
New Triazolyl N^N Bidentate Rh(III), Ir(III), Ru(II) and Os(II) Complexes: Synthesis and Characterization, Probing Possible Relations between Cytotoxicity with Transfer Hydrogenation Efficacy and Interaction with Model Biomolecules

Abstract
:1. Introduction
2. Results and Discussion
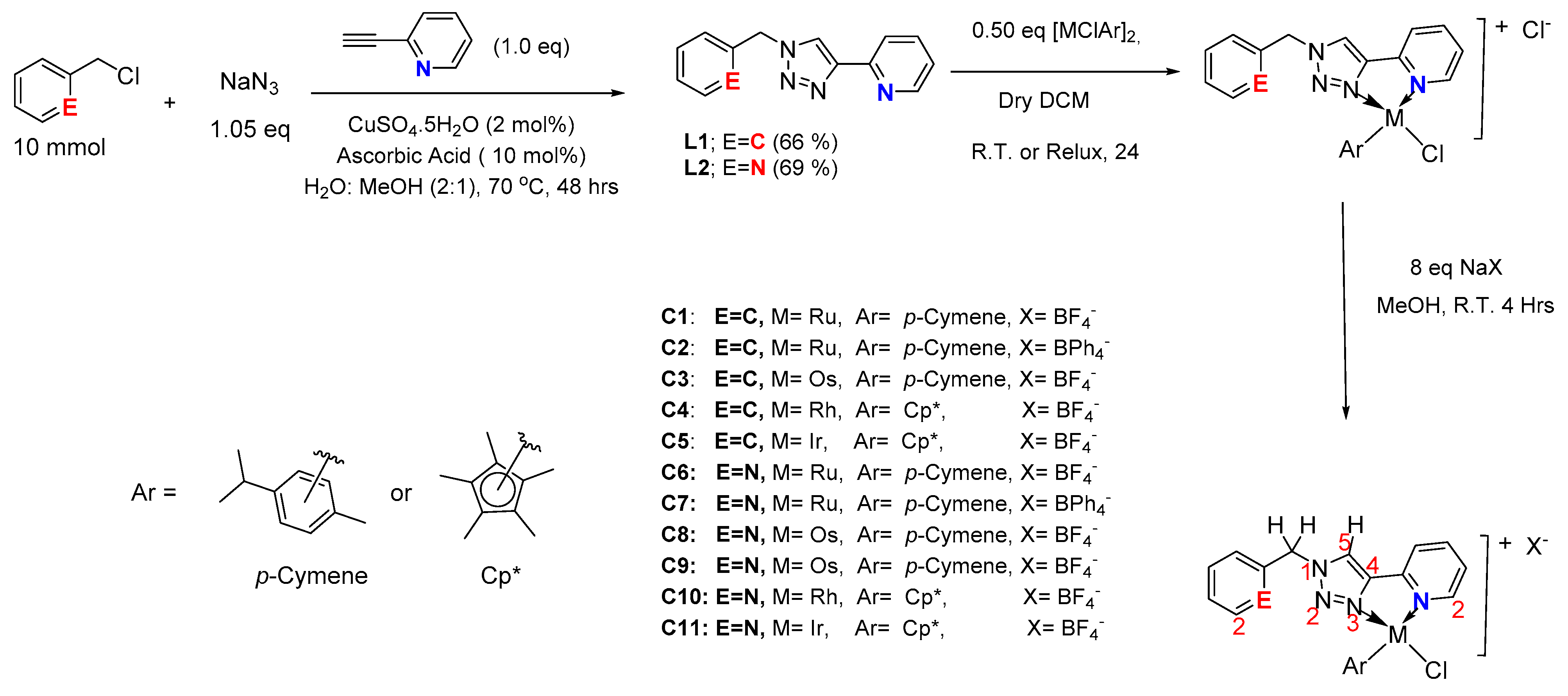
2.1. Synthesis and Characterization of the Ligands and Their Respective Complexes
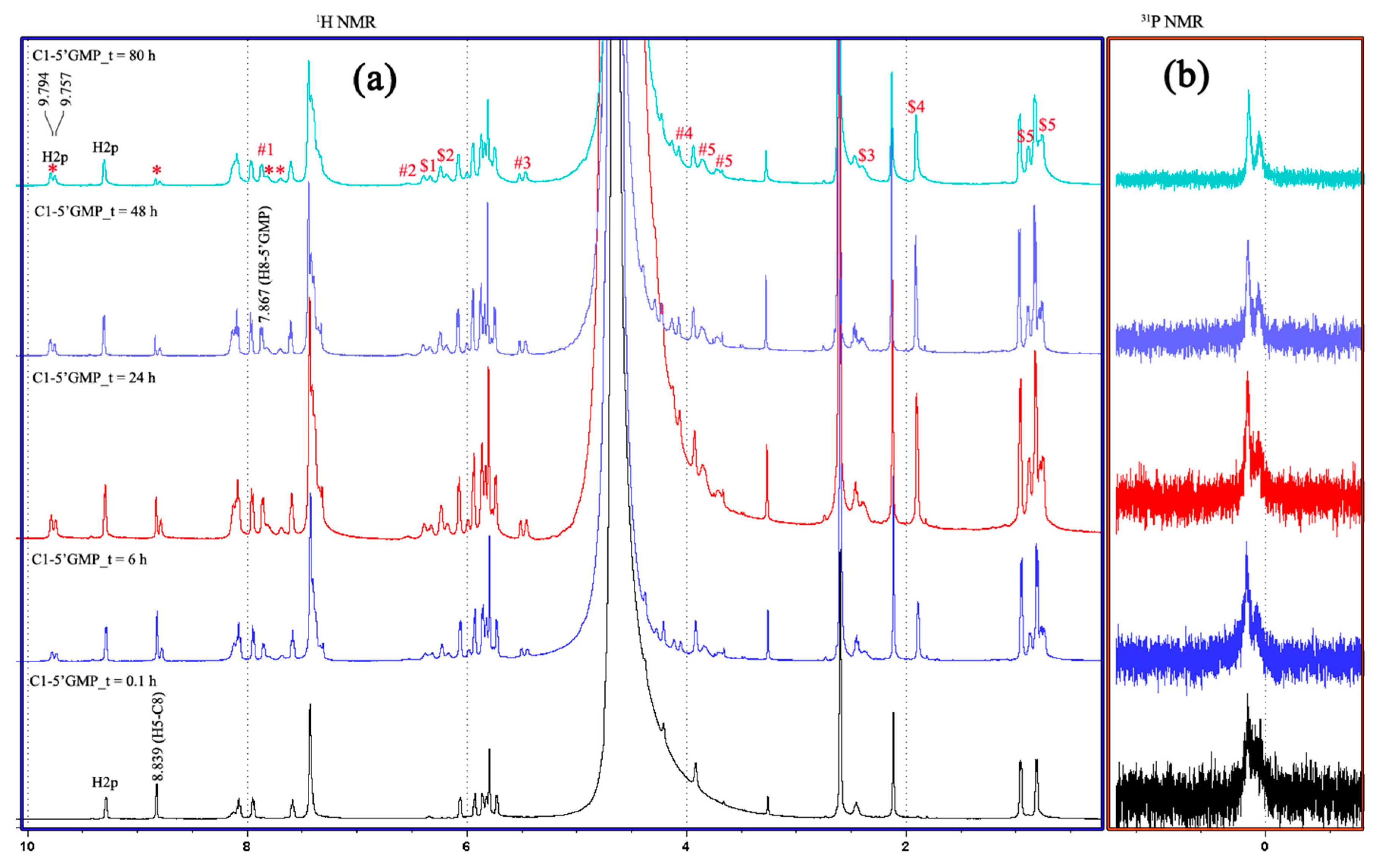
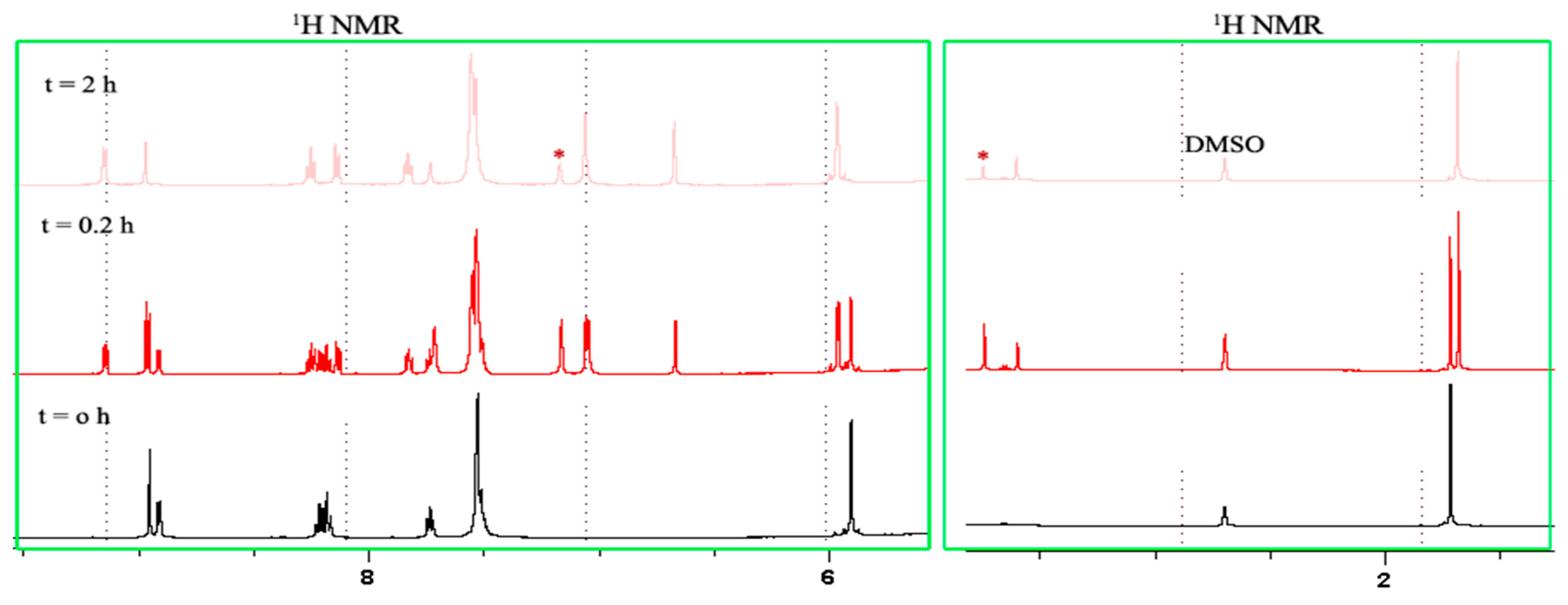
2.2. NMR Studies on the Stability and Behaviour of the Complexes in an Aqueous Model System
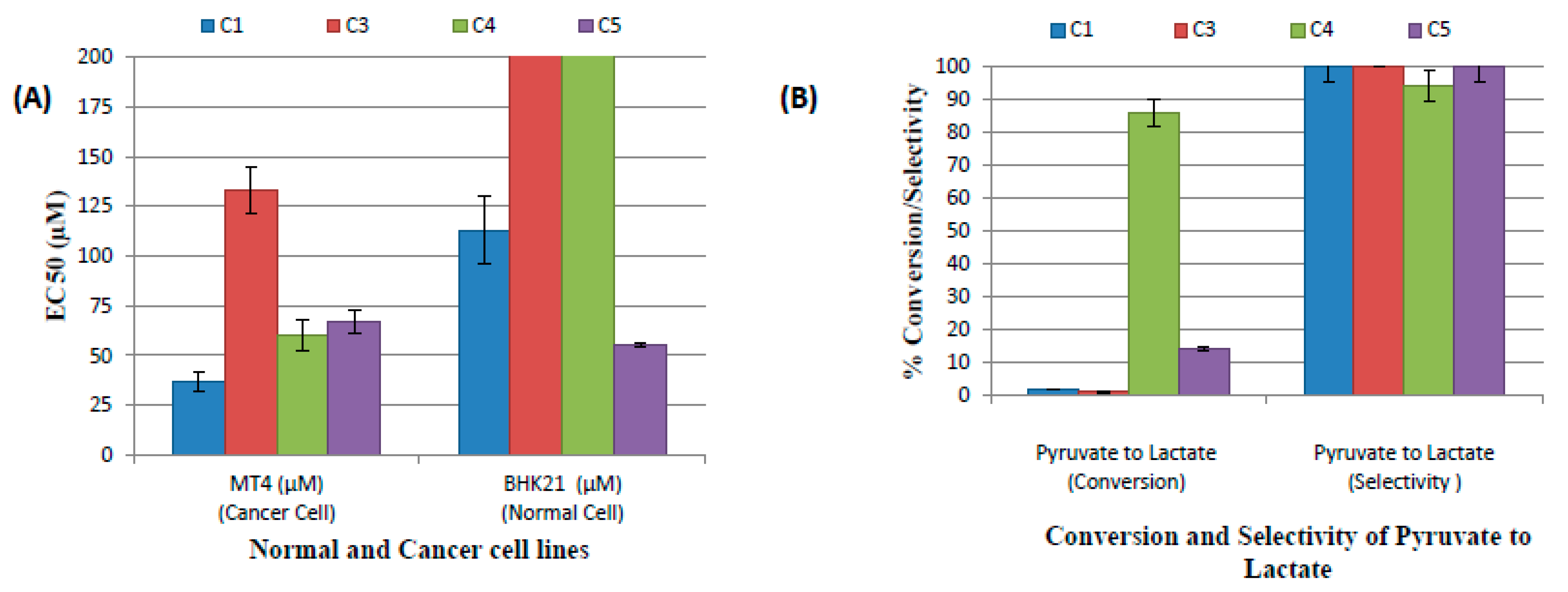
2.3. Catalytic Evaluation
2.4. Cytotoxicity Evaluation
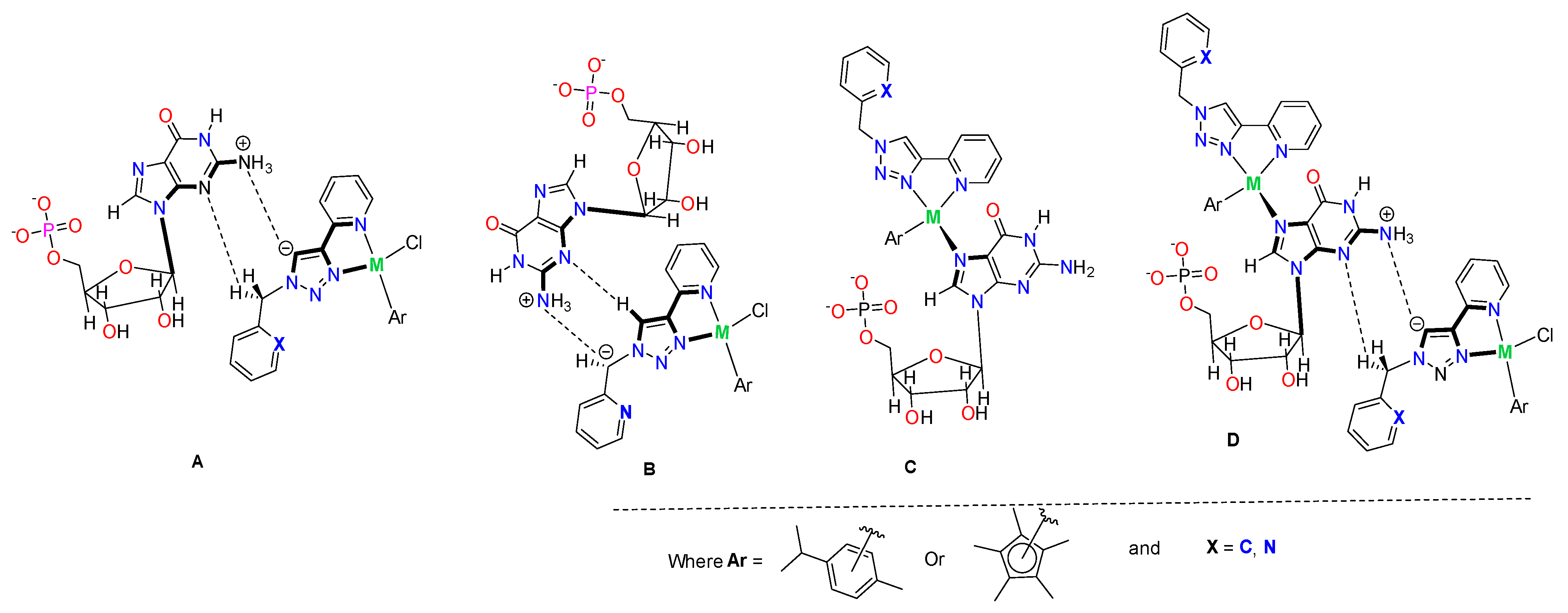
2.5. Interactions of the Complexes with Guanosine-5′-monophosphate (5′-GMP) (DNA Nucleoside)
2.6. Interaction of the Complexes with Protein and Amino Acids
3. Experimental Section
3.1. Materials and Instruments
3.2. General Procedure for the Ligand Synthesis and Isolation
3.2.1. 2-(1-Benzyl-1H-1,2,3-triazol-4-yl)pyridine (L1)
3.2.2. 2-((4-(Pyridin-2-yl)-1H-1,2,3-triazol-1-yl)methyl)pyridine (L2)
3.3. General Procedure for the Synthesis of Complexes C1-C11
3.3.1. [Chlorido(2-(1-benzyl-(3-κN)-1,2,3-triazol-4-yl)-(2′-κN)-pyridine)(η6-p-cymene)ruthenium(II)] tetrafluoroborate (C1)
3.3.2. [Chlorido(2-(1-benzyl-(3-κN)-1,2,3-triazol-4-yl)-(2′-κN)-pyridine)(η6-p-cymene)ruthenium(II)] tetraphenylborate (C2)
3.3.3. [Chlorido(2-(1-benzyl-(3-κN)-1,2,3-triazol-4-yl)-(2′-κN)-pyridine)(η6-p-cymene)osmium(II)] tetrafluoroborate (C3)
3.3.4. [Chlorido(2-(1-benzyl-(3-κN)-1,2,3-triazol-4-yl)-(2′-κN)-pyridine)(η6-pentamethylcypentadiene)rhodium(III)] tetrafluoroborate (C4)
3.3.5. [Chlorido(2-(1-benzyl-(3-κN)-1,2,3-triazol-4-yl)-(2′-κN)-pyridine)(η6-pentamethylcypentadiene)iridium(III)] tetrafluoroborate (C5)
3.3.6. [Chlorido(2-((4-(pyridine-2-yl)-(3-κN)-1,2,3-trizol-1-yl)methyl)-(2′-κN)-pyridine)(η6-p-cymene)ruthenium(II)] tetrafluoroborate (C6)
