
Development of Improved High-Performance Liquid Chromatography Method for the Determination of Residual Caprylic Acid in Formulations of Human Immunoglobulins


Abstract
:1. Introduction
2. Results and Discussion
2.1. Development of HPLC Method and Sample Preparation
2.2. Validation
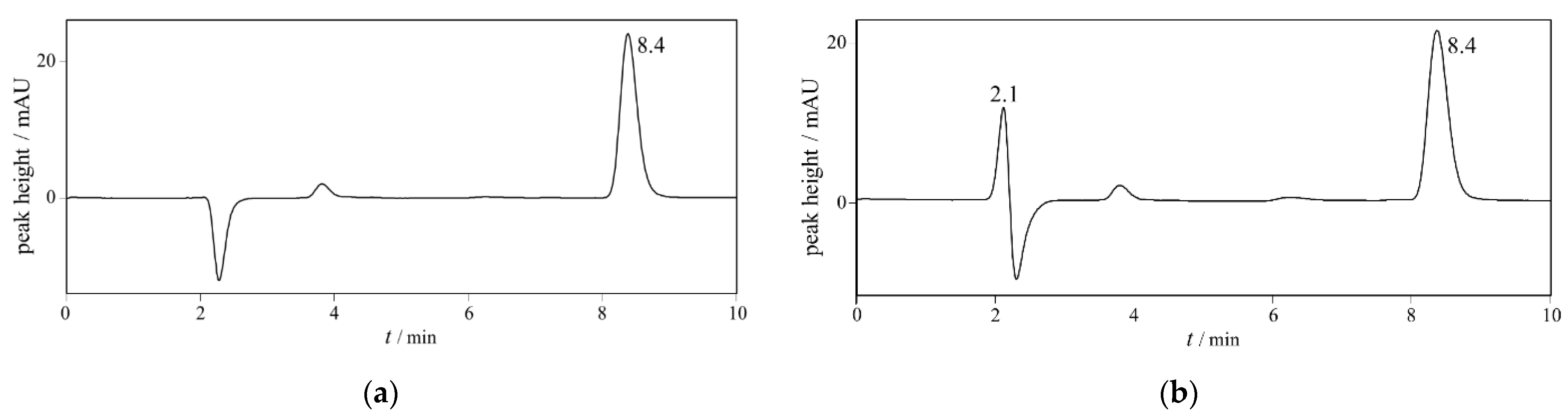
2.2.1. Specificity
2.2.2. Linearity
2.2.3. Precision and Accuracy
2.2.4. Limit of Detection (LOD) and Limit of Quantification (LOQ)
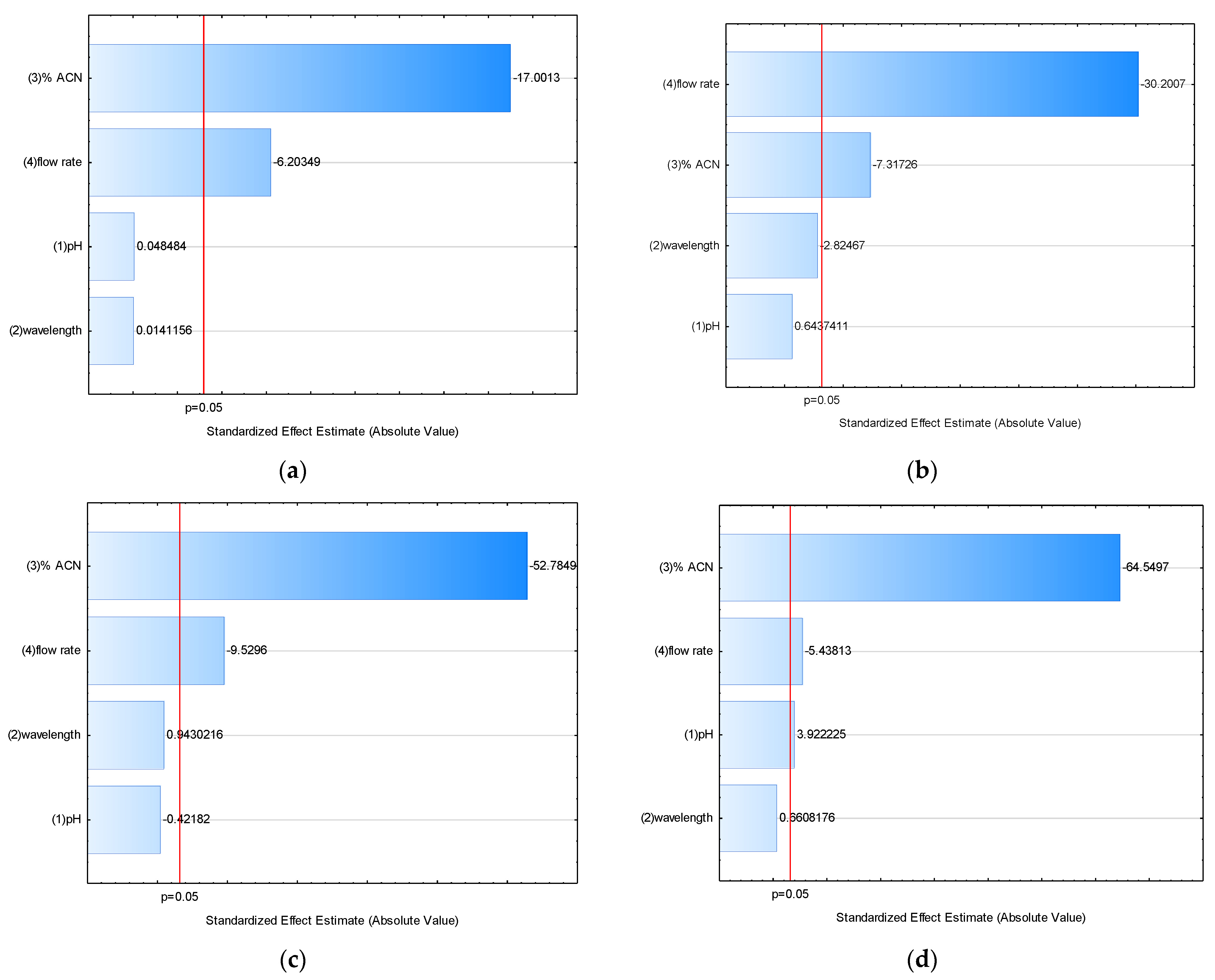
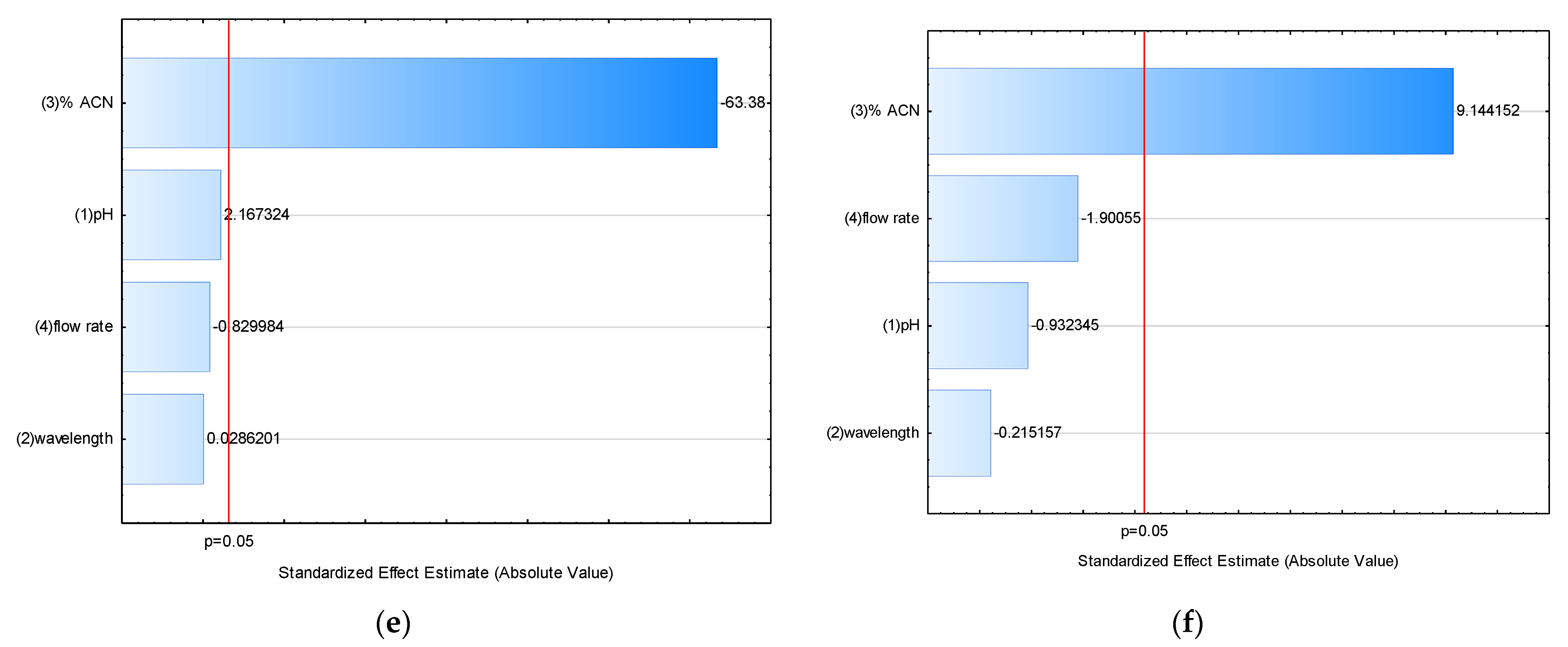
2.2.5. Robustness
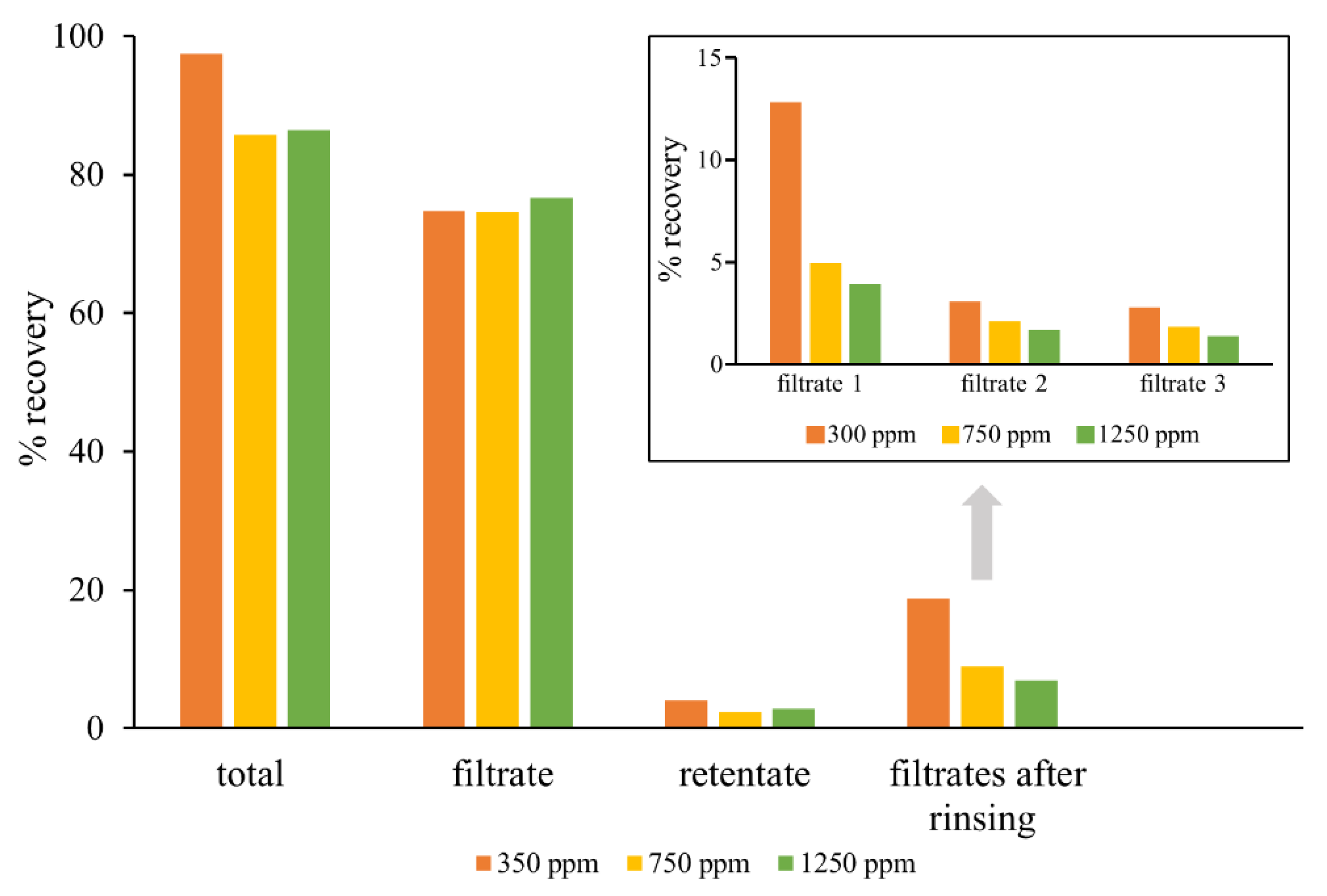
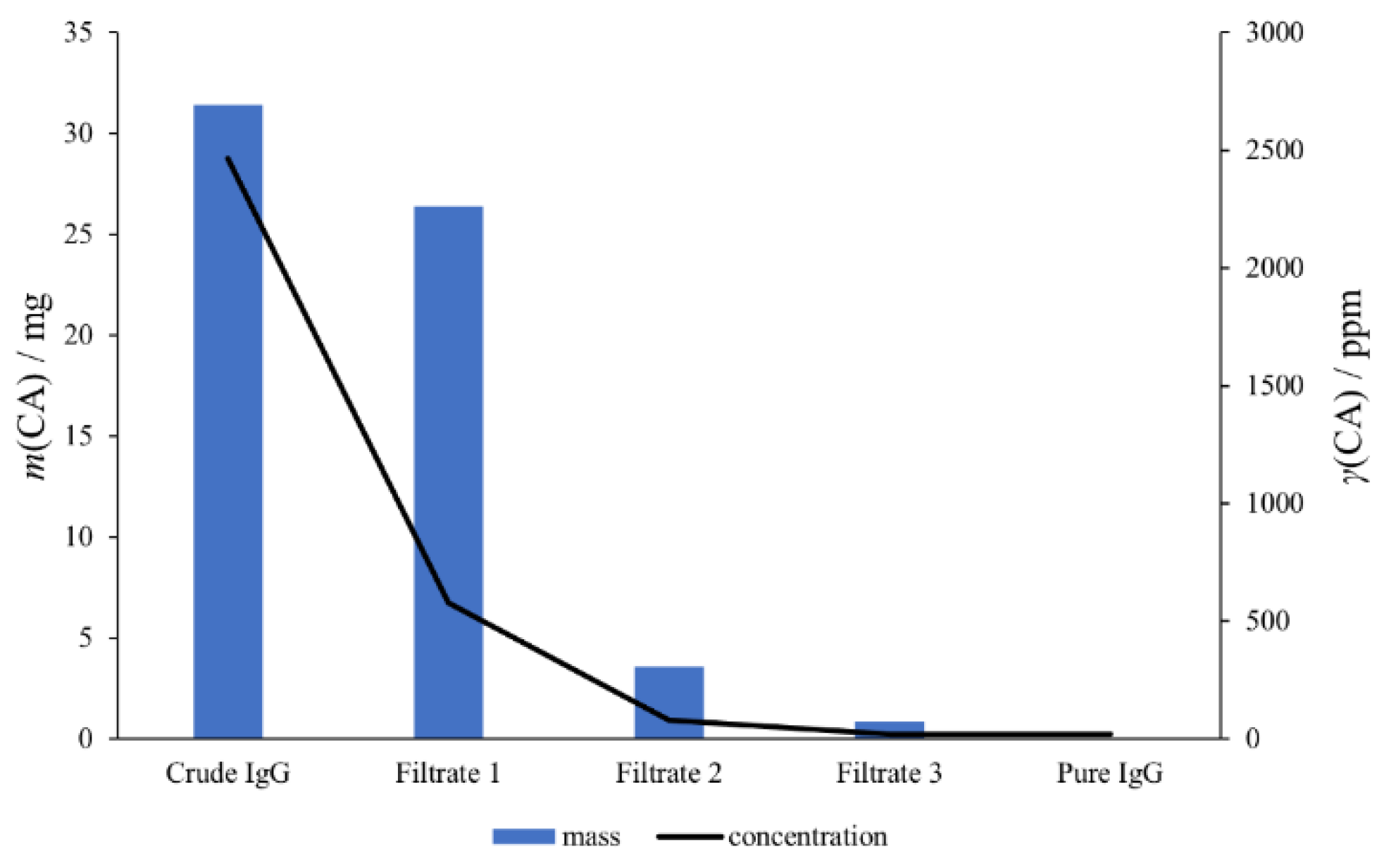
2.3. Application of Developed Method
3. Materials and Methods
3.1. Reagents and Chemicals
3.2. Standard Solutions and Quality Control Sample Preparation
3.3. HPLC Instrumentation and Conditions
3.4. Validation Parameters of Developed Method
3.4.1. Specificity
3.4.2. Linearity
3.4.3. Precision and Accuracy
3.4.4. Limit of Detection (LOD) and Limit of Quantification (LOQ)
3.4.5. Robustness
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Casadevall, A.; Dadachova, E.; Pirofski, L.A. Passive Antibody Therapy for Infectious Diseases. Nat. Rev. Microbiol. 2004, 2, 695–703. [Google Scholar] [CrossRef]
