3. Materials and Methods
NMR spectra were recorded on a Bruker DPX spectrometer (400 MHz), and chemical shifts are quoted in ppm relative to tetramethylsilane as internal standard using the following abbreviations: s, singlet, d, doublet, at, apparent triplet, as, apparent singlet and m, multiplet. Liquid Chromatography Mass Spectrometry (LCMS) was accomplished using a ThermoFisher Scientific Accela LC system coupled to a ThermoFisher Scientific LTQ Fleet Ion Trap Mass Spectrometer (ThermoFisher Scientific, Loughborough, UK). Melting points were recorded on a TA Instruments DSC Q2000 instrument heating at 10 °C/min (Hertfordshire, UK). FTIR spectra were recorded on a ThermoFisher Scientific Nicolet iS10 instrument. Thin-layer chromatography was performed using ALUGRAM SIL G precoated plates (Macherey-Nagel, Germany). The purity of deprotected samples was achieved using an Agilent 1100 series HPLC with a ThermoFisher Scientific Hypersil Gold Column (50 × 4.6 mm, 3 µm) eluting 0.1% (v/v) formic acid in water/MeCN (85:15), 2 mL/min, UV absorbance at 254 nm.
All culture media was “Oxoid” from ThermoFisher Scientific, Basingstoke, UK. Nadifloxacin was purchased from AOKChem (Shanghai, China) as a racemate and was used without further purification. Per-O-acetylated and 2,3,4,6-tetra-O-benzyl glucose and galactose were purchased from Carbosynth (Berkshire, UK). All other reagents were purchased from Sigma-Aldrich (Poole, Dorset, UK) or Fisher Scientific (Loughborough, UK).
Synthesis of 1,2-trans glycosides promoted by 10 molar equivalents of TMSOTf–General method.
Nadifloxacin (1) (5 g, 13.9 mmoles) was suspended in anhydrous acetonitrile (250 mL), and to this TMSOTf (2.51 mL, 13.9 mmoles) was added, and the solution was stirred. Following this were the additions of the per-O-acetylated glycoside (either 1,2,3,5-tri-O-acetyl-α-L-arabinofuranoside, 1,2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside, 1,2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside, 1,2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside or 1,2,3,5-tri-O-acetyl-β-D-xylofuranoside, 13.9 mmoles) followed by TMSOTf (22.59 mL, 124.9 mmoles). Reactions were stirred at room temperature under an atmosphere of nitrogen for 24 h. The reaction mixtures were then diluted with anhydrous dichloromethane (250 mL), washed with sat. NaHCO3 (2 × 100 mL) and brine (2 × 100 mL), dried over MgSO4 and concentrated in vacuo. The products were purified on silica columns and eluted with toluene (A) and acetonitrile (B) at 14% B to 36% B over 5 column volumes and maintained at 36% for 10-column volumes.
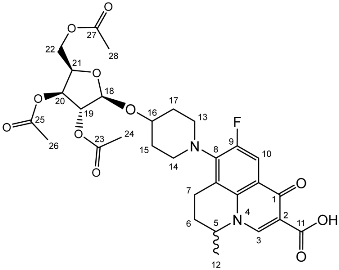
Nadifloxacin 2,3,5-tri-O-acetyl-α-L-arabinofuranoside (2a)
Nadifloxacin (1) (5 g, 13.9 mmoles) was added to anhydrous acetonitrile and reacted with 1 molar equivalent of TMSOTf, 1,2,3,5-tetra-O-acetyl-α-L-arabinofuranoside (4.53, 13.9 mmoles), followed by 9 molar equivalents of TMSOTf overnight to yield the product (2a) as a white powder.
Yield (4.81 g, 56%), m.p. 93–106 °C, 1H NMR (400 MHz, CDCl3) δ: 8.69 (1H, s, C3-H), 7.98 (1H, d, J = 12.5 Hz, C10-H), 5.25–5.21 (1H, m, C18-H), 5.13–5.08 (1H, m, C21-H), 5.02 (1H, d, J = 5.0 Hz, C19-H), 4.56 (1H, dd, J = 7.0, 3.5 Hz, C5-H), 4.44 (1H, dt, J = 11.5, 2.5 Hz, C22-Ha), 4.34–4.28 (1H, m, C20-H), 4.28–4.21 (1H, m, C22-Hb), 3.31 (1H, m, C7-Ha), 2.95–2.79 (1H, m, C7-Hb), 2.22–2.18 (2H, m, C6-H2), 2.16–2.09 (9H, m, acetyl CH3), 2.07–1.63 (4H, m, C15-H2 and C17-H2), 1.52 (3H, d, J = 6.5 Hz). 13C NMR (100 MHz, CDCl3) δ: 206.98 ketone (C=O), 170.62, 170.19, 169.84 (acetyl C=O), 167.26 (carbonyl C=O), 147.35 (C3), 137.83, 133.35 (aromatic C), 110.66 (C10), 107.63 (C18), 81.71 (C21), 80.32 (C20), 76.80 (C19), 63.29 (C22), 57.94 (C5), 32.71 (C15, C17), 30.91 (C16), 25.97 (C6), 21.44, 20.78, 20.75 (acetyl CH3), 20.20 (C5-CH3), 18.83 (C7). FT-IR ν cm−1: 2936.38 (w, carboxyl OH), 1738.96 (carboxylic or ester C=O), 1621.49 (aromatic C=C). MS +ESI found 619.2125 (MH+) C30H36FN2O11 required 619.2298.
