
Dihydropyridines Potentiate ATP-Induced Currents Mediated by the Full-Length Human P2X5 Receptor


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
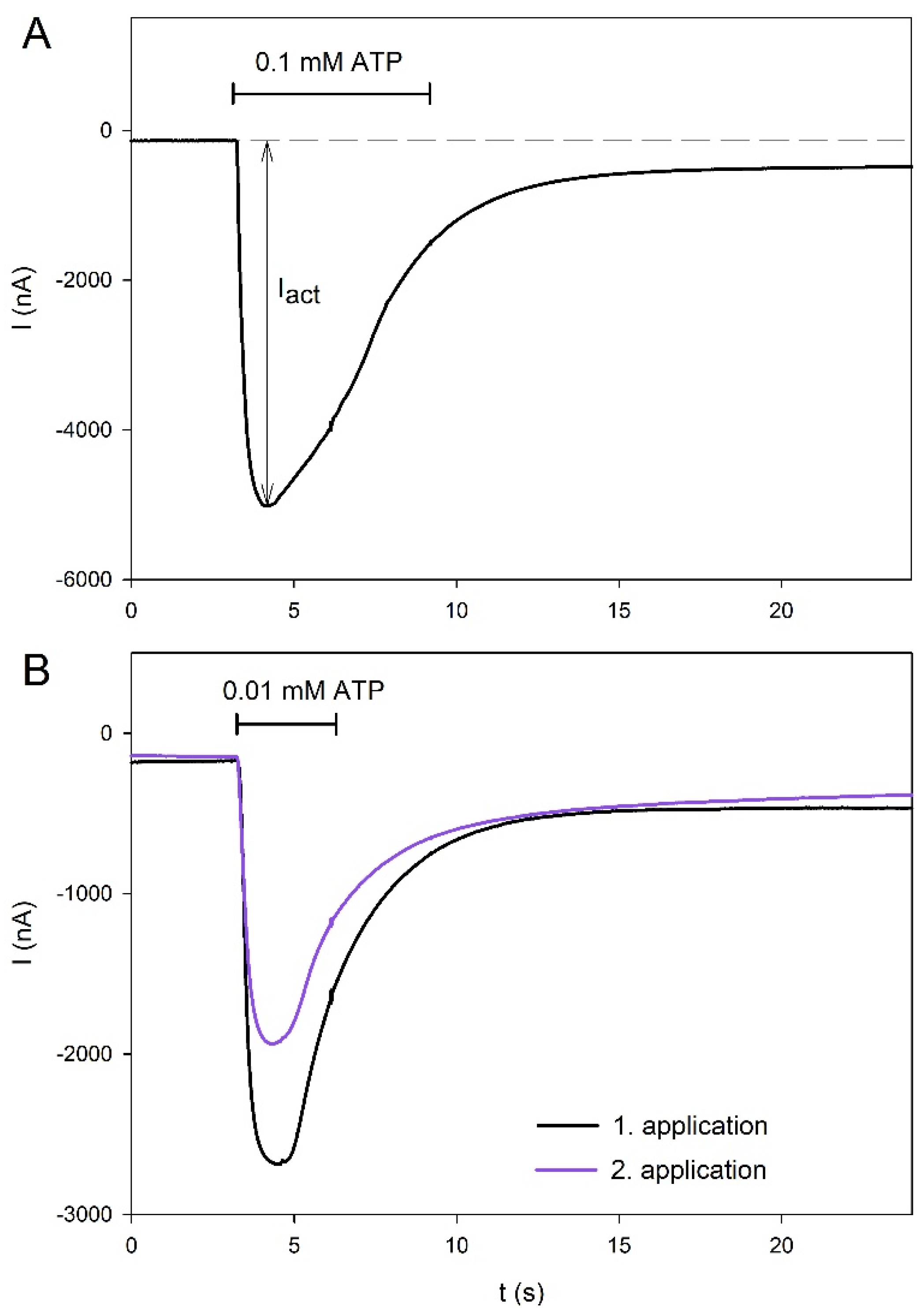
2.1. Agonist-Dependent Activation of hP2X5FL Receptors
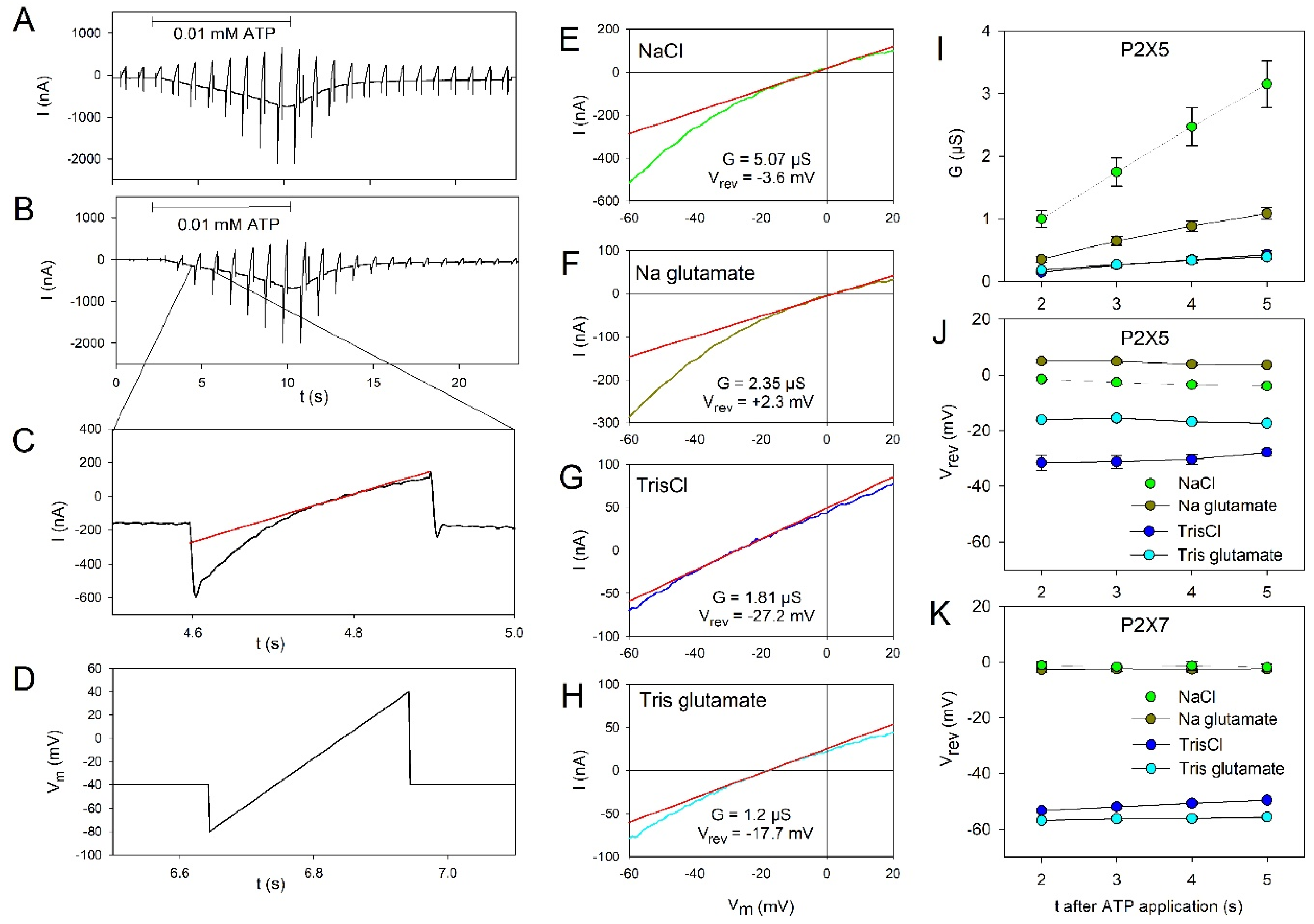
2.2. Permeation Behavior of hP2X5FL Receptor Ion Channels
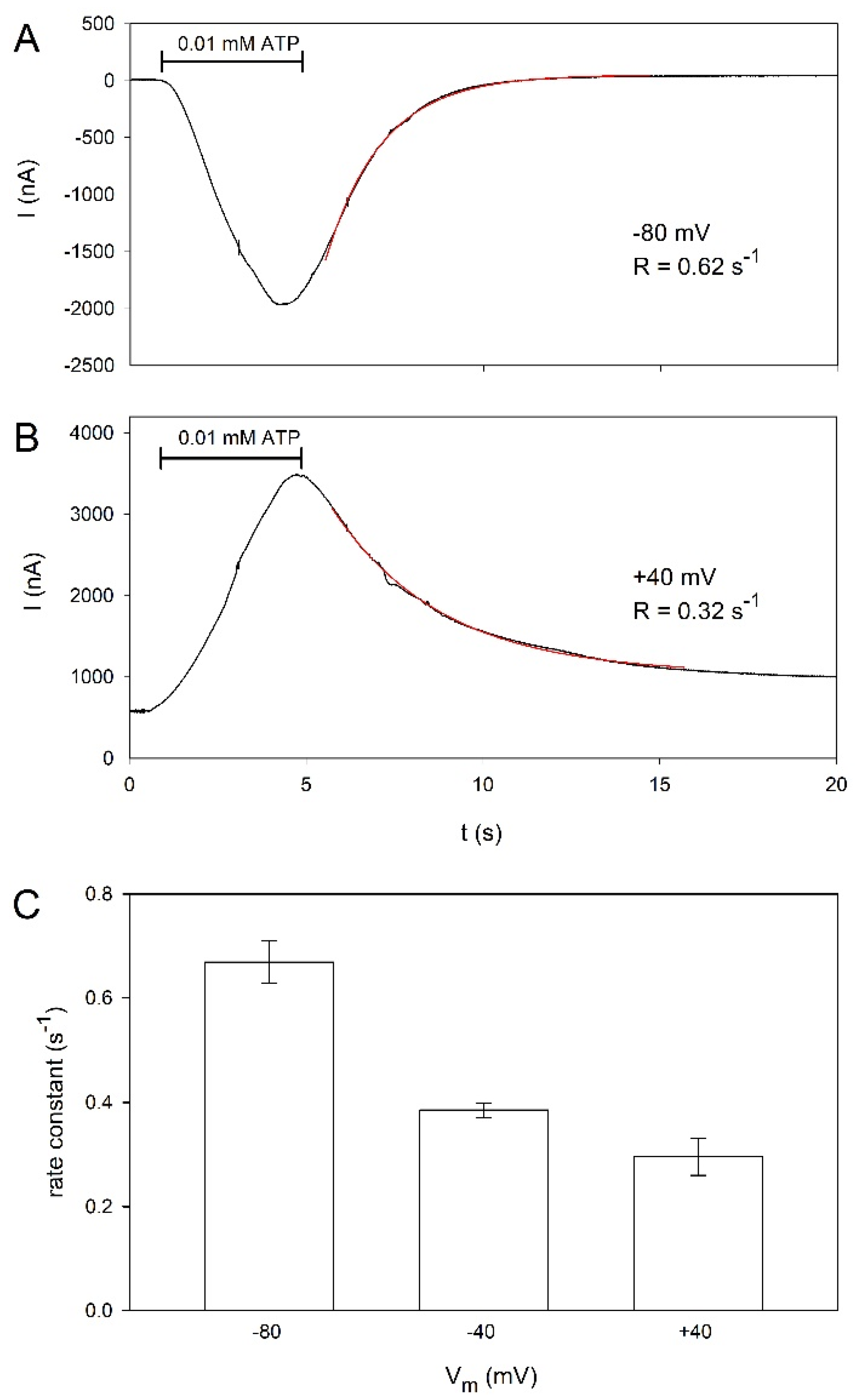
2.3. Voltage Dependence of hP2X5FL Receptor Ion Channels
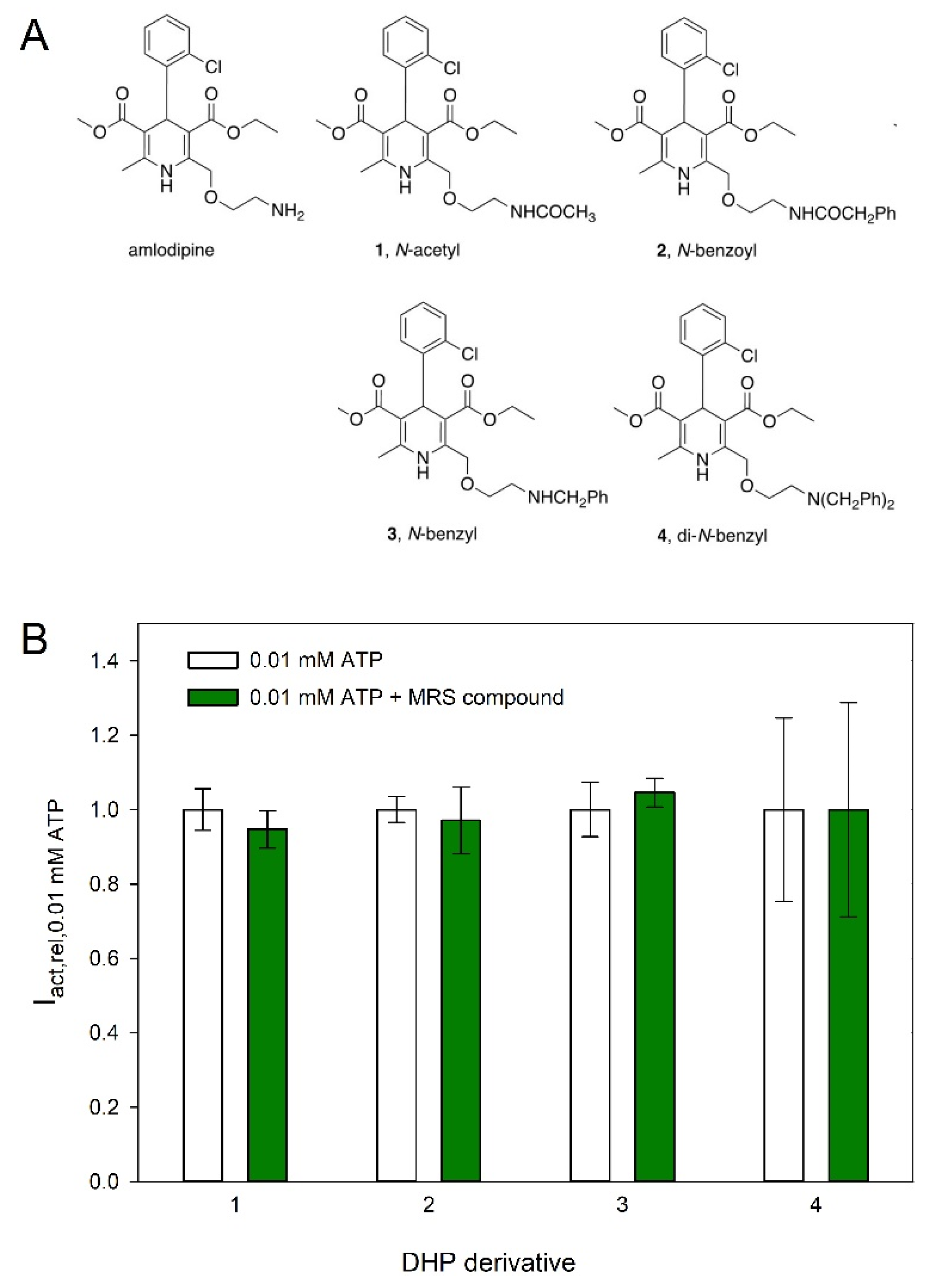
2.4. Effect of Dihydropyridines on hP2X5FL-Mediated Currents
3. Discussion
3.1. Activation and Kinetics of hP2X5FL-Mediated Currents
3.2. Permeation Characteristics of hP2X5FL
3.3. Effect of Dihydropyridines on hP2X5FL
4. Materials and Methods
4.1. Reagents
4.2. Construction of Full-Length hP2X5 mRNA
4.3. Expression and of hP2X5FL in Oocytes of Xenopus Laevis
4.4. Voltage Clamp Measurements
4.5. Data Analysis and Presentation
4.6. Chemical Synthesis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Illes, P.; Müller, C.E.; Jacobson, K.A.; Grutter, T.; Nicke, A.; Fountain, S.J.; Kennedy, C.; Schmalzing, G.; Jarvis, M.F.; Stojilkovic, S.S.; et al. Update of P2X receptor properties and their pharmacology: IUPHAR Review 30. Br. J. Pharmacol. 2021, 178, 489–514. [Google Scholar] [CrossRef]
- Bhattacharya, A. Recent advances in CNS P2X7 physiology and pharmacology: Focus on neuropsychiatric disorders. Front. Pharmacol. 2018, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.P.; Dillon, M.P.; Kitt, M.M.; Gever, J.R. The discovery and development of gefapixant. Auton. Neurosci. 2021, 235, 102859. [Google Scholar] [CrossRef] [PubMed]