- Abraham, J. Passive Antibody Therapy in COVID-19. Nat. Rev. Immunol. 2020, 20, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Pirofski, L.A. The Convalescent Sera Option for Containing COVID-19. J. Clin. Investig. 2020, 130, 1545–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Uddin, S.M.; Ali, A.; Anjum, F.; Ali, R.; Shalim, E.; Khan, M.; Ahmed, I.; Muhaymin, S.M.; Bukhari, U.; et al. Production of Hyperimmune Anti-SARS-CoV-2 Intravenous Immunoglobulin from Pooled COVID-19 Convalescent Plasma. Immunotherapy 2021, 13, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Buchacher, A.; Iberer, G. Purification of Intravenous Immunoglobulin G from Human Plasma-Aspects of Yield and Virus Safety. Biotechnol. J. 2006, 1, 148–163. [Google Scholar] [CrossRef]
- Russo, C.; Callegaro, L.; Lanza, E.; Ferrone, S. Purification of IgG Monoclonal Antibody by Caprylic Acid Precipitation. J. Immunol. Methods 1983, 65, 269–271. [Google Scholar] [CrossRef]
- Temponi, M.; Kageshita, T.; Perosa, F.; Ono, K.; Okada, H.; Ferrone, S. Purification of Murine IgG Monoclonal Antibodies by Precipitation with Caprylic Acid: Comparison with Other Methods of Purification. Hybridoma 1989, 8, 85–95. [Google Scholar] [CrossRef]
- Eliasson, M.; Olsson, A.; Palmcrantz, E.; Wiberg, K.; Inganas, M.; Guss, B.; Lindberg, M.; Uhlen, M. Chimeric IgG-Binding Receptors Engineered from Staphylococcal Protein A and Streptococcal Protein G. J. Biol. Chem. 1988, 263, 4323–4327. [Google Scholar] [CrossRef]