Nadifloxacin 2,3,4,6-tetra-O-acetyl-β-d-galactopyranoside (2b)
Nadifloxacin (1) (5 g, 13.9 mmoles) was added to anhydrous acetonitrile and reacted with 1 molar equivalent of TMSOTf, 1,2,3,4,6-penta-O-acetyl-β-D-galactopyranoside (5.6 g, 13.9 mmoles) followed by 9 molar equivalents of TMSOTf overnight to yield the product (2b) as a white powder.
Yield (4.98 g, 52%). m.p. 151–165 °C, 1H NMR (400 MHz, CDCl3) δ: 8.63 (1H, s, C3-H), 7.92 (1H, d, J = 12.0 Hz, C10-H), 5.39–5.31 (1H, m, C21-H), 5.19 (1H, at, J = 9.0 Hz, C19-H), 5.02–4.96 (1H, m, C20-H), 4.57 (1H, d, J = 8.0 Hz, C18-H), 4.53–4.45 (1H, m, C5-H), 4.15 (1H, dd, J = 11.5, 7.0 Hz, C23-Ha), 4.06 (1H, dd, J = 11.5, 7.0 Hz, C23-Hb), 3.87 (1H, at, J = 7.0 Hz, C22-H), 3.30–2.90 (5 H, m, C7-Ha, C13-H2, C14-H2), 2.79 (1 H, dt, J = 18.0, 8.5 Hz, C7-Hb), 2.14 (2H, m, C6-H2), 2.04 (2H, s, acetyl CH3), 2.01 (2H, s, acetyl CH3), 1.99 (3 H, s, acetyl CH3), 1.94 (3 H, s, acetyl CH3), 1.89–1.55 (4H, m, C15-H2, C17-H2), 1.46 (3H, d, J = 6.5 Hz, C12-H3). 13C NMR (100 MHz, CDCl3) δ: 170.41, 170.28, 170.19, 167.25 (carboxyl C=O), 146.35 (C3), 133.61 (aromatic C), 110.81, 110.59 (C10), 100.33 (C18), 70.86 (C20), 70.66 (C22), 68.98 (C19), 66.99 (C21), 61.23 (C23), 57.94 (C5), 25.97 (C6), 20.81, 20.71, 20.62 (Acetyl CH3), 20.23 (C12). FT-IR ν cm−1: 2976.02 (w, carboxyl OH), 1748.35 (s, ketone C=O), 1731.64 (carboxylic or ester C=O), 1624.15 (aromatic C=C). MS +ESI found 691.2141 (MH+) C33H40FN2O13 required 691.2509.
Nadifloxacin 2,3,4,6-tetra-O-acetyl-β-d-glucopyranoside (2c)
Nadifloxacin (1) (5 g, 13.9 mmoles) was added to anhydrous acetonitrile and reacted with 1 molar equivalent of TMSOTf, 1,2,3,4,6-penta-O-acetyl-β-D-glucopyranoside (5.6 g, 13.9 mmoles) followed by 9 molar equivalents of TMSOTf overnight to yield the product (2c) as a white powder.
Yield (2.73 g, 29%). m.p. 156–159 °C, 1H NMR (400 MHz, CDCl3) δ: 8.68 (1H, s, C3-H), 7.96 (1H, d, J = 12.0 Hz, C10-H), 5.22 (1H, dd, J = 9.5, 2.0 Hz, C20-H), 5.08 (1H, dd, J = 9.5, 2.0 Hz, C21-H), 5.04–4.96 (1H, m, C19-H), 4.67 (1H, d, J = 8.0 Hz, C18-H), 4.57 (1H, dt, J = 7.0, 3.5 Hz, C5-H), 4.25 (1H, dd, J = 12.5, 5.0 Hz, C23-Ha), 4.17–4.10 (1H, m, C23-Hb), 3.72 (1H, ddd, J = 10.0, 4.5, 2.5 Hz, C22-H), 3.33–2.94 (5 H, m, C7-Ha, C13-H2, C14-H2), 2.84 (1H, ddt, J = 17.0, 10.5, 5.5 Hz, C7-Hb), 2.15 (2H, m, C6-H2), 2.07 (3H, acetyl CH3), 2.04 (3H, acetyl CH3), 2.02 (3H, acetyl CH3), 2.00 (3H, acetyl CH3), 1.50 (3H, d, J = 6.5 Hz, C12-H3). 13C NMR (100 MHz, CDCl3) δ: 207.66 (C1), 170.78, 170.38, 169.51, 167.64 (carboxyl C=O), 146.45 (C3), 133.64 (aromatic C), 107.39 (C10), 99.17 (C18), 72.76 (C20), 71.71 (C22), 71.44 (C19), 68.43 (C21), 61.98 (C23), 57.99 (C5), 30.89 (C6), 20.1 (acetyl CH3), 20.14 (C12). FT-IR ν cm−1: 2973.23 (w, carboxyl OH), 1748.42 (s, ketone C=O), 1725.81 (carboxylic or ester C=O), 1672.21 (aromatic C=C). MS +ESI found 691.2930 (MH+) C33H40FN2O13 required 691.2509.