- Gelin, C.F.; Bhattacharya, A.; Letavic, M.A. P2X7 receptor antagonists for the treatment of systemic inflammatory disorders. Prog. Med. Chem. 2020, 59, 63–99. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Yamashita, T.; Sasaki, A.; Nakata, E.; Kohno, K.; Masuda, T.; Tozaki-Saitoh, H.; Imai, T.; Kuraishi, Y.; Tsuda, M.; et al. A novel P2X4 receptor-selective antagonist produces anti-allodynic effect in a mouse model of herpetic pain. Sci. Rep. 2016, 6, 32461. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, S.E.; Lu, W.; Oosterheert, W.; Shekhar, M.; Tajkhorshid, E.; Gouaux, E. X-ray structures define human P2X3 receptor gating cycle and antagonist action. Nature 2016, 538, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Le, K.T.; Paquet, M.; Nouel, D.; Babinski, K.; Seguela, P. Primary structure and expression of a naturally truncated human P2X ATP receptor subunit from brain and immune system. FEBS Lett. 1997, 418, 195–199. [Google Scholar] [CrossRef]
- Ussar, S.; Lee, K.Y.; Dankel, S.N.; Boucher, J.; Haering, M.F.; Kleinridders, A.; Thomou, T.; Xue, R.; Macotela, Y.; Cypess, A.M.; et al. ASC-1, PAT2, and P2RX5 are cell surface markers for white, beige, and brown adipocytes. Sci. Transl. Med. 2014, 6, 247ra103. [Google Scholar] [CrossRef]
- Berchtold, S.; Ogilvie, A.L.J.; Bogdan, C.; Muhlzurbes, P.; Ogilvie, A.; Schuler, G.; Steinkasserer, A. Human monocyte derived dendritic cells express functional P2X and P2Y receptors as well as ecto-nucleotidases. FEBS Lett. 1999, 458, 424–428. [Google Scholar] [CrossRef]
- Janssens, R.; Boeynaems, J.M. Effects of extracellular nucleotides and nucleosides on prostate carcinoma cells. Br. J. Pharmacol. 2001, 132, 536–546. [Google Scholar] [CrossRef]
- Burnstock, G.; Di Virgilio, F. Purinergic signalling and cancer. Purinergic Signal. 2013, 9, 491–540. [Google Scholar] [CrossRef] [PubMed]
- de Rijke, B.; van Horssen-Zoetbrood, A.; Beekman, J.M.; Otterud, B.; Maas, F.; Woestenenk, R.; Kester, M.; Leppert, M.; Schattenberg, A.V.; de Witte, T.; et al. A frameshift polymorphism in P2X5 elicits an allogeneic cytotoxic T lymphocyte response associated with remission of chronic myeloid leukemia. J. Clin. Investig. 2005, 115, 3506–3516. [Google Scholar] [CrossRef]