- Roque, A.C.A.; Silva, C.S.O.; Taipa, M.Â. Affinity-Based Methodologies and Ligands for Antibody Purification: Advances and Perspectives. J. Chromatogr. A 2007, 1160, 44–55. [Google Scholar] [CrossRef]
- Bergmann-Leitner, E.S.; Mease, R.M.; Duncan, E.H.; Khan, F.; Waitumbi, J.; Angov, E. Evaluation of Immunoglobulin Purification Methods and Their Impact on Quality and Yield of Antigen-Specific Antibodies. Malar. J. 2008, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Herrera, M.; Meneses, F.; Gutiérrez, J.M.; León, G. Development and Validation of a Reverse Phase HPLC Method for the Determination of Caprylic Acid in Formulations of Therapeutic Immunoglobulins and Its Application to Antivenom Production. Biologicals 2009, 37, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Kurtović, T.; Balija, M.L.; Brgles, M.; Sviben, D.; Tunjić, M.; Cajner, H.; Marchetti-Deschmann, M.; Allmaier, G.; Halassy, B. Refinement Strategy for Antivenom Preparation of High Yield and Quality. PLoS Negl. Trop. Dis. 2019, 13, e0007431. [Google Scholar] [CrossRef] [PubMed]
- Kurtović, T.; Brgles, M.; Balija, M.L.; Steinberger, S.; Sviben, D.; Marchetti-Deschmann, M.; Halassy, B. Streamlined Downstream Process for Efficient and Sustainable (Fab’)2 Antivenom Preparation. J. Venom. Anim. Toxins Incl. Trop. Dis. 2020, 26, 1–11. [Google Scholar] [CrossRef]