3.3.7. [Chlorido(2-((4-(pyridine-2-yl)-(3-κN)-1,2,3-trizol-1-yl)methyl)-(2′-κN)-pyridine)(η6-p-cymene)ruthenium(II)] tetraphenylborate (C7)
3.3.8. [Chlorido(2-((4-(pyridine-2-yl)-(3-κN)-1,2,3-trizol-1-yl)methyl)-(2′-κN)-pyridine)(η6-p-cymene)osmium(II)] tetrafluoroborate (C8)
3.3.9. [Chlorido(2-((4-(pyridine-2-yl)-(3-κN)-1,2,3-trizol-1-yl)methyl)-(2′-κN)-pyridine)(η6-p-cymene)osmium(II)] tetrafluoroborate (C9)
3.3.10. [Chlorido(2-((4-(pyridine-2-yl)-(3-κN)-1,2,3-trizol-1-yl)methyl)-(2′-κN)-pyridine)(η6-pentamethylcypentadiene)rhodium(III)] tetrafluoroborate (C10)
3.3.11. [Chlorido(2-((4-(pyridine-2-yl)-(3-κN)-1,2,3-trizol-1-yl)methyl)-(2′-κN)-pyridine)(η6-pentamethylcypentadiene)iridium(III)] tetrafluoroborate (C11)
3.4. NMR Studies on the Stability and Behaviour of the Complexes in an Aqueous Model System
3.5. Catalytic Evaluation
3.6. Cytotoxicity Evaluation
3.7. DNA Model Interaction with Guanosine-5′-monophosphate (5′-GMP)
3.8. Protein Interaction
3.9. Amino Acids Interaction with Complexes C1 and C5
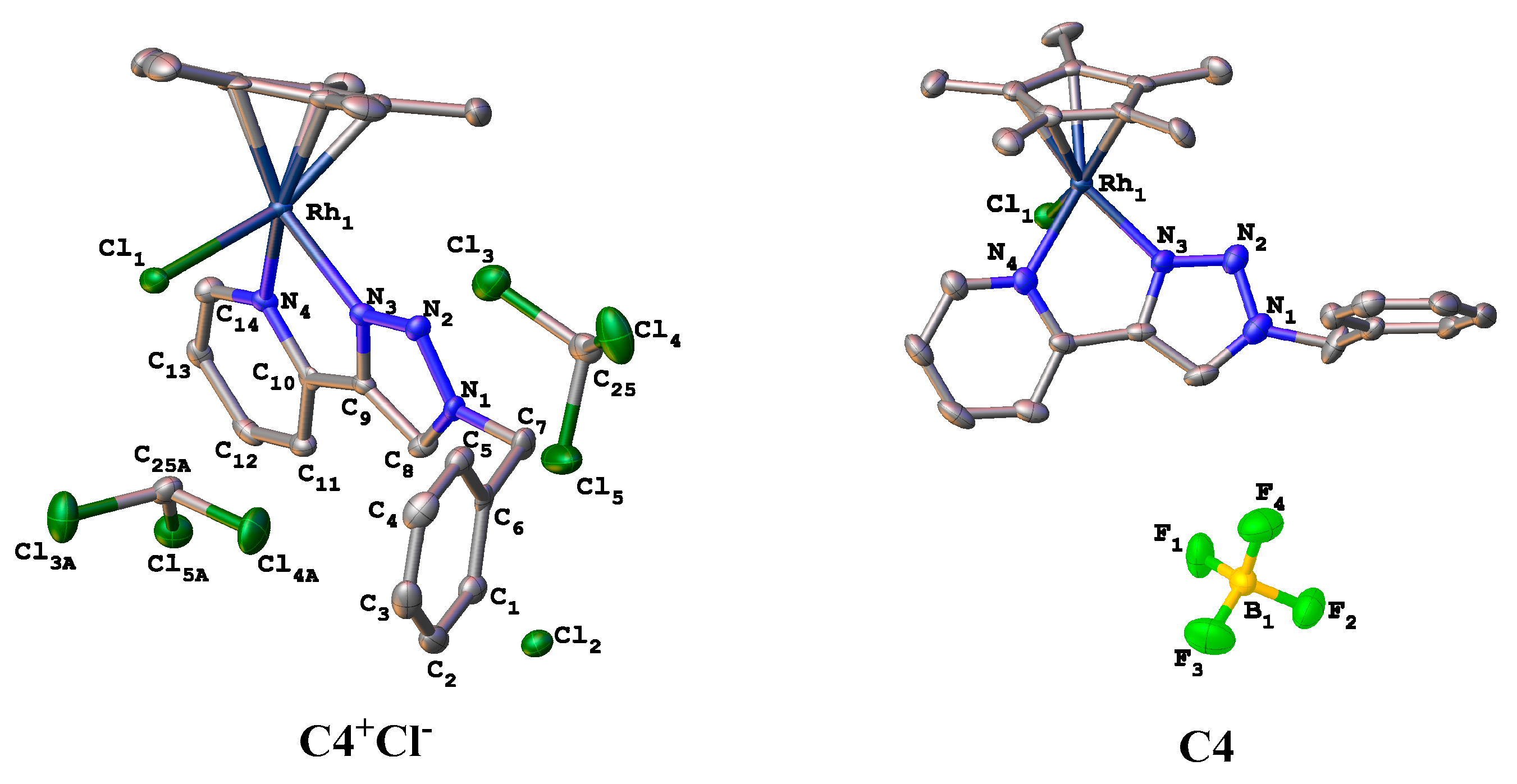
3.10. Crystal Structure Determination for C4 and Its Cl− Counterion Analogue
3.11. Determining Calculated Partition Coefficient (clogP)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bertram, J.S. The Molecular Biology of Cancer. Mol. Aspects Med. 2001, 21, 167–223. [Google Scholar] [CrossRef]
- American Cancer Society. Global Cancer Facts & Figures, 4th ed.; American Cancer Society: Atlanta, GA, USA, 2018; pp. 1–74. [Google Scholar]
- Mareel, M.; Leroy, A. Clinical, Cellular, and Molecular Aspects of Cancer Invasion. Physiol. Rev. 2003, 83, 337–376. [Google Scholar]
- Martínez-Alonso, M.; Busto, N.; Jalón, F.A.; Manzano, B.R.; Leal, J.M.; Rodríguez, A.M.; García, B.; Espino, G. Derivation of Structure—Activity Relationships from the Anticancer Properties of Ruthenium(II) Arene Complexes with 2-Aryldiazole Ligands. Inorg. Chem. 2014, 53, 11274–11288. [Google Scholar]