Nadifloxacin 2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside (2d)
Nadifloxacin (1) (5 g, 13.9 mmoles) was added to anhydrous acetonitrile and reacted with 1 molar equivalent of TMSOTf, 1,2,3,4,6-penta-O-acetyl-D-mannopyranoside (5.6 g, 13.9 mmoles) followed by 9 molar equivalents of TMSOTf overnight to yield the product (2d) as a white powder.
Yield (2.20 g, 23%). m.p. 190–204 °C 1H NMR (400 MHz, CDCl3) δ: 8.63 (1H, s, C3-H), 7.92 (1H, d, J = 12.0 Hz, C10-H), 5.34 (1H, dd, J = 10.5, 3.0 Hz, C20-H), 5.24 (1H, dd, J = 10.0, 3.0 Hz, C21-H), 5.20–5.14 (1H, m, C19-H), 4.99–4.95 (1H, m, C18-H), 4.55–4.44 (1H, m, C5-H), 4.23 (1H, dd, J = 12.0, 4.5 Hz, C23-Ha), 4.12–4.01 (2H, m, C23-Hb, C22-H), 3.50–2.93 (3H, m, C7-Ha, C13-H2, C14-H2), 2.81 (1H, dt, J = 17.5, 9.5 Hz, C7-Hb), 2.11 (5H, m,C6-H2, acetyl CH3), 2.04 (acetyl CH3), 2.00 (acetyl CH3), 1.95 (acetyl CH3), 1.91–1.52 (4H, m, C15-H2, C17-H2) 1.47 (3H, d, J = 6.5 Hz, C12-H3). 13C NMR (100 MHz, CDCl3) δ: 177.36 (C11) 170.59, 170.22, 170.01, 169.72 (carboxyl C=O), 146.39 (C3), 133.65 (aromatic C), 110.79, 110.56 (C10), 107.66 (aromatic C) 95.97 (C18), 70.10 (C19), 69.01 (C20), 68.77 (C22), 66.31 (C21), 62.56 (C23), 57.96 (C5), 25.95 (C6), 20.95, 20.75, 20.73 (acetyl CH3), 20.24 (C12), 18.84 (C7). FT-IR ν cm−1: 2956.60 (w, carboxyl OH), 1737.23 (carboxylic or ester C=O), 1617.60 (aromatic C=C). MS +ESI found 691.2372 (MH+) C33H40FN2O13 required 691.2509.
Nadifloxacin 2,3,5-tri-O-acetyl-β-d-xylofuranoside (2e)
Nadifloxacin (1) (5 g, 13.9 mmoles) was added to anhydrous acetonitrile and reacted with 1 molar equivalent of TMSOTf, 1,2,3,5-tetra-O-acetyl-β-D-xylofuranoside (4.53, 13.9 mmoles) followed by 9 molar equivalents of TMSOTf overnight to yield the product (2e) as a white powder.
Yield (4.72 g, 55%). m.p. 84–95 °C, 1H NMR (400 MHz, CDCl3) δ: 8.70 (1H, s, C3-H), 7.98 (1H, d, J = 12.5 Hz, C10-H), 5.34 (1 H, dd, J = 6.0, 2.0 Hz, C20), 5.22–5.19 (1H, m, C18-H), 5.15–5.12 (1H, m, C19-H), 4.65–4.50 (2H, m, C5-H, C21-H), 4.37–4.11 (2H, m, C22-H2), 3.37–3.01 (5H, m, C7-Ha, C13-H2, C14-H2), 2.90 (1H, ddd, J = 18.0, 11.5, 7.0 Hz, C7-Hb), 2.23–1.69 (m, C6, acetyl CH3, C15-H2, C17-H2), 1.52 (3H, d, J = 7.0 Hz). 13C NMR (100 MHz, CDCl3) δ: 177.85 (C11), 170.78, 170.14, 169.94, 167.32 (acetyl C=O), 146.76 (C3), 138.33, 134.19 (aromatic C), 110.60, 110.36 (C10), 104.69 (C18), 81.38 (C19), 78.47 (C21), 75.27 (C20), 63.44 (C22), 58.49 (C5), 26.24 (C6), 21.00, 20.86, (acetyl C), 20.28 (C12), 19.25 (C13 or C14). FT-IR ν cm−1: 2936.38 (w, carboxyl OH), 1738.96 (carboxylic or ester C=O), 1621.49 (aromatic C=C). MS +ESI found 619.2062 (MH+) C30H36FN2O11 required 619.2298.