- Overes, I.M.; de Rijke, B.; van Horssen-Zoetbrood, A.; Fredrix, H.; de Graaf, A.O.; Jansen, J.H.; van Krieken, J.H.; Raymakers, R.A.; van der Voort, R.; de Witte, T.M.; et al. Expression of P2X5 in lymphoid malignancies results in LRH-1-specific cytotoxic T-cell-mediated lysis. Br. J. Haematol. 2008, 141, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Overes, I.M.; Levenga, T.H.; Vos, J.C.; van Horssen-Zoetbrood, A.; van der Voort, R.; De Mulder, P.H.; de Witte, T.M.; Dolstra, H. Aberrant expression of the hematopoietic-restricted minor histocompatibility antigen LRH-1 on solid tumors results in efficient cytotoxic T cell-mediated lysis. Cancer Immunol. Immunother. 2009, 58, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Kotnis, S.; Bingham, B.; Vasilyev, D.V.; Miller, S.W.; Bai, Y.; Yeola, S.; Chanda, P.K.; Bowlby, M.R.; Kaftan, E.J.; Samad, T.A.; et al. Genetic and functional analysis of human P2X5 reveals a distinct pattern of exon 10 polymorphism with predominant expression of the nonfunctional receptor isoform. Mol. Pharmacol. 2010, 77, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Bo, X.N.; Jiang, L.H.; Wilson, H.L.; Kim, M.; Burnstock, G.; Surprenant, A.; North, R.A. Pharmacological and biophysical properties of the human P2X5 receptor. Mol. Pharmacol. 2003, 63, 1407–1416. [Google Scholar] [CrossRef]
- Le, K.T.; Boue-Grabot, E.; Archambault, V.; Seguela, P. Functional and biochemical evidence for heteromeric ATP-gated channels composed of P2X1 and P2X5 subunits. J. Biol. Chem. 1999, 274, 15415–15419. [Google Scholar] [CrossRef]
- Duckwitz, W.; Hausmann, R.; Aschrafi, A.; Schmalzing, G. P2X5 subunit assembly requires scaffolding by the second transmembrane domain and a conserved aspartate. J. Biol. Chem. 2006, 281, 39561–39572. [Google Scholar] [CrossRef]