- Bruijnincx, P.C.; Sadler, P.J. New Trends for Metal Complexes with Anticancer Activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.W.-Y.; Ma, D.-L.; Wong, E.L.-M.; Che, C.-M. Some Uses of Transition Metal Complexes as Anti-Cancer and Anti-HIV Agents. Dalton Trans. 2007, 43, 4884–4892. [Google Scholar]
- Coverdale, J.P.C.; Romero-Canelón, I.; Sanchez-Cano, C.; Clarkson, G.J.; Habtemariam, A.; Wills, M.; Sadler, P.J. Asymmetric Transfer Hydrogenation by Synthetic Catalysts in Cancer Cells. Nat. Chem. 2018, 10, 347–354. [Google Scholar] [CrossRef]
- Alderden, R.A.; Hall, M.D.; Hambley, T.W. The Discovery and Development of Cisplatin. J. Chem. Educ. 2006, 83, 728–734. [Google Scholar] [CrossRef]
- Karges, J.; Stokes, R.W.; Cohen, S.M. Metal Complexes for Therapeutic Applications. Trends Chem. 2021, 3, 523–534. [Google Scholar] [CrossRef]
- Soldevila-Barreda, J.J.; Romero-Canelón, I.; Habtemariam, A.; Sadler, P.J. Transfer Hydrogenation Catalysis in Cells as a New Approach to Anticancer Drug Design. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Romero-canelo, I.; Soldevila-barreda, J.J.; Song, J.; Coverdale, J.P.C.; Clarkson, G.J.; Kasparkova, J.; Habtemariam, A.; Wills, M.; Brabec, V.; et al. Transfer Hydrogenation and Antiproliferative Activity of Tethered Half-Sandwich Organoruthenium Catalysts. Organometallics 2018, 37, 1555–1566. [Google Scholar]
- San-Millán, I.; Brooks, G.A. Reexamining Cancer Metabolism: Lactate Production for Carcinogenesis Could Be the Purpose and Explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Haghdoost, M.M.; Guard, J.; Golbaghi, G.; Castonguay, A. Anticancer Activity and Catalytic Potential of Ruthenium(II) − Arene Complexes with N,O-Donor Ligands. Inorg. Chem. 2018, 57, 7558–7567. [Google Scholar] [CrossRef]
- Singh, A.K.; Pandey, D.S.; Xu, Q.; Braunstein, P. Recent Advances in Supramolecular and Biological Aspects of Arene Ruthenium(II) Complexes. Coord. Chem. Rev. 2014, 270–271, 31–56. [Google Scholar] [CrossRef]