Deprotection of per-O-acetylated nadifloxacin glycosides–General method
The isolated glycosides (2a-e) were de-O-acetylated using a mixture of anhydrous dichloromethane and methanol (1:6) and potassium carbonate (50% w/w) over 30–60 min. The mixtures were adjusted to approximately pH 3 by the addition of formic acid and were purified on C18 columns and eluted with 0.1% (v/v) formic acid in water (A) and acetonitrile (B) isocratically at 7:3.
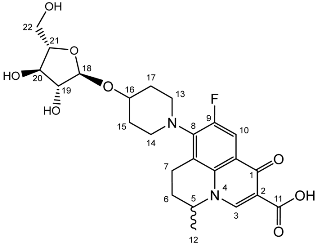
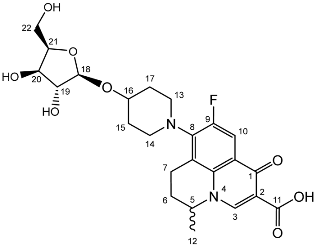
Nadifloxacin α-l-arabinofuranoside (3a)
Nadifloxacin 2,3,5-tri-O-acetyl-α-L-arabinofuranoside (2a) (2.04 g, 3.31 mmoles) was dissolved in anhydrous dichloromethane and methanol, reacted with potassium carbonate (1.02 g) and purified to afford the product (3a) as an off-white powder.
Yield (1.12 g, 69%). mp: 167–178 °C, 1H NMR (400 MHz, DMSO-d6) δ: 8.94 (1H, s, C3-H), 7.81 (1H, d, J = 12.5 Hz, C10-H), 4.95–4.91 (1H, m, C18-H), 4.91–4.82 (1H, m, C5-H), 3.83–3.79 (1H, m, C19-H), 3.76 (1H, m, C21-H), 3.64 (1H, m, C20-H), 3.61–3.54 (1H, m, C22-Ha), 3.42 (1H, dd, J = 12.5, 6.0 Hz, C22-Hb), 3.37–3.07 (5H, m, C7-Ha, C13-H2 and C14-H2), 3.03–2.81 (1H, m, C7-Hb), 2.16–1.86 (3H, m, C6-H2, C15-H2 or C17-H2), 1.82–1.50 (2H, m, C15-H2 or C17-H2), 1.41 (3H, d, J = 6.5 Hz, C12). 13C NMR (100 MHz, DMSO-d6) δ: 206.32 (ketone C=O), 176.49, 166.35 (carboxyl C=O), 147.56 (C3), 133.81 (aromatic C), 109.06 (C10), 106.17 (C18), 83.54 (C21), 82.58 (C19), 77.09 (C20), 61.25 (C22), 57.10 (C5), 49.40 (C13 and C14), 33.70 (C15 or C17), 30.64 (C6), 19.62 (C12), 18.63 (C7). FT-IR ν cm−1: 3343.32 (m, alcohol OH), 2936.72 (w, carboxyl OH), 1716.43 (carboxylic or ester C=O), 1618.00 (aromatic C=C). MS +ESI found 493.1568 (MH+) C24H30FN2O8 required 493.1986. HPLC analysis: retention time: 5.912 min, 97.4%.
Nadifloxacin β-d-galactopyranoside (3b)
Nadifloxacin 2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside (2b) (2 g, 2.90 mmoles) was dissolved in anhydrous dichloromethane and methanol, reacted with potassium carbonate (1 g) and purified to afford the product (3b) as an off-white powder.