- Lebing, W.; Remington, K.M.; Schreiner, C.; Paul, H.I. Properties of a New Intravenous Immunoglobulin (IGIV-C, 10%) Produced by Virus Inactivation with Caprylate and Column Chromatography. Vox Sang. 2003, 84, 193–201. [Google Scholar] [CrossRef] [PubMed]
- El-Ekiaby, M.; Vargas, M.; Sayed, M.; Gorgy, G.; Goubran, H.; Radosevic, M.; Burnouf, T. Minipool Caprylic Acid Fractionation of Plasma Using Disposable Equipment: A Practical Method to Enhance Immunoglobulin Supply in Developing Countries. PLoS Negl. Trop. Dis. 2015, 9, e0003501. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Champagne, J.; Toueille, M.; Gantier, R.; Burnouf, T. Dedicated Removal of Immunoglobulin (Ig)A, IgM, and Factor (F)XI/Activated FXI from Human Plasma IgG. Transfusion 2014, 54, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Alonso, W.; Vandeberg, P.; Lang, J.; Yuziuk, J.; Silverstein, R.; Stokes, K.; McBride, D.; Cruz, M.; Burns, D.; Merritt, W.K.; et al. Immune Globulin Subcutaneous, Human 20% Solution (Xembify®), a New High Concentration Immunoglobulin Product for Subcutaneous Administration. Biologicals 2020, 64, 34–40. [Google Scholar] [CrossRef]