- Matsheku, A.C.; Chen, M.Y.H.; Jordaan, S.; Prince, S.; Smith, G.S.; Makhubela, B.C.E. Acridine-Containing RuII, OsII, RhIII and IrIII Half-Sandwich Complexes: Synthesis, Structure and Antiproliferative Activity. Appl. Organomet. Chem. 2017, 31, 1–14. [Google Scholar] [CrossRef]
- Rausch, M.; Dyson, P.J.; Nowak-Sliwinska, P. Recent Considerations in the Application of RAPTA-C for Cancer Treatment and Perspectives for Its Combination with Immunotherapies. Adv. Ther. 2019, 2, 1900042. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Dyson, P.J. Bioorganometallic Chemistry—from Teaching Paradigms to Medicinal Applications. Chem. Soc. Rev. 2009, 38, 391–401. [Google Scholar] [CrossRef]
- Rono, C.K.; Chu, W.K.; Darkwa, J.; Meyer, D.; Makhubela, B.C.E. Triazolyl RuII, RhIII, OsII, and IrIII Complexes as Potential Anticancer Agents: Synthesis, Structure Elucidation, Cytotoxicity, and DNA Model Interaction Studies. Organometallics 2019, 38, 3197–3211. [Google Scholar] [CrossRef]
- Schulze, B.; Schubert, U.S. Beyond Click Chemistry—Supramolecular Interactions of 1,2,3-Triazoles. Chem. Soc. Rev. 2014, 43, 2522–2571. [Google Scholar] [CrossRef]
- Wong, C.M.; Vuong, K.Q.; Gatus, M.R.D.; Hua, C.; Bhadbhade, M.; Messerle, B.A. Catalyzed Tandem C-N/C-C Bond Formation for the Synthesis of Tricyclic Indoles Using Ir(III) Pyrazolyl-1,2,3-Triazolyl Complexes. Organometallics 2012, 31, 7500–7510. [Google Scholar] [CrossRef]
- Riedl, C.A.; Flocke, L.S.; Hejl, M.; Roller, A.; Klose, M.H.M.; Jakupec, M.A.; Kandioller, W.; Keppler, B.K. Introducing the 4 - Phenyl-1,2,3-Triazole Moiety as a Versatile Sca Ff Old for the Development of Cytotoxic Ruthenium(II) and Osmium(II) Arene Cyclometalates. Inorg. Chem. 2017, 56, 528–541. [Google Scholar] [CrossRef]