Yield (1.12 g, 74%), m.p. 149–161 °C. 1H NMR (400 MHz, DMSO-d6) δ: 8.95 (1 H, d, J = 3.0 Hz, C3-H), 7.84 (1 H, dd, J = 12.5, 3.0 Hz, C10-H), 4.96–4.83 (1H, m, C5-H), 4.28–4.2 (1H, m, C18-H), 3.66–3.60 (1H, m, C20-H), 3.58–3.43 (2H, m, C23-H2), 3.44–3.20 (2H, m, C19-H, C21-H, C22-H), 3.11 (1H, d, J = 16.5 Hz, C7-Ha), 3.02–2.83 (1H, m, C7-Hb), 2.20–1.97 (2H, m, C6-H2), 2.02–1.53 (4H, m, C15-H2 and C17-H2), 1.41 (3H, d, J = 4.5 Hz, C12-H3). 13C NMR (100 MHz, DMSO-d6) δ: 166.08 (carboxyl C=O), 147.25 (C3), 133.46 (aromatic C), 108.99 (C10), 101.56 (C18), 75.09 (C22), 73.19, 70.39 (C19 and C21), 67.95 (C20), 60.45 (C23), 57.02 (C5), 33.66 (C15 and C17), 24.80 (C6), 19.55 (C12), 18.53 (C7). FT-IR ν cm−1: 3383 (w, alcohol C-OH), 2925 (w, carboxylic C-OH), 1698 (m, ketone C=O), 1628 (m, aromatic C=C), 1448 (m, alkyl C-H), 1021 (s, C-O). MS + ESI found 523.2897 (MH+) C25H32FN2O9 required 523.2092. HPLC analysis: retention time: 3.042 min, purity: 97.3%.
Nadifloxacin β-d-glucopyranoside (3c)
Nadifloxacin 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (2c) (2.6 g, 3.77 mmoles) was dissolved in anhydrous dichloromethane and methanol, reacted with potassium carbonate (1.3 g) and purified to afford the product (3c) as an off-white powder.
Yield (1.26 g, 64%), m.p. 162–163 °C, 1H NMR (400 MHz, DMSO-d6) δ: 8.94 (1H, s, C3-H), 7.83 (1H, d, J = 12.0 Hz, C10-H), 4.93–4.80 (1H, m, C5-H), 4.30 (1H, d, J = 7.0 Hz, C18-H), 3.88 (2H, s, C13-H2 or C14-H2), 3.67 (1H, d, J = 11.5 Hz, C23-Ha), 3.43 (1H, d, J = 9.5 Hz, C23-Hb), 3.23 (2H, s, C13-H2 or C14-H2), 3.20–3.06 (4H, m, C20-H, C21-H, C22-H and C7-Ha), 2.96 (1H, at, J = 7.0 Hz, C19-H), 2.94–2.85 (1H, m, C7-Hb), 2.20–1.98 (2H, m, C6-H2), 1.85–1.48 (4H, m, C15-H2 and C17-H2), 1.41 (3H, d, J = 5.5Hz, C12-H3). 13C NMR (100 MHz, DMSO-d6) δ: 166.08 (carboxyl C=O), 147.10 (C3), 133.48 (aromatic C), 133.16 (aromatic C), 106.24 (C10), 100.41 (C18), 76.8 (C20, C22), 73.52 (C19), 70.45 (C21), 61.06 (C23), 57.04 (C5), 49 (C13 or C14) 33.60 (C15 and C16),19.56 (C12), 18.60 (C7). FT-IR ν cm−1: 3386 (w, b, alcohol C-OH), 2925 (w, carboxylic C-OH), 1703 (m, ketone C=O), 1628 (m, aromatic C=C), 1448 (m, alkyl C-H), 1021 (s, C-O). MS +ESI found 523.2073 (MH+) C25H32FN2O9 required 523.2092. HPLC analysis: retention time: 2.537 min, purity: 97.7%.
Nadifloxacin α-d-mannopyranoside (3d)
Nadifloxacin 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside (2d) (2.0 g, 2.90 mmoles) was dissolved in anhydrous dichloromethane and methanol, reacted with potassium carbonate (1.0 g) and purified to afford the product (3d) as an off-white powder.
Yield (1.10 g, 73%). m.p. 186 °C, 1H NMR (400 MHz, DMSO-d6) δ: 8.94 (1H, s, C3-H), 7.84 (1H, d, J = 12.5 Hz, C10-H), 4.92–4.81 (2H, m, C5-Hand C18-H), 3.83 (2H, as, C13-H2 or C14-H2), 3.70–3.62 (1H, m, C23-Ha), 3.62–3.58 (1H, m, C19-H), 3.55–3.30 (5H, m, C20-H, C21-H, C22-H, C23-Ha), 3.25–3.09 (1H, m, C7-Ha), 3.02–2.82 (1H, m, C7-Hb), 2.16–1.88 (4H, m, C6-H2 and C15-H2 or C17-H2), 1.80–1.52 (2H, m, C15-H2 or C17-H2), 1.46–1.35 (3H, m, C12-H3). 13C NMR (100 MHz, DMSO-d6) δ: 165.62 (aromatic C), 147.46 (C3), 133.75 (aromatic C), 109.03 (C10), 98.23 (C18), 74.27, 70.89, 70.77, 67.05 (C20, C21 and C22), 61.37 (C23), 56.95 (C5), 33.39 (C15 and C17), 19.58 (C12), 18.52 (C7). FT-IR ν cm−1: 3429.13, 3343.67 (m, alcohol OH), 2937.98 (w, carboxyl OH), 1722.74 (carboxylic or ester C=O), 1616.53 (aromatic C=C). MS +ESI found 523.2296 (MH+) C25H32FN2O9 required 523.2092. HPLC analysis: retention time: 6.923 min, purity: 98.1%.
