
Accurate Physical Property Predictions via Deep Learning


Abstract
:1. Introduction
2. Results
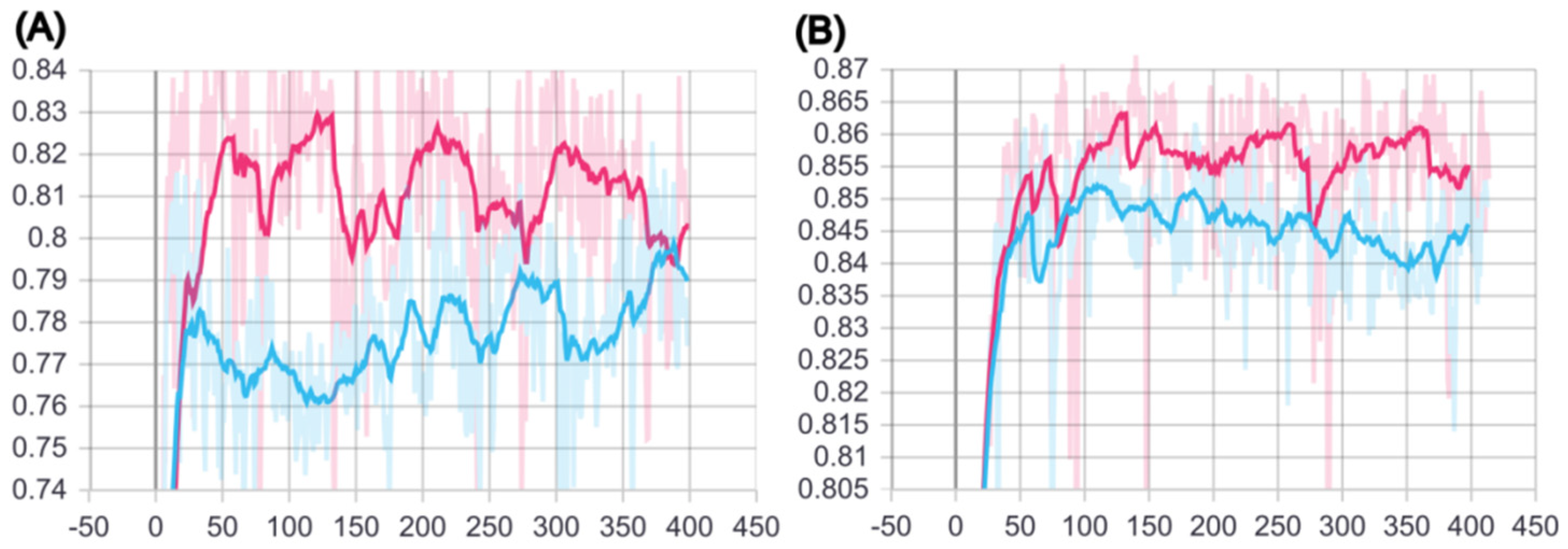
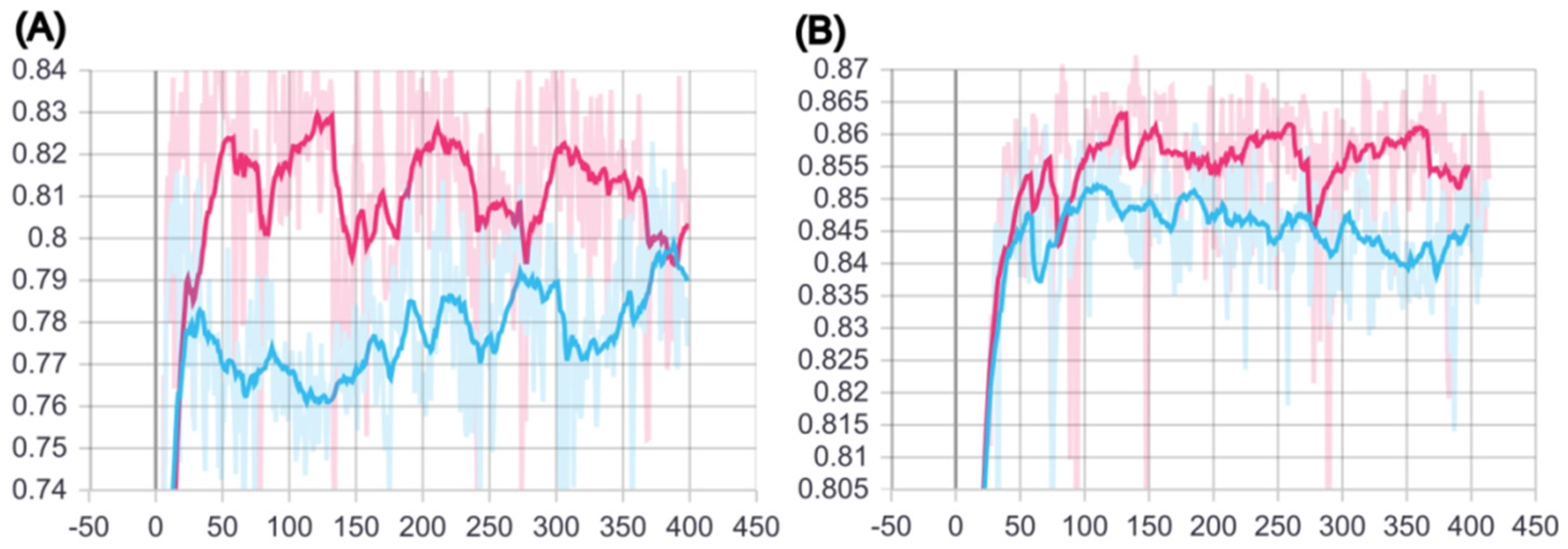
2.1. Training of BCSA Model
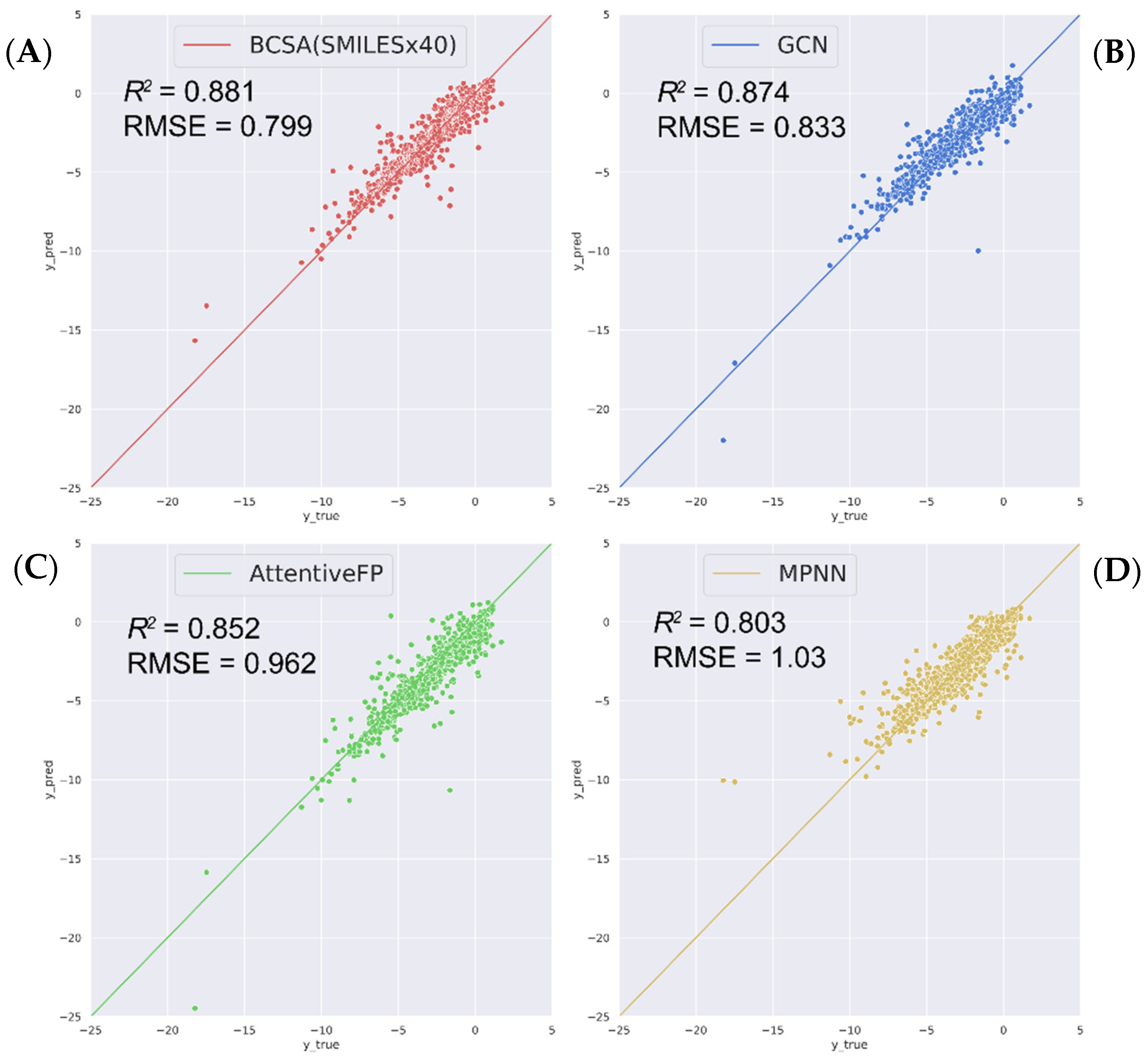
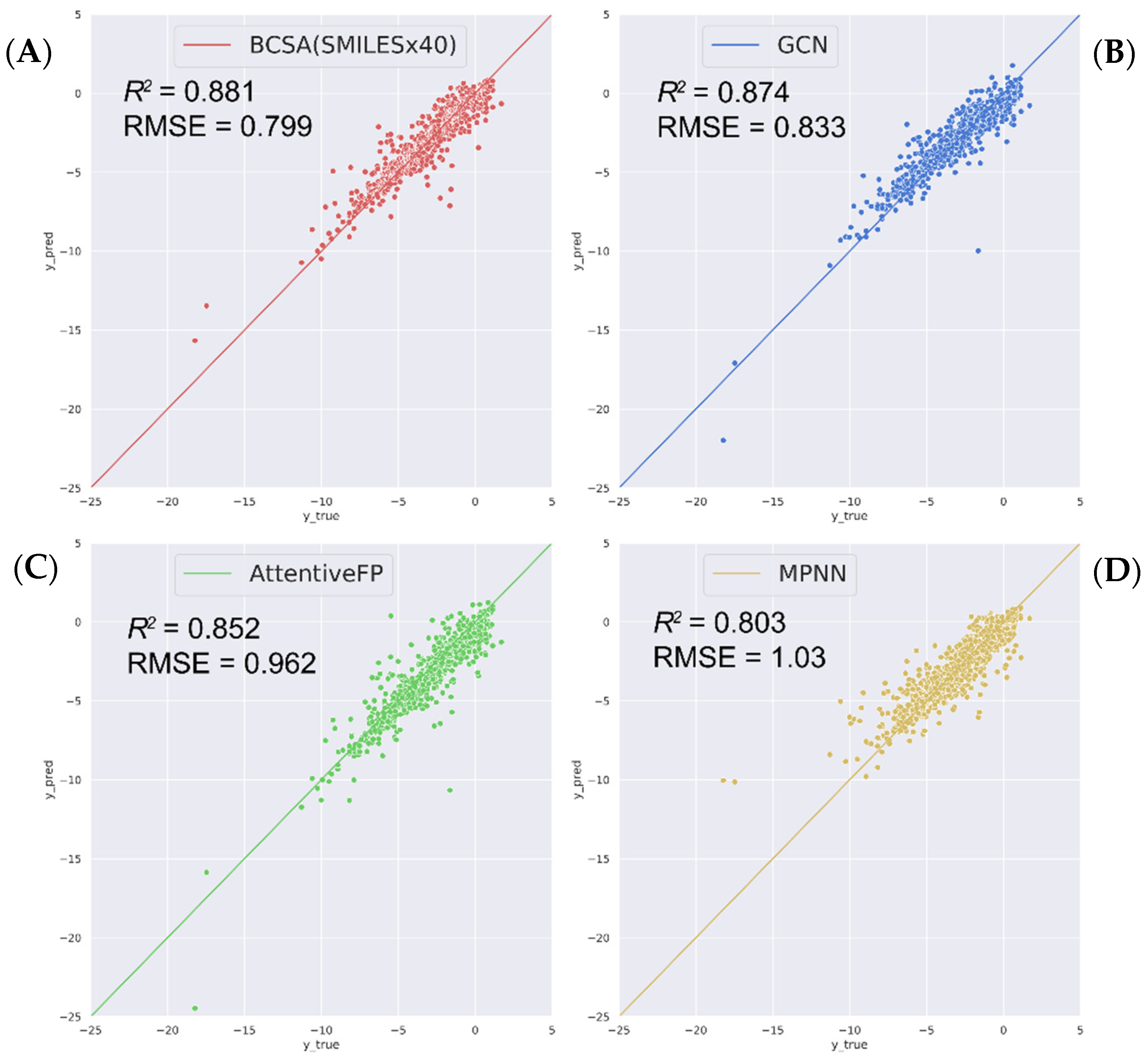
2.2. Compare with State-of-the-Art Models
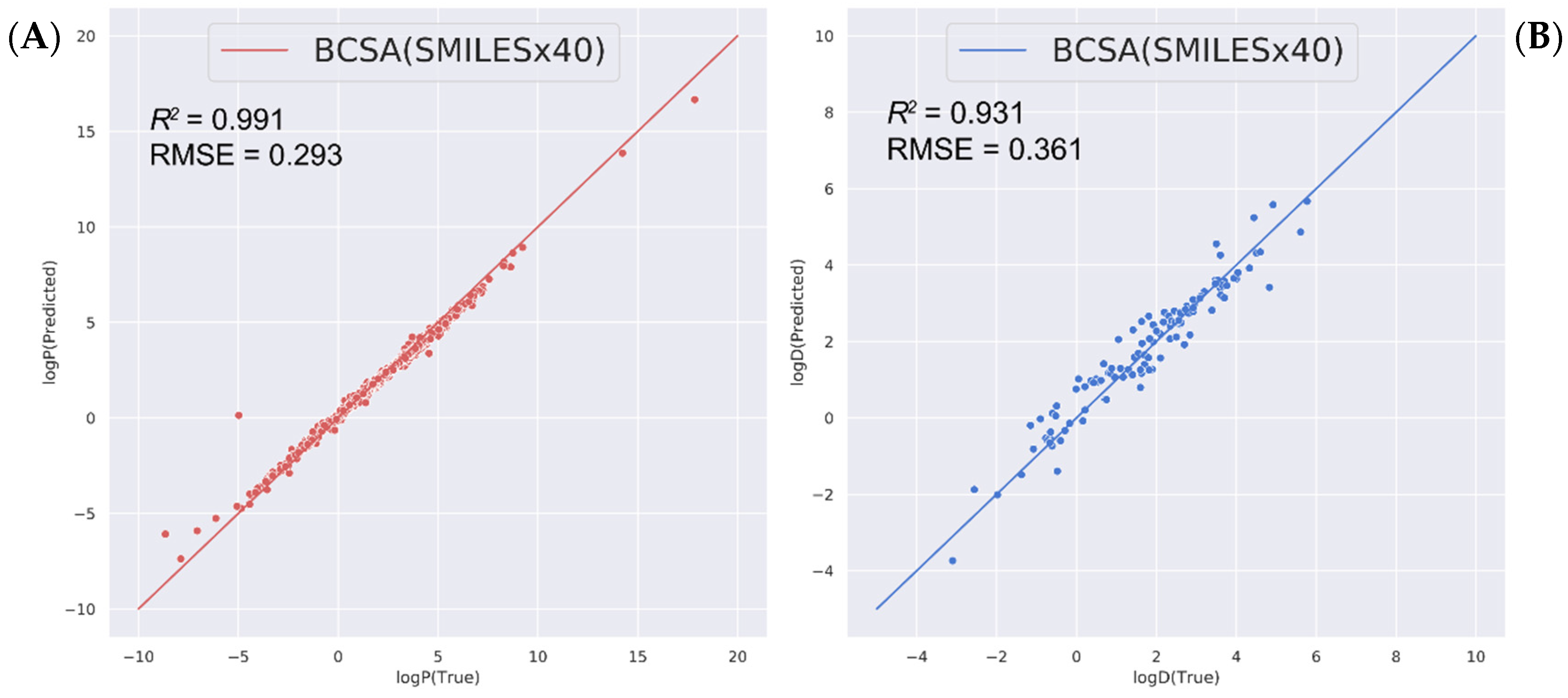
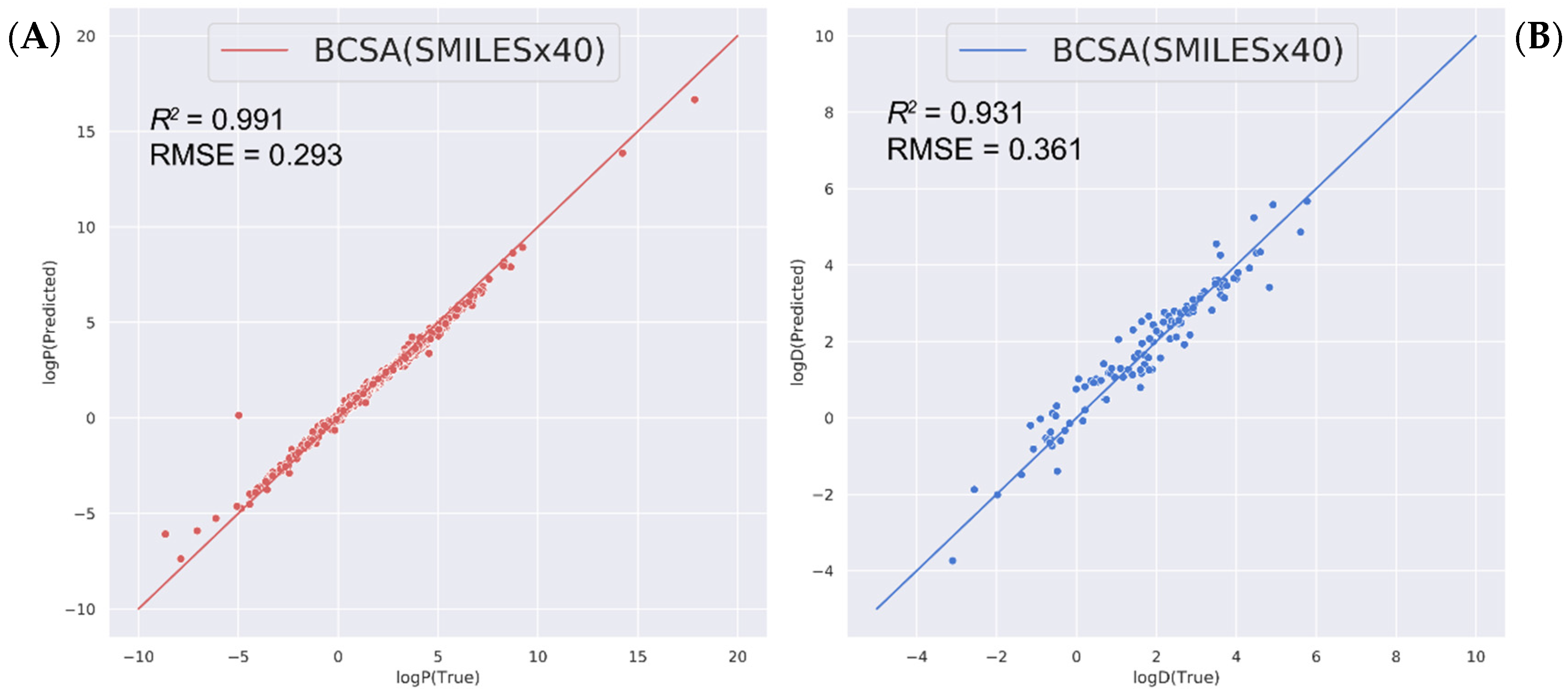
2.3. Predicting Other Related Physicochemical Properties
3. Discussion
4. Materials and Methods
4.1. Molecular Dataset and Processing
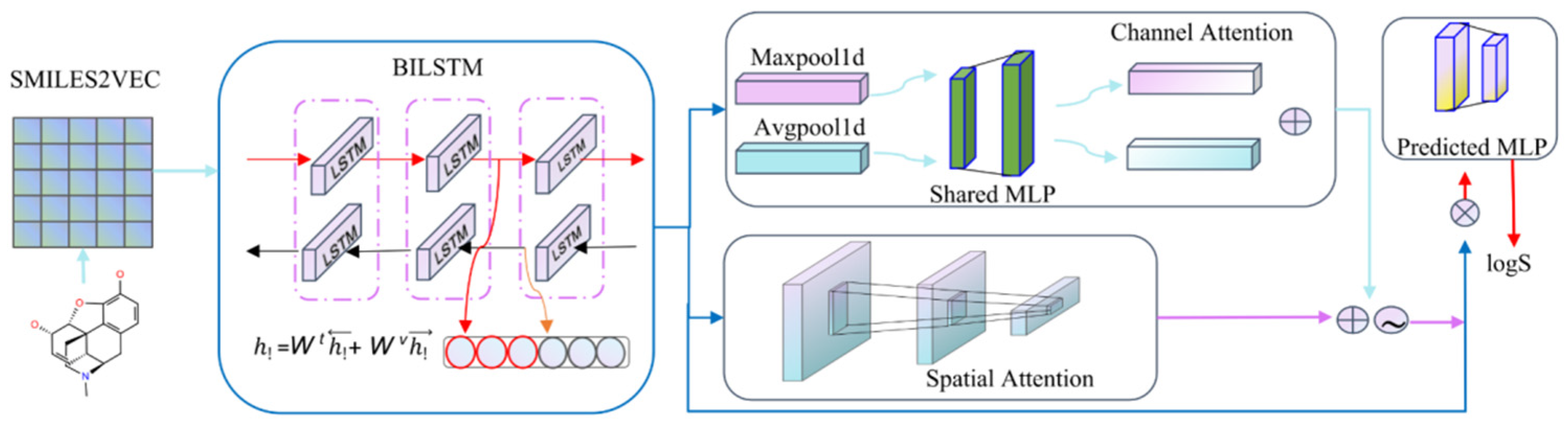
4.2. Model Building
4.3. Hyperparameter Search
4.4. Evaluation Metrics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Merkwirth, C.; Lengauer, T. Automatic generation of complementary descriptors with molecular graph networks. J. Chem. Inf. Modeling 2005, 45, 1159–1168. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [PubMed] [Green Version]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient estimation of word representations in vector space. arXiv 2013, arXiv:1301.3781. [Google Scholar]
- Pennington, J.; Socher, R.; Manning, C.D. Glove: Global vectors for word representation. In Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, 25–29 October 2014; pp. 1532–1543. [Google Scholar]