- Overes, I.M.; Fredrix, H.; Kester, M.G.; Falkenburg, J.H.; van der Voort, R.; de Witte, T.M.; Dolstra, H. Efficient activation of LRH-1-specific CD8+ T-cell responses from transplanted leukemia patients by stimulation with P2X5 mRNA-electroporated dendritic cells. J. Immunother. 2009, 32, 539–551. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Garcia-Guzman, M.; Soto, F.; Laube, B.; Stühmer, W. Molecular cloning and functional expression of a novel rat heart P2X purinoceptor. FEBS Lett. 1996, 388, 123–127. [Google Scholar] [CrossRef]
- Collo, G.; North, R.A.; Kawashima, E.; Merlo-Pich, E.; Neidhart, S.; Surprenant, A.; Buell, G. Cloning of P2X5 and P2X6 receptors and the distribution and properties of an extended family of ATP-gated ion channels. J. Neurosci. 1996, 16, 2495–2507. [Google Scholar] [CrossRef] [PubMed]
- Aschrafi, A.; Sadtler, S.; Niculescu, C.; Rettinger, J.; Schmalzing, G. Trimeric architecture of homomeric P2X2 and heteromeric P2X1+2 receptor subtypes. J. Mol. Biol. 2004, 342, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Abramowski, P.; Ogrodowczyk, C.; Martin, R.; Pongs, O. A truncation variant of the cation channel P2RX5 is upregulated during T cell activation. PLoS ONE 2014, 9, e104692. [Google Scholar] [CrossRef]
- Harper, J.L.; Camerini-Otero, C.S.; Li, A.H.; Kim, S.A.; Jacobson, K.A.; Daly, J.W. Dihydropyridines as inhibitors of capacitative calcium entry in leukemic HL-60 cells. Biochem. Pharmacol. 2003, 65, 329–338. [Google Scholar] [CrossRef]
- Roh, E.J.; Keller, J.M.; Olah, Z.; Iadarola, M.J.; Jacobson, K.A. Structure-activity relationships of 1,4-dihydropyridines that act as enhancers of the vanilloid receptor 1 (TRPV1). Bioorg. Med. Chem. 2008, 16, 9349–9358. [Google Scholar] [CrossRef]