- Vandeberg, P.; Cruz, M.; Diez, J.M.; Merritt, W.K.; Santos, B.; Trukawinski, S.; Wellhouse, A.; Jose, M.; Willis, T. Production of Anti-SARS-CoV-2 Hyperimmune Globulin from Convalescent Plasma. Transfusion 2021, 61, 1705–1709. [Google Scholar] [CrossRef]
- Cheng, J.H.; Wu, Y.W.; Wang, C.Y.; Wu, S.S.; Hong, C.L.; Chan, K.W.; Liao, L.X.; Cao, X.; Wang, B.; Burnouf, T. Process Steps for the Fractionation of Immunoglobulin (Ig) G Depleted of IgA, Isoagglutinins, and Devoid of in Vitro Thrombogenicity. Blood Transfus. 2021, 19, 467–478. [Google Scholar] [CrossRef]
- Steinbuch, M.; Audran, R. The Isolation of IgG from Mammalian Sera with the Aid of Caprylic Acid. Arch. Biochem. Biophys. 1969, 134, 279–284. [Google Scholar] [CrossRef]
- Parkkinen, J.; Rahola, A.; Von Bonsdorff, L.; Tölö, H.; Törmä, E. A Modified Caprylic Acid Method for Manufacturing Immunoglobulin G from Human Plasma with High Yield and Efficient Virus Clearance. Vox Sang. 2006, 90, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Lukačević, S.M.; Kurtović, T.; Balija, M.L.; Brgles, M.; Steinberger, S.; Marchetti-Deschmann, M.; Halassy, B. Quality-Related Properties of Equine Immunoglobulins Purified by Different Approaches. Toxins 2020, 12, 798. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.; Massaldi, H. A Model Mechanism for Protein Precipitation by Caprylic Acid: Application to Plasma Purification. Biotechnol. Appl. Biochem. 