- Ling, W.; Luís, T.; Marujo, L.; Astudillo, R.F.; Amir, S.; Dyer, C.; Black, A.W.; Trancoso, I. Finding function in form: Compositional character models for open vocabulary word representation. arXiv 2015, arXiv:1508.02096. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. Adv. Neural Inf. Processing Syst. 2017, 30, 6000–6010. [Google Scholar]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Gers, F.A.; Schmidhuber, J.; Cummins, F. Learning to forget: Continual prediction with LSTM. Neural Comput. 2000, 12, 2451–2471. [Google Scholar] [CrossRef]
- Sutskever, I.; Vinyals, O.; Le, Q.V. Sequence to sequence learning with neural networks. Adv. Neural Inf. Processing Syst. 2014, 27. [Google Scholar]
- Girshick, R.; Donahue, J.; Darrell, T.; Malik, J. Rich feature hierarchies for accurate object detection and semantic segmentation. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Columbus, OH, USA, 23–28 June 2014; pp. 580–587. [Google Scholar]
- Ren, S.; He, K.; Girshick, R.; Sun, J. Faster r-cnn: Towards real-time object detection with region proposal networks. Adv. Neural Inf. Processing Syst. 2015, 28, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rathod, V.; Sun, C.; Zhu, M.; Korattikara, A.; Fathi, A.; Fischer, I.; Wojna, Z.; Song, Y.; Guadarrama, S. Speed/accuracy trade-offs for modern convolutional object detectors. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017; pp. 7310–7311. [Google Scholar]
- Tan, M.; Le, Q. Efficientnet: Rethinking model scaling for convolutional neural networks. In Proceedings of the 36th International Conference on Machine Learning, Long Beach, CA, USA, 9–15 June 2019; pp. 6105–6114. [Google Scholar]