- Garbacz, G.; Ko, B.; Koziolek, M.; Weitschies, W.; Klein, S. Brief / Technical Note An Automated System for Monitoring and Regulating the PH of Bicarbonate Buffers. AAPS PharmSciTechn. 2013, 14, 2–7. [Google Scholar]
- Hoorn, E.J. Intravenous Fluids: Balancing Solutions. J. Nephrol. 2017, 30, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Mirabelli, C.K.; Johnson, R.K.; Sung, C.; Faucette, L.; Muirhead, K.; Crooke, S.T. Evaluation of the in Vivo Antitumor Activity and in Vitro Cytotoxic Properties of Auranofin, a Coordinated Gold Compound, in Murine Tumor Models. Cancer Res. 1985, 45, 32–39. [Google Scholar]
- Simon, T.M.; Kunishima, D.H.; Vibert, G.J.; Lorber, A. Screening Trial with the Coordinated Gold Compound Auranofin Using Mouse Lymphocytic Leukemia P3881. Cancer Res. 1981, 41, 94–97. [Google Scholar]
- Haghdoost, M.M.; Golbaghi, G.; Guard, J.; Sielanczyk, S.; Patten, S.A.; Castonguay, A. Synthesis, Characterization and Biological Evaluation of Cationic Organoruthenium(II) Fluorene Complexes: Influence of the Nature of the Counteranion. Dalton Trans. 2019, 48, 13396–13405. [Google Scholar] [CrossRef]
- Golbaghi, G.; Haghdoost, M.M.; Yancu, D.; de los Santos, Y.L.; Doucet, N.; Patten, S.A.; Sanderson, J.T.; Castonguay, A. Organoruthenium(II) Complexes Bearing an Aromatase Inhibitor: Synthesis, Characterization, in Vitro Biological Activity and in Vivo Toxicity in Zebrafish Embryos. Organometallics 2019, 38, 702–711. [Google Scholar] [CrossRef]
- Loughrey, B.T.; Healy, P.C.; Parsons, P.G.; Williams, M.L. Selective Cytotoxic Ru(II) Arene Cp* Complex Salts [R-PhRuCp*]+X- for X = BF4-, PF6-, and BPh4-. Inorg. Chem. 2008, 47, 8589–8591. [Google Scholar] [CrossRef]
- Martĺnez-Alonso, M.; Sanz, P.; Ortega, P.; Espino, G.; Jalón, F.A.; Martín, M.; Rodrĺguez, A.M.; López, J.A.; Tejel, C.; Manzano, B.R. Analysis of Ion Pairing in Solid State and Solution in P-Cymene Ruthenium Complexes. Inorg. Chem. 2020, 59, 14171–14183. [Google Scholar] [CrossRef]