Nadifloxacin β-d-xylofuranoside (3e)
Nadifloxacin 2,3,5-tri-O-acetyl-β-D-xylofuranoside (2e) (2.31 g, 3.74 mmoles) was dissolved in anhydrous dichloromethane and methanol, reacted with potassium carbonate (1.16 g) and purified to afford the product (3e) as an off-white powder.
Yield (1.10 g, 66%). m.p.145 °C, 1H NMR (400 MHz, DMSO-d6) δ: 9.01 (1H, s, C3-H), 7.90 (1H, d, J = 12.5 Hz, C10-H), 5.00–4.91 (1H, m, C5-H), 4.34 (1H, d, J = 7.5 Hz, C18-H), 3.75 (1H, m, C22-Ha), 3.54–3.26 (1H, m, C21-H), 3.22–3.10 (3H, m, C20-H, C22-Hb, C7-Ha), 3.07–2.93 (2H, m, C19-H and C7-Hb), 2.26–1.92 (4H, m, C6-H2, C15-H2 or C17-H2), 1.87–1.61 (2 H, m, C15-H2 or C17-H2), 1.48 (3H, d, J = 4.0 Hz, C12-CH3). 13C NMR (100 MHz, DMSO-d6) δ: 147.79 (C3), 133.75 (aromatic C), 109.09 (C10), 102.3 (C18), 76.65 (C20), 73.28 (C19), 69.62 (C21), 65.65 (C22), 33.77 (C15 and C17), 24.91 (C6), 19.65 (C12), 18.85 (C7). FT-IR ν cm−1: 3335.48 (m, alcohol OH), 2933.13 (w, carboxyl OH), 1614.51 (aromatic C=C). MS + ESI found 493.1762 (MH+) C24H30FN2O8 required 493.1986. HPLC analysis: retention time: 8.713 min, purity: 98.7%.
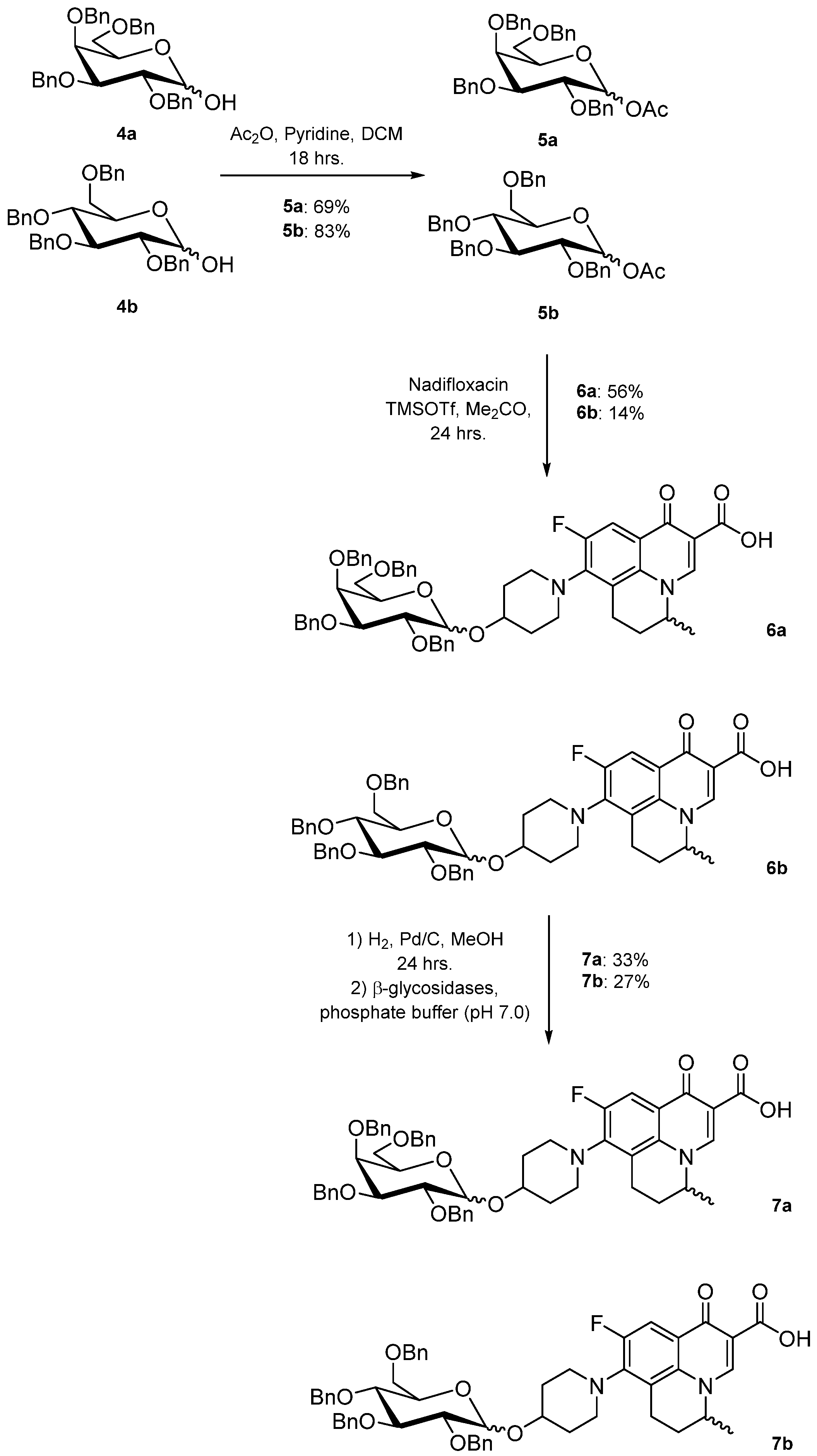
Synthesis of 1-O-acetyl-2,3,4,6-tetra-O-benzyl-d-glycosides (5a, 5b)–General method
2,3,4,6-Tetra-O-benzyl-D-galactopyranose (4a) and 2,3,4,6-tetra-O-benzyl-D-glucopyranose (4b) (20 g, 37 mmoles) were each dissolved in anhydrous dichloromethane (100 mL). To these solutions pyridine (6.4 mL, 74.4 mmoles) and