- Goh, G.B.; Hodas, N.O.; Siegel, C.; Vishnu, A. Smiles2vec: An interpretable general-purpose deep neural network for predicting chemical properties. arXiv 2017, arXiv:1712.02034. [Google Scholar]
- Cui, Q.; Lu, S.; Ni, B.; Zeng, X.; Tan, Y.; Chen, Y.D.; Zhao, H. Improved prediction of aqueous solubility of novel compounds by going deeper with deep learning. Front. Oncol. 2020, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Zheng, S.; Song, Y.; Chen, J.; Li, C.; Xie, J.; Yang, H.; Chen, H.; Yang, Y. MolRep: A deep representation learning library for molecular property prediction. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wieder, O.; Kohlbacher, S.; Kuenemann, M.; Garon, A.; Ducrot, P.; Seidel, T.; Langer, T. A compact review of molecular property prediction with graph neural networks. Drug Discov. Today Technol. 2020, 37, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, E.N.; Sur, D.; Wu, Z.; Husic, B.E.; Mai, H.; Li, Y.; Sun, S.; Yang, J.; Ramsundar, B.; Pande, V.S. PotentialNet for molecular property prediction. ACS Cent. Sci. 2018, 4, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural message passing for quantum chemistry. In Proceedings of the International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; pp. 1263–1272. [Google Scholar]
- Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 2019, 63, 8749–8760. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, G.; Zhang, Z.; Yang, C.; Liu, Z.; Wang, L.; Li, C.; Sun, M. Graph neural networks: A review of methods and applications. arXiv 2018, arXiv:1812.08434. [Google Scholar] [CrossRef]
- Gomes, J.; Ramsundar, B.; Feinberg, E.N.; Pande, V.S. Atomic convolutional networks for predicting protein-ligand binding affinity. arXiv 2017, arXiv:1703.10603. [Google Scholar]
- Coley, C.W.; Jin, W.; Rogers, L.; Jamison, T.F.; Jaakkola, T.S.; Green, W.H.; Barzilay, R.; Jensen, K.F. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 2019, 10, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Schütt, K.T.; Kindermans, P.-J.; Sauceda, H.E.; Chmiela, S.; Tkatchenko, A.; Müller, K.-R. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions. arXiv 2017, arXiv:1706.08566. [Google Scholar]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Philip, S.Y. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2020, 32, 4–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.; Yoon, S. Deepcci: End-to-end deep learning for chemical-chemical interaction prediction. In Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Boston, MA, USA, 20–23 August 2017; pp. 203–212. [Google Scholar]
- Feng, Q.; Dueva, E.; Cherkasov, A.; Ester, M. Padme: A deep learning-based framework for drug-target interaction prediction. arXiv 2018, arXiv:1807.09741. [Google Scholar]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular transformer: A model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Jo, J.; Kwak, B.; Choi, H.-S.; Yoon, S. The message passing neural networks for chemical property prediction on SMILES. Methods 2020, 179, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.; Schmidhuber, J. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw. 2005, 18, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Bjerrum, E.J. SMILES enumeration as data augmentation for neural network modeling of molecules. arXiv 2017, arXiv:1703.07076. [Google Scholar]
- Nirmalakhandan, N.N.; Speece, R.E. Prediction of aqueous solubility of organic chemicals based on molecular structure. Environ. Sci. Technol. 1988, 22, 328–338. [Google Scholar] [CrossRef]
- Bodor, N.; Harget, A.; Huang, M.J. Neural network studies. 1. Estimation of the aqueous solubility of organic compounds. J. Am. Chem. Soc. 1991, 113, 9480–9483. [Google Scholar] [CrossRef]
- Huuskonen, J. Estimation of aqueous solubility for a diverse set of organic compounds based on molecular topology. J. Chem. Inf. Comput. Sci. 2000, 40, 773–777. [Google Scholar] [CrossRef]
- Llinas, A.; Glen, R.C.; Goodman, J.M. Solubility challenge: Can you predict solubilities of 32 molecules using a database of 100 reliable measurements? J. Chem. Inf. Modeling 2008, 48, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Nunes, C.; Vyas, S.; Jonnalagadda, S. Prediction of solubility