- Koch, A.; Tamez, P.; Pezzuto, J.; Soejarto, D.J. Evaluation of Plants Used for Antimalarial Treatment by the Maasai of Kenya. Ethnopharmacol. 2005, 101, 95–99. [Google Scholar] [CrossRef]
- Liu, Z.; Salassa, L.; Habtemariam, A.; Pizarro, A.M.; Clarkson, G.J.; Sadler, P.J. Contrasting Reactivity and Cancer Cell Cytotoxicity of Isoelectronic Organometallic Iridium(III) Complexes. Inorg. Chem. 2011, 50, 5777–5783. [Google Scholar] [CrossRef] [Green Version]
- Odani, A.; Shimata, R.; Masuda, I.H.; Ja, O.Y. Platinum DNA Intercalator-Mononucleotide Adduct Formation. Cooperativity between Aromatic Ring Stacking and Electrostatic Interactions. Inorg. Chem. 1991, 30, 2133–2138. [Google Scholar] [CrossRef]
- Komeda, S.; Ohishi, H.; Yamane, H.; Harikawa, M.; Sakaguchi, K.; Chikuma, M. An NMR study and crystal structure of [{cis-Pt(NH3)2(9EtG-κN7)}2(µ-pz)][NO3]3 (9EtG = 9-ethylguanine) as a model compound for the 1,2-intrastrand GG crosslink. J. Chem. Soc. Dalton Trans. 1999, 17, 2959–2962. [Google Scholar] [CrossRef]
- Komeda, S.; Lutz, M.; Spek, A.L.; Yamanaka, Y.; Sato, T.; Chikuma, M.; Reedijk, J. A Novel Isomerization on Interaction of Antitumor-Active Azole-Bridged Dinuclear Platinum(II) Complexes with 9-Ethylguanine. Platinum(II) Atom Migration from N2 to N3 on 1,2,3-Triazole. J. Am. Chem. Soc. 2002, 124, 4738–4746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Liu, J.; Tian, X.; Hu, Z.; Chen, X. Study of the Interaction of Kaempferol with Bovine Serum Albumin. J. Mol. Struct. 2004, 691, 197–202. [Google Scholar] [CrossRef]
- Ojha, B.; Das, G. The interaction of 5-(Alkoxy)naphthalen-1-amine with bovine serum albumin and Its effect on the conformation of protein. J. Phys. Chem. B 2010, 114, 3979–3986. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Atoms | Bond Distance (Å) | Atoms Involved | Bond Angle (°) |
|---|---|---|---|---|
| C4+Cl− | Rh1-N3 | 2.115 (3) | N2-N3-Rh1 | 133.6 (3) |
| Rh1-N4 | 2.135 (5) | C9-N3-Rh1 | 115.4 (3) | |