2012, 59, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Trapp, A.; Faude, A.; Hörold, N.; Schubert, S.; Faust, S.; Grob, T.; Schmidt, S. Multiple Functions of Caprylic Acid-Induced Impurity Precipitation for Process Intensification in Monoclonal Antibody Purification. J. Biotechnol. 2018, 279, 13–21. [Google Scholar] [CrossRef]
- Brodsky, Y.; Zhang, C.; Yigzaw, Y.; Vedantham, G. Caprylic Acid Precipitation Method for Impurity Reduction: An Alternative to Conventional Chromatography for Monoclonal Antibody Purification. Biotechnol. Bioeng. 2012, 109, 2589–2598. [Google Scholar] [CrossRef]
- Yu, M.W.; Finlayson, J.S. Quantitative Determination of the Stabilizers Octanoic Acid and N-acetyl-dl-tryptophan in Human Albumin Products. J. Pharm. Sci. 1984, 73, 82–86. [Google Scholar] [CrossRef]
- Athavale, M.; Fernandes, A.; Kaundinya, J.; Daftary, G. Validation of a Simple, Rapid and Cost Effective Method for the Estimation of Caprylic Acid and Sodium Caprylate from Biological Products Using NEFA-C Kit. Biologicals 2010, 38, 321–324. [Google Scholar] [CrossRef]
- Validation of Analytical Procedures: Text and Methodology, ICH Q2 (R1). In Proceedings of the International Conference on Harmonization (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, November 2005; Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 10 February 2022).