acetic anhydride (7.52 mL, 74.4 mmoles) were added. The mixtures were stirred under nitrogen, monitored by TLC (hexane–ether 1:1) and stirred at room temperature for approximately 18 h. The organic solutions were extracted with 1M HCl (3 × 100 mL), sat. NaHCO3 (3 × 100 mL) and brine (3 × 100 mL). The organic fractions were dried over MgSO4 and concentrated in vacuo to afford an oil. The galactoside (5a) was precipitated from methanol to afford a white powder. The glucoside (5b) was used as a crude oil.
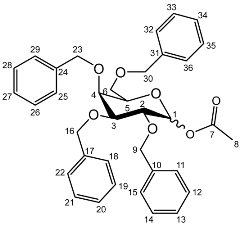
1-O-Acetyl-2,3,4,6-tetra-O-benzyl-d-galactopyranoside (5a)
![Molecules 27 01504 i011]()
Yield (14.8 g, 69%), m.p. 102–104 °C, isolated as a mixture of anomers, α:β 1:1.59,
1H NMR (400 MHz, CDCl
3) δ: 7.38–7.22 (m, benzyl C-H), 6.38 (d,
J = 3.5 Hz, α-C1-H), 5.57 (d,
J = 8.0 Hz, β-C1-H), 4.95 (d,
J = 11.5 Hz, α-benzyl CH
2), 4.94 (d,
J = 11.5 Hz, β-benzyl CH
2), 4.84 (d,
J = 11.5 Hz, β-benzyl CH
2), 4.82 (d,
J = 11.5 Hz, α-benzyl CH
2), 4.74 (d,
J = 11.5 Hz, α-benzyl CH
2), 4.75–4.69 (m, benzyl CH
2), 4.62 (d,
J = 11.5 Hz, β-benzyl CH
2), 4.57 (d,
J = 11.5 Hz, α-benzyl CH
2), 4.46 (d,
J = 11.5 Hz, α-benzyl CH
2), 4.43 (d,
J = 11.5 Hz, β-benzyl CH
2), 4.39 (d,
J = 11.5 Hz, α-benzyl CH
2), 4.38 (d,
J = 11.5 Hz, β-benzyl CH
2), 4.16 (dd,
J = 10.0, 3.5 Hz, α-C2-H), 4.06–3.99 (m, C5-H), 3.94 (m, β-C2-H), 3.89 (dd,
J = 10.0, 2.5 Hz, α-C3-H), 3.72–3.66 (m, β-C4-H), 2.11 (s, α-C8-H
3), 2.03 (s, β-C8-H
3).