| Rh1-Cl | 2.3871 (11) | C10-N4-Rh1 | 117.1 (3) | |
| N3-C9 | 1.364 (5) | N3-N2-N1 | 105.1 (3) | |
| N4-C10 | 1.358 (5) | N4-C10-C9 | 113.7 (3) | |
| N1-C8 | 1.338 (5) | C10-C9-N3 | 117.7 (3) | |
| N1-C7 | 1.474 (5) | N3-Rh1-N4 | 76.19 (13) | |
| C4 | Rh1-N3 | 2.085 (5) | N2-N3-Rh1 | 133.9 (4) |
| Rh1-N4 | 2.129 (5) | C9-N3-Rh1 | 115.3 (4) | |
| Rh1-Cl | 2.384 (2) | C10-N4-Rh1 | 115.6 (4) | |
| N3-C9 | 1.363 (8) | N3-N2-N1 | 104.9 (5) | |
| N4-C10 | 1.354 (8) | N4-C10-C9 | 113.9 (6) | |
| N1-C8 | 1.344 (9) | C10-C9-N3 | 117.2 (6) | |
| N1-C7 | 1.448 (9) | N3-Rh1-N4 | 76.9 (2) |
| Catalyst | Base | Conversion (%) | TON | Selectivity (%) | Structures of the Complexes |
|---|---|---|---|---|---|
| No catalyst | None | 6 ± 1 | 6 | 0 |  |
| No catalyst | NaHCO3 | 52 ± 2 | 52 | 0 | |
| C1 | None | 2 ± 0.2 | 2 | 100 | |
| C1 | NaHCO3 | 0.1 ± 0.02 | ≤0.1 | 100 | |
| C1 * | None | 5 ± 1 | 5 | >99 | |
| C3 | None | 1 ± 0.3 | 1 | 100 | |
| C3 | NaHCO3 | 0.1 ± 0.03 | ≤0.1 | 100 | |
| C3 * | None | 3 ± 1 | 3 | >99 | |
| C4 | None | 86 ± 5 | 86 | 94 | |
| C4 | NaHCO3 | 70 ± 2 | 70 | 80 | |
| C4 | Et3N | 60 ± 4 | 60 | 44 | |
| C4 * | None | 90 ± 5 | 90 | >99 | |
| C5 | None | 14 ± 2 | 14 | 100 | |
| C5 | NaHCO3 | 0.1 ± 0.02 | ≤0.1 | 100 | |
| C5 * | None | 20 ± 3 | 20 | >99 |
| Compounds | EC50 (µM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| A549 | SI | HEK293 | SI | Hela | SI | MT4 | SI | BHK21 | |
| L1 | >1300 | n/d | >1300 | ≤1 | 168 ± 31 | n/d | >1300 | n/d | >1300 |
| C1 | >500 | n/d | >500 | <1 | >500 | n/d | 37 ± 5 | n/d | 113 ± 17 |
| C2 | 14 ± 4.0 | n/d | 11 ± 1.5 | 14 | 15 ± 1.7 | n/d | 9 ± 1 | n/d | 151 ± 0.6 |
| C3 | >500 | n/d | >500 | ≤1 | >500 | n/d | 133 ± 12 | n/d | >500 |
| C4 | >500 | n/d | >500 | ≤1 | ≥300 | n/d | 60 ± 8 | n/d | >500 |
| C5 | >500 | n/d | >500 | <1 | >500 | n/d | 67 ± 6 | n/d | 55 ± 1 |
| L2 | >1300 | n/d | >1300 | ≤1 | 1070 ± 134 | n/d | N.R. | n/d | >1300 |