- Edraki, N.; Mehdipour, A.R.; Khoshneviszadeh, M.; Miri, R. Dihydropyridines: Evaluation of their current and future pharmacological applications. Drug Discov. Today 2009, 14, 1058–1066. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Kim, Y.C.; King, B.F. In search of selective P2 receptor ligands: Interaction of dihydropyridine derivatives at recombinant rat P2X2 receptors. J. Auton. Nerv. Syst. 2000, 81, 152–157. [Google Scholar] [CrossRef]
- Jiang, J.; Li, A.H.; Jang, S.Y.; Chang, L.; Melman, N.; Moro, S.; Ji, X.; Lobkovsky, E.B.; Clardy, J.C.; Jacobson, K.A. Chiral resolution and stereospecificity of 6-phenyl-4-phenylethynyl-1,4-dihydropyridines as selective A3 adenosine receptor antagonists. J. Med. Chem. 1999, 42, 3055–3065. [Google Scholar] [CrossRef]
- Striessnig, J.; Zernig, G.; Glossmann, H. Human red-blood-cell Ca2+-antagonist binding sites. Evidence for an unusual receptor coupled to the nucleoside transporter. Eur. J. Biochem. 1985, 150, 67–77. [Google Scholar] [CrossRef]
- Flittiger, B.; Klapperstück, M.; Schmalzing, G.; Markwardt, F. Effects of protons on macroscopic and single-channel currents mediated by the human P2X7 receptor. Biochim. Biophys. Acta Biomembr. 2010, 1798, 947–957. [Google Scholar] [CrossRef]
- Kubick, C.; Schmalzing, G.; Markwardt, F. The effect of anions on the human P2X7 receptor. Biochim. Biophys. Acta Biomembr. 2011, 1808, 2913–2922. [Google Scholar] [CrossRef][Green Version]
- Stolz, M.; Klapperstück, M.; Kendzierski, T.; Detro-Dassen, S.; Panning, A.; Schmalzing, G.; Markwardt, F. Homodimeric anoctamin-1, but not homodimeric anoctamin-6, is activated by calcium increases mediated by the P2Y1 and P2X7 receptors. Pflügers Arch. 2015, 467, 2121–2140. [Google Scholar] [CrossRef]
- Bretschneider, F.; Klapperstück, M.; Löhn, M.; Markwardt, F. Nonselective cationic currents elicited by extracellular ATP in human B-lymphocytes. Pflügers Arch. 1995, 429, 691–698. [Google Scholar] [CrossRef]