parameters and miscibility of pharmaceutical compounds by molecular dynamics simulations. J. Phys. Chem. B 2011, 115, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Lusci, A.; Pollastri, G.; Baldi, P. Deep architectures and deep learning in chemoinformatics: The prediction of aqueous solubility for drug-like molecules. J. Chem. Inf. Modeling 2013, 53, 1563–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Totton, T.; Frenkel, D. Computational methodology for solubility prediction: Application to the sparingly soluble solutes. J. Chem. Phys. 2017, 146, 214110. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Kramer, S.T.; Fang, M.; Qiu, Y.; Wu, Z.; Xu, D. A self-attention based message passing neural network for predicting molecular lipophilicity and aqueous solubility. J. Cheminform. 2020, 12, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panapitiya, G.; Girard, M.; Hollas, A.; Murugesan, V.; Wang, W.; Saldanha, E. Predicting aqueous solubility of organic molecules using deep learning models with varied molecular representations. arXiv 2021, arXiv:2105.12638. [Google Scholar]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Li, M.; Zhou, J.; Hu, J.; Fan, W.; Zhang, Y.; Gu, Y.; Karypis, G. DGL-LifeSci: An open-source toolkit for deep learning on graphs in life science. arXiv 2021, arXiv:2106.14232. [Google Scholar] [CrossRef]