| C6 | N.R. | n/d | >500 | - | 388 ± 70.9 | n/d | 158 ± 44.2 | n/d | N.R. |
| C7 | 53 ± 6.0 | n/d | 17 ± 4.8 | 4 | 26 ± 0.8 | n/d | 20 ± 4.8 | n/d | 67 ± 7.3 |
| C8 | >500 | n/d | >500 | ≤1 | N.R. | n/d | N.R. | n/d | >500 |
| C9 | >500 | n/d | >400 | - | N.R. | n/d | N.R. | n/d | N.R. |
| C10 | N.R. | n/d | >500 | - | N.R. | n/d | >500 | n/d | N.R. |
| C11 | >500 | n/d | >500 | - | 2.5 ± 1 | n/d | 111 ± 7 | n/d | N.R. |
| Auranofin | 12 ± 2 | n/d | <0.1 | ≤1 | <2 | n/d | <2 | n/d | <0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, W.K.; Rono, C.K.; Makhubela, B.C.E. New Triazolyl N^N Bidentate Rh(III), Ir(III), Ru(II) and Os(II) Complexes: Synthesis and Characterization, Probing Possible Relations between Cytotoxicity with Transfer Hydrogenation Efficacy and Interaction with Model Biomolecules. Molecules 2022, 27, 2058. https://doi.org/10.3390/molecules27072058
Chu WK, Rono CK, Makhubela BCE. New Triazolyl N^N Bidentate Rh(III), Ir(III), Ru(II) and Os(II) Complexes: Synthesis and Characterization, Probing Possible Relations between Cytotoxicity with Transfer Hydrogenation Efficacy and Interaction with Model Biomolecules. Molecules. 2022; 27(7):2058. https://doi.org/10.3390/molecules27072058
Chicago/Turabian StyleChu, William K., Charles K. Rono, and Banothile C. E. Makhubela. 2022. "New Triazolyl N^N Bidentate Rh(III), Ir(III), Ru(II) and Os(II) Complexes: Synthesis and Characterization, Probing Possible Relations between Cytotoxicity with Transfer Hydrogenation Efficacy and Interaction with Model Biomolecules" Molecules 27, no. 7: 2058. https://doi.org/10.3390/molecules27072058
APA StyleChu, W. K., Rono, C. K., & Makhubela, B. C. E. (2022). New Triazolyl N^N Bidentate Rh(III), Ir(III), Ru(II) and Os(II) Complexes: Synthesis and Characterization, Probing Possible Relations between Cytotoxicity with Transfer Hydrogenation Efficacy and Interaction with Model Biomolecules. Molecules, 27(7), 2058. https://doi.org/10.3390/molecules27072058