- Riedel, T.; Schmalzing, G.; Markwardt, F. Influence of extracellular monovalent cations on pore and gating properties of P2X7 receptor-operated single channels currents. Biophys. J. 2007, 93, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Silberberg, S.D.; Swartz, K.J. Subtype-specific control of P2X receptor channel signaling by ATP and Mg2+. Proc. Natl. Acad. Sci. USA 2013, 110, E3455–E3463. [Google Scholar] [CrossRef] [PubMed]
- Klapperstück, M.; Büttner, C.; Schmalzing, G.; Markwardt, F. Functional evidence of distinct ATP activation sites at the human P2X7 receptor. J. Physiol. 2001, 534, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Ruppelt, A.; Ma, W.Y.; Borchardt, K.; Silberberg, S.D.; Soto, F. Genomic structure, developmental distribution and functional properties of the chicken P2X5 receptor. J. Neurochem. 2001, 77, 1256–1265. [Google Scholar] [CrossRef]
- Schneider, M.; Prudic, K.; Pippel, A.; Klapperstück, M.; Braam, U.; Müller, C.E.; Schmalzing, G.; Markwardt, F. Interaction of purinergic P2X4 and P2X7 receptor subunits. Front. Pharmacol. 2017, 8, 860. [Google Scholar] [CrossRef]
- Lewis, C.J.; Ennion, S.J.; Evans, R.J. P2 purinoceptor-mediated control of rat cerebral (pial) microvasculature; contribution of P2X and P2Y receptors. J. Physiol. 2000, 527, 315–324. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Kim, Y.C.; Wildman, S.S.; Mohanram, A.; Harden, T.K.; Boyer, J.L.; King, B.F.; Burnstock, G. A pyridoxine cyclic phosphate and its 6-azoaryl derivative selectively potentiate and antagonize activation of P2X1 receptors. J. Med. Chem. 1998, 41, 2201–2206. [Google Scholar] [CrossRef] [PubMed]