- Wang, J.B.; Cao, D.S.; Zhu, M.F.; Yun, Y.H.; Xiao, N.; Liang, Y.Z. In silico evaluation of logD7. 4 and comparison with other prediction methods. J. Chemom. 2015, 29, 389–398. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, H.; Su, Z.; Xu, Y. Chinese comments sentiment classification based on word2vec and SVMperf. Expert Syst. Appl. 2015, 42, 1857–1863. [Google Scholar] [CrossRef]
- Goldberg, Y.; Levy, O. word2vec Explained: Deriving mikolov et al.’s negative-sampling word-embedding method. arXiv 2014, arXiv:1402.3722. [Google Scholar]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Woo, S.; Park, J.; Lee, J.-Y.; Kweon, I.S. Cbam: Convolutional block attention module. In Proceedings of the European Conference on Computer Vision (ECCV), Munich, Germany, 8–14 September 2018; pp. 3–19. [Google Scholar]
- Nair, V.; Hinton, G.E. Rectified linear units improve restricted boltzmann machines. In Proceedings of the ICML, Haifa, Israel, 21–24 June 2010. [Google Scholar]
- Snoek, J.; Larochelle, H.; Adams, R.P. Practical bayesian optimization of machine learning algorithms. Adv. Neural Inf. Processing Syst. 2012, 25, 2951–2959. [Google Scholar]
- Bergstra, J.; Bardenet, R.; Bengio, Y.; Kégl, B. Algorithms for hyper-parameter optimization. Adv. Neural Inf. Processing Syst. 2011, 24, 2546–2554. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Possible Values | The Best Found |
|---|---|---|
| batch_size | (512,1024) | 1024 |
| vocab_size | (120,150) | 120 |
| Smiles_max_len | (150,200) | 150 |