- Saadati, N.; Abdullah, M.P.; Zakaria, Z.; Sany, S.B.T.; Rezayi, M.; Hassonizadeh, H. Limit of Detection and Limit of Quantification Development Procedures for Organochlorine Pesticides Analysis in Water and Sediment Matrices. Chem. Cent. J. 2013, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Şengül, Ü. Comparing Determination Methods of Detection and Quantification Limits for Aflatoxin Analysis in Hazelnut. J. Food Drug Anal. 2016, 24, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Sanagi, M.M.; Ling, S.L.; Nasir, Z.; Hermawan, D.; Wan Ibrahim, W.A.; Naim, A.A. Comparison of Signal-to-Noise, Blank Determination, and Linear Regression Methods for the Estimation of Detection and Quantification Limits for Volatile Organic Compounds by Gas Chromatography. J. AOAC Int. 2009, 92, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, A.; Gupta, V. Methods for the Determination of Limit of Detection and Limit of Quantitation of the Analytical Methods. Chron. Young Sci. 2011, 2, 21. [Google Scholar] [CrossRef]
- Vander Heyden, Y.; Nijhuis, A.; Smeyers-Verbeke, J.; Vandeginste, B.G.M.; Massart, D.L. Guidance for Robustness/Ruggedness Tests in Method Validation. J. Pharm. Biomed. Anal. 2001, 24, 723–753. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Reviewer Guidance: Validation of Chromatographic Method; US Food and Drug Ad-Ministration: Rockville, MD, USA, 1994.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Chromatographic Condition |
|---|---|
| Mobile phase | 0.05% TFA in water–ACN (50:50, v/v) |
| Flow rate | 0.2 mL/min |
| Mode | Isocratic |
| Run time | 10 min |
| Injection volume | 5 μL |
| Detection wavelength | 210 nm |
| Temperature | Ambient temperature |
| df | Sum of Squares | Mean Square | F | Significance F * | |
|---|---|---|---|---|---|
| Regression | 1 | 6.62 × 1012 | 6.62 × 1012 | 68,106.87 | 2.96 × 10−66 |
| Residual | 40 | 3.89 × 109 | 97,259,462 | ||
| Total | 41 | 6.63 × 1012 |
| Nominal Amount of CA in ppm | Measured Amount of CA in ppm (Mean ± SD) | % RSD | % Recovery (Mean ± SD) | |
|---|---|---|---|---|
| Intra-day precision a | ||||
| 1st day | 300 | 303.06 ± 1.74 | 0.575 | 101.02 ± 0.58 |
| 750 | 753.68 ± 5.30 | 0.703 | 100.49 ± 0.71 | |
| 1250 | 1249.33 ± 10.36 | 0.829 | 99.94 ± 0.83 | |
| 2nd day | 300 | 297.08 ± 1.09 | 0.669 | 99.03 ± 0.36 |
| 750 | 739.37 ± 3.93 | 0.671 | 98.58 ± 0.52 | |
| 1250 | 1243.27 ± 7.98 | 0.642 | 99.46 ± 0.64 | |
| 3rd day | 300 | 302.31 ± 2.56 | 0.848 | 100.77 ± 0.85 |
| 750 | 740.83 ± 3.38 | 0.457 | 98.78 ± 0.49 | |
| 1250 | 1252.12 ± 8.67 | 0.692 | 100.17 ± 0.69 | |
| Inter-day precision b | 300 | 301.04 ± 3.22 | 1.069 | 100.35 ± 1.07 |
| 750 | 744.39 ± 7.64 | 1.027 | 99.25 ± 1.02 | |
| 1250 | 1248.24 ± 9.32 | 0.746 | 99.86 ± 0.75 |
| S/N | SD of Regression Line * | SD of y-Intercept * | |
|---|---|---|---|
| LOD | 8 ppm | 10.3 ± 1.1 ppm | 7.0 ± 0.7 ppm |
| LOQ | 25 ppm | 31.3 ± 3.4 ppm | 21.1 ± 2.2 ppm |
| Experiment | RT | A | N | R | k’ | Tailing Factor |
