresses bladder cancer cell growth in vitro and in vivo. J. Transl. Med. 2007, 5, 1–15. [Google Scholar] [CrossRef]
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012, 481, 335–340. [Google Scholar] [CrossRef]
- Burli, R.W.; Luckhurst, C.A.; Aziz, O.; Matthews, K.L.; Yates, D.; Lyons, K.A.; Beconi, M.; McAllister, G.; Breccia, P.; Stott, A.J.; et al. Design, synthesis, and biological evaluation of potent and selective class IIa histone deacetylase (HDAC) inhibitors as a potential therapy for Huntington’s disease. J. Med. Chem. 2013, 56, 9934–9954. [Google Scholar] [CrossRef]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | R1 | R2 | n | Yield (%) | IC50 (µM) | Imax (%) |
|---|---|---|---|---|---|---|
| 1 | Bn | - | 1 | 32 | ND | 20.0 |
| 2 | 4-F-Bn | - | 1 | 13 | ND | 11.0 |
| 3 | 3-F-Bn | - | 1 | 15 | ND | 24.0 |
| 4 | 3F-4F-Bn | - | 1 | 44 | ND | 23.0 |
| 5 | 2-NO2-Bn | - | 1 | 20 | ND | 17.0 |
| 6 | 4-CH3-Bn | - | 1 | 69 | ND | 22.0 |
| 7 | CH2-(3-pyridinyl) | - | 4 | 23 | ND | 17.0 |
| 8 | 4-F-Bn | - | 4 | 44 | 3.44 ± 1.11 | 90.6 |
| 9 | 3F-4F-Bn | - | 4 | 51 | 36.9 ± 1.12 | 96.8 |
| 10 | 4-phenyl-Bn | - | 4 | 25 | 14.2 ± 1.14 | 89.7 |
| 11 | (CH2)2-piperazinyl | 4-OCH3-Phenyl | - | 30 | ND | 3.0 |
| 12 | (CH2)2-piperazinyl | 4-F-Phenyl | - | 40 | ND | 3.0 |
| 13 | - | 4-OCH3-Phenyl | - | 46 | 37.26 ± 1.21 | 95.8 |
| 14 | - | 4-F-Phenyl | - | 53 | 37.85 ± 1.71 | 100 |
| 15 | - | 2-Furyl | - | 30 | ND | 1.5 |
| Belinostat | - | - | - | - | 2.6 ± 1.40 | 100 |
| IC50 (µM) | Selective Fold (Ratio) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| HDAC3 (Class I) | HDAC8 (Class I) | HDAC4 (Class IIa) | HDAC6 (Class IIb) | HDAC11 (Class IV) | HDAC3/ HDAC6 | HDAC4/ HDAC6 | HDAC8/ HDAC6 | HDAC11/ HDAC6 | |
| 8 | 2.7 ± 0.08 | 0.8 ± 0.01 | >10 | 0.3 ± 0.02 | >3 | 7.94 | >29.4 | 2.32 | >29.4 |
| Belinostat | 0.6 ± 0.09 | 0.3 ± 0.02 | 2.7 ± 0.12 | 0.2 ± 0.01 | >10 | 3.0 | 13.5 | 1.5 | >50 |
| Enzyme | Dock Score (kcal/mol) | H-Bond Interacting Residues | |||
|---|---|---|---|---|---|
| Compound 8 | Belinostat | Compound 8 | Belinostat | ||
| Class I | |||||
| HDAC3 | −7.20 | −9.78 | His172, Asp259, Tyr298 | Gly143, Cys145, His172 | |
| HDAC8 | −7.70 | −8.13 | Lys33, Cys153, Asp178, Phe208 | Gly140, Cys153, Asp178, Gln263 | |
| Class IIa | |||||
| HDAC4 | −6.85 | −8.62 | Leu943, Gly975, His976 | His802, His803, Phe871, Asp934, Pro942 | |
| Class IIb | |||||
| HDAC6 | −7.92 | −8.00 | His573, Gly582, His614, Tyr745 | His573, His574, Asp612, His614 | |
| Class IV | |||||
| HDAC11 | −7.33 | −6.56 | His610, Gly619, His651 | Cys621, Asp649 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bui, H.T.B.; Nguyen, P.H.; Pham, Q.M.; Tran, H.P.; Tran, D.Q.; Jung, H.; Hong, Q.V.; Nguyen, Q.C.; Nguyen, Q.P.; Le, H.T.; et al. Target Design of Novel Histone Deacetylase 6 Selective Inhibitors with 2-Mercaptoquinazolinone as the Cap Moiety. Molecules 2022, 27, 2204. https://doi.org/10.3390/molecules27072204
Bui HTB, Nguyen PH, Pham QM, Tran HP, Tran DQ, Jung H, Hong QV, Nguyen QC, Nguyen QP, Le HT, et al. Target Design of Novel Histone Deacetylase 6 Selective Inhibitors with 2-Mercaptoquinazolinone as the Cap Moiety. Molecules. 2022; 27(7):2204. https://doi.org/10.3390/molecules27072204
Chicago/Turabian StyleBui, Hue Thi Buu, Phuong Hong Nguyen, Quan Minh Pham, Hoa Phuong Tran, De Quang Tran, Hosun Jung, Quang Vinh Hong, Quoc Cuong Nguyen, Quy Phu Nguyen, Hieu Trong Le, and et al. 2022. "Target Design of Novel Histone Deacetylase 6 Selective Inhibitors with 2-Mercaptoquinazolinone as the Cap Moiety" Molecules 27, no. 7: 2204. https://doi.org/10.3390/molecules27072204
APA StyleBui, H. T. B., Nguyen, P. H., Pham, Q. M., Tran, H. P., Tran, D. Q., Jung, H., Hong, Q. V., Nguyen, Q. C., Nguyen, Q. P., Le, H. T., & Yang, S.-G. (2022). Target Design of Novel Histone Deacetylase 6 Selective Inhibitors with 2-Mercaptoquinazolinone as the Cap Moiety. Molecules, 27(7), 2204. https://doi.org/10.3390/molecules27072204