| hidden_size | (16,32,64) | 64 |
| number_layers | 3–5 | 3 |
| dropout | 0–0.6 | 0.12215 |
| mlp_hidden_size | (32,64) | 32 |
| learning_rate | 0.01–0.001 | 0.00966 |
| Dataset | (Higher is Better) | (Lower is Better) | |||
|---|---|---|---|---|---|
| R2 | Spearman | RMSE | MAE | ||
| Source data | validation | 0.8714 | 0.9294 | 0.8085 | 0.5671 |
| Test | 0.8365 | 0.9185 | 0.9513 | 0.6435 | |
| SMILES × 20 | validation | 0.8790 | 0.9352 | 0.8233 | 0.5512 |
| Test | 0.8779 | 0.9339 | 0.8181 | 0.5493 | |
| SMILES × 40 | validation | 0.8828 | 0.9375 | 0.8025 | 0.5207 |
| Test | 0.8813 | 0.9361 | 0.7997 | 0.5226 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, Y.; Wang, S.; Bai, B.; Chan, H.C.S.; Yuan, S. Accurate Physical Property Predictions via Deep Learning. Molecules 2022, 27, 1668. https://doi.org/10.3390/molecules27051668
Hou Y, Wang S, Bai B, Chan HCS, Yuan S. Accurate Physical Property Predictions via Deep Learning. Molecules. 2022; 27(5):1668. https://doi.org/10.3390/molecules27051668
Chicago/Turabian StyleHou, Yuanyuan, Shiyu Wang, Bing Bai, H. C. Stephen Chan, and Shuguang Yuan. 2022. "Accurate Physical Property Predictions via Deep Learning" Molecules 27, no. 5: 1668. https://doi.org/10.3390/molecules27051668
APA StyleHou, Y., Wang, S., Bai, B., Chan, H. C. S., & Yuan, S. (2022). Accurate Physical Property Predictions via Deep Learning. Molecules, 27(5), 1668. https://doi.org/10.3390/molecules27051668