|---|---|---|---|---|---|---|
| 1 | 12.30 ± 0.01 | 377.79 ± 5.43 | 5064 ± 54 | 18.38 ± 0.13 | 4.15 ± 0.01 | 1.197 ± 0.012 |
| 2 | 10.18 ± 0.05 | 312.55 ± 4.55 | 4632 ± 32 | 18.13 ± 0.02 | 4.21 ± 0.02 | 1.179 ± 0.005 |
| 3 | 10.17 ± 0.01 | 302.32 ± 4.26 | 4712 ± 30 | 17.88 ± 0.13 | 4.10 ± 0.01 | 1.176 ± 0.006 |
| 4 | 12.35 ± 0.03 | 369.27 ± 4.39 | 5115 ± 33 | 19.09 ± 0.04 | 4.27 ± 0.02 | 1.183 ± 0.005 |
| 5 | 6.03 ± 0.02 | 293.72 ± 2.93 | 2744 ± 24 | 10.64 ± 0.15 | 2.15 ± 0.01 | 1.222 ± 0.004 |
| 6 | 7.24 ± 0.01 | 358.67 ± 2.87 | 3029 ± 9 | 11.56 ± 0.07 | 2.17 ± 0.01 | 1.224 ± 0.002 |
| 7 | 7.23 ± 0.01 | 354.06 ± 2.57 | 3051 ± 4 | 11.06 ± 0.14 | 2.13 ± 0.01 | 1.234 ± 0.002 |
| 8 | 6.02 ± 0.01 | 292.91 ± 2.37 | 2732 ± 12 | 10.98 ± 0.04 | 2.16 ± 0.01 | 1.225 ± 0.004 |
| Factor | E on RT | E on A | E on N | E on R | E on k’ | E on Tailing Factor |
|---|---|---|---|---|---|---|
| pH | 0.01317 | 1.3764 | −15.92 | 0.44417 * | 0.06942 | −0.0043 |
| Wavelength (nm) | 0.00383 | −6.0396 | 35.58 | 0.07483 | 0.00092 | −0.0010 |
| %ACN (v/v) | −4.61700 * | −15.6454 * | −1991.75 * | −7.30983 * | −2.03008 * | 0.0425 * |
| Flow rate (mL/min) | −1.68467 * | −64.5738 * | −359.58 * | −0.61583 * | −0.02658 | −0.0088 |
| Factors | Low Level | Nominal Level | High Level |
|---|---|---|---|
| pH | 2.2 | 2.4 | 2.6 |
| Wavelength (nm) | 208 | 210 | 212 |
| %ACN (v/v) | 45 | 50 | 55 |
| Flow rate (mL/min) | 0.18 | 0.2 | 0.22 |
| Experiment Number | pH | Wavelength (nm) | %ACN (v/v) | Flow Rate (mL/min) |
|---|---|---|---|---|
| 1 | 2.2 | 208 | 45 | 0.18 |
| 2 | 2.6 | 208 | 45 | 0.22 |
| 3 | 2.2 | 212 | 45 | 0.22 |
| 4 | 2.6 | 212 | 45 | 0.18 |
| 5 | 2.2 | 208 | 55 | 0.22 |
| 6 | 2.6 | 208 | 55 | 0.18 |
| 7 | 2.2 | 212 | 55 | 0.18 |
| 8 | 2.6 | 212 | 55 | 0.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Štimac, A.; Kurtović, T.; Pavlović, N.; Halassy, B. Development of Improved High-Performance Liquid Chromatography Method for the Determination of Residual Caprylic Acid in Formulations of Human Immunoglobulins. Molecules 2022, 27, 1665. https://doi.org/10.3390/molecules27051665
Štimac A, Kurtović T, Pavlović N, Halassy B. Development of Improved High-Performance Liquid Chromatography Method for the Determination of Residual Caprylic Acid in Formulations of Human Immunoglobulins. Molecules. 2022; 27(5):1665. https://doi.org/10.3390/molecules27051665
Chicago/Turabian StyleŠtimac, Adela, Tihana Kurtović, Nediljko Pavlović, and Beata Halassy. 2022. "Development of Improved High-Performance Liquid Chromatography Method for the Determination of Residual Caprylic Acid in Formulations of Human Immunoglobulins" Molecules 27, no. 5: 1665. https://doi.org/10.3390/molecules27051665
APA StyleŠtimac, A., Kurtović, T., Pavlović, N., & Halassy, B. (2022). Development of Improved High-Performance Liquid Chromatography Method for the Determination of Residual Caprylic Acid in Formulations of Human Immunoglobulins. Molecules, 27(5), 1665. https://doi.org/10.3390/molecules27051665