- Bidula, S.; Nadzirin, I.B.; Cominetti, M.; Hickey, H.; Cullum, S.A.; Searcey, M.; Schmid, R.; Fountain, S.J. Structural basis of the negative allosteric modulation of 5-BDBD at human P2X4 receptors. Mol. Pharmacol. 2022, 101, 33–44. [Google Scholar] [CrossRef]
- Ryten, M.; Dunn, P.M.; Neary, J.T.; Burnstock, G. ATP regulates the differentiation of mammalian skeletal muscle by activation of a P2X5 receptor on satellite cells. J. Cell Biol. 2002, 158, 345–355. [Google Scholar] [CrossRef]
- Birdsong, W.T.; Fierro, L.; Williams, F.G.; Spelta, V.; Naves, L.A.; Knowles, M.; Marsh-Haffner, J.; Adelman, J.P.; Almers, W.; Elde, R.P.; et al. Sensing muscle ischemia: Coincident detection of acid and ATP via interplay of two ion channels. Neuron 2010, 68, 739–749. [Google Scholar] [CrossRef]
- Burnstock, G.; Kennedy, C. P2X receptors in health and disease. Adv. Pharmacol. 2011, 61, 333–372. [Google Scholar] [CrossRef]
- Kim, H.; Walsh, M.C.; Takegahara, N.; Middleton, S.A.; Shin, H.I.; Kim, J.; Choi, Y. The purinergic receptor P2X5 regulates inflammasome activity and hyper-multinucleation of murine osteoclasts. Sci. Rep. 2017, 7, 196. [Google Scholar] [CrossRef]
- Kim, H.; Kajikawa, T.; Walsh, M.C.; Takegahara, N.; Jeong, Y.H.; Hajishengallis, G.; Choi, Y. The purinergic receptor P2X5 contributes to bone loss in experimental periodontitis. BMB Rep. 2018, 51, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xie, A.H.; Liu, Z.; Li, D.; Ning, B.; Thakkar, S.; Tong, W.; Xu, J. Linking pharmacogenomic information on drug safety and efficacy with ethnic minority populations. Pharmaceutics 2020, 12, 1021. [Google Scholar] [CrossRef] [PubMed]
- Compan, V.; Ulmann, L.; Stelmashenko, O.; Chemin, J.; Chaumont, S.; Rassendren, F. P2X2 and P2X5 subunits define a new heteromeric receptor with P2X7-like properties. J. Neurosci. 2012, 32, 4284–4296. [Google Scholar] [CrossRef]
- Gloor, S.; Pongs, O.; Schmalzing, G. A vector for the synthesis of cRNAs encoding Myc epitope-tagged proteins in Xenopus laevis oocytes. Gene 1995, 160, 213–217. [Google Scholar] [CrossRef]
- Weiner, M.P.; Costa, G.L.; Schoettlin, W.; Cline, J.; Mathur, E.; Bauer, J.C. Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene 1994, 151, 119–123. [Google Scholar] [CrossRef]
- Klapperstück, M.; Büttner, C.; Böhm, T.; Schmalzing, G.; Markwardt, F. Characteristics of P2X7 receptors from human B lymphocytes expressed in Xenopus oocytes. Biochim. Biophys. Acta 2000, 1467, 444–456. [Google Scholar] [CrossRef]
- Pippel, A.; Stolz, M.; Woltersdorf, R.; Kless, A.; Schmalzing, G.; Markwardt, F. Localization of the gate and selectivity filter of the full-length P2X7 receptor. Proc. Natl. Acad. Sci. USA 2017, 114, E2156–E2165. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiller, I.C.; Jacobson, K.A.; Wen, Z.; Malisetty, A.; Schmalzing, G.; Markwardt, F. Dihydropyridines Potentiate ATP-Induced Currents Mediated by the Full-Length Human P2X5 Receptor. Molecules 2022, 27, 1846. https://doi.org/10.3390/molecules27061846
Schiller IC, Jacobson KA, Wen Z, Malisetty A, Schmalzing G, Markwardt F. Dihydropyridines Potentiate ATP-Induced Currents Mediated by the Full-Length Human P2X5 Receptor. Molecules. 2022; 27(6):1846. https://doi.org/10.3390/molecules27061846
Chicago/Turabian StyleSchiller, Ida C., Kenneth A. Jacobson, Zhiwei Wen, Aparna Malisetty, Günther Schmalzing, and Fritz Markwardt. 2022. "Dihydropyridines Potentiate ATP-Induced Currents Mediated by the Full-Length Human P2X5 Receptor" Molecules 27, no. 6: 1846. https://doi.org/10.3390/molecules27061846
APA StyleSchiller, I. C., Jacobson, K. A., Wen, Z., Malisetty, A., Schmalzing, G., & Markwardt, F. (2022). Dihydropyridines Potentiate ATP-Induced Currents Mediated by the Full-Length Human P2X5 Receptor. Molecules, 27(6), 1846. https://doi.org/10.3390/molecules27061846