13C NMR (100 MHz, DMSO-d6) δ: 169.63, 169.41 (carboxyl C=O), 138.64, 138.52, 138.42, 138.22, 138.06, 137.80, 137.77 (benzyl
ipso C), 128.63–127.36 (benzyl CH), 94.30 (β-C1), 90.81 (α-C1), 82.42 (α-C5), 78.61 (α-C3), 78.19 (β-C2), 75.42 (α-C2), 75.35, 74.95, 74.72 (benzyl CH2), 74.08 (β-C4), 73.61, 73.54, 73.40, 73.40 (benzyl CH2), 73.04 (β-C5), 72.89 (benzyl CH2), 68.41 (α-C6), 67.94 (β-C6), 21.24 (α-C8), 21.06 (β-C8). FT-IR ν cm
−1: 3059.70, 3026.72 (w, aromatic C-H), 1747.65 (m, ester C=O), 1081.32, 1045.51, 1024.29 (s, ether C-O). MS + ESI found 621.3333 (M + K
+) C
36H
38KO
7 required 621.2249.
1-O-Acetyl-2,3,4,6-tetra-O-benzyl-d-glucopyranoside (5b)
![Molecules 27 01504 i012]()
Yield (18.0 g, 83%). Isolated as a mixture of anomers, α:β 1:1.23,
1H NMR (400 MHz, CDCl
3) δ: 7.38–7.22 (m, benzyl C-H), 7.16–7.10 (m, benzyl C-H), 6.35 (d,
J = 3.5 Hz, α-C1-H), 5.60 (d,
J = 8.0 Hz, β-C1-H), 4.99–4.44 (m, benzyl CH2), 3.99–3.90 (m, glucose H), 3.89–3.83 (m, glucose H), 3.79–3.53 (m, α-, β-C2-H and α-, β-C6-H
2), 2.13 (s, α-C8-H
3), 2.05 (s, β-C8-H
3).
13C NMR (100 MHz, DMSO-d6) δ: 169.47, 169.31 (C7), 138.64, 138.11, 138.02, 137.89, 137.82, 137.60 (aromatic C), 128.58–127.59 (aromatic C-H), 94.05 (α-C1), 84.82, 81.68, 81.07, 78.88, 77.22, 76.95 (glucose C), 75.72 (benzyl CH
2), 75.40 (glucose C), 75.30, 75.04, 73.55, 73.22 (benzyl CH
2), 72.83 (glucose C), 68.09 (C6), 21.10 (C8). FT-IR ν cm
−1: 3088.08, 3062.95, 3030.18 (w, aromatic C-H), 1755.44 (m, ester C=O), 1072.79, 1027.24 (s, ether C-O). MS-ESI found 621.5834 (M + K
+) KC36H38O7 required 621.2249.
Synthesis of benzyl protected nadifloxacin α-galactoside (6a) and α-glucoside (6b)–General method
Nadifloxacin (1) (5 g, 13.9 mmoles) was suspended in anhydrous acetone (250 mL), and to this TMSOTf was added (2.51 mL, 13.9 mmoles), and the solution was stirred. To this the 1-O-acetyl-2,3,4,6-tetra-O-benzyl-glycoside (5a or 5b) (16.1 g, 13.9 mmoles) was added followed by TMSOTf (22.59 mL, 124.9 mmoles). Reactions were stirred at room temperature under an atmosphere of nitrogen for 24 h. The reaction mixtures were then diluted with anhydrous dichloromethane (250 mL), washed with sat. NaHCO3 (2 × 100 mL) and brine (2 × 100 mL), dried over MgSO4 and concentrated in vacuo. The products (6a) or (6b) were purified on silica columns and eluted with toluene (A) and acetonitrile (B) at 14% B to 36% B over 5 column volumes and maintained at 36% for 10-column volumes.
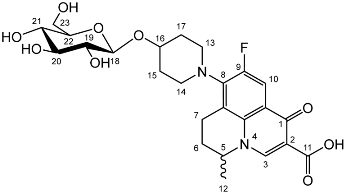
Nadifloxacin 2,3,4,6-tetra-O-benzyl-α,β-d-galactopyranoside (6a)
![Molecules 27 01504 i013]()
Yield (6.83 g, 56%). m.p. 61–65 °C, isolated as a mixture of anomers, α:β 3:5,
1H NMR (400 MHz, CDCl
3) δ: 8.63 (1H, s, C3-H), 7.93 (1H, d,
J = 12.0 Hz, C10-H), 7.36–7.15 (m, benzyl C-H), 4.98–4.94 (m, α-C18-H), 4.94–4.84 (m, benzyl CH
2), 4.83–4.46 (m, benzyl CH
2, C5-H), 4.44 (d,
J = 8.5 Hz, β-C18-H), 4.41–4.30 (m, benzyl CH
2), 4.03–3.95 (m, α-C19-H and C22-H), 3.95–3.88 (C21-H), 3.82 (d,
J = 9.5 Hz, C22-H), 3.77 (d,
J = 9.0 Hz, β-C19-H), 3.52–3.44 (m, C20-H and C23-H
2), 3.21 (d,
J = 17.0 Hz, C7-Ha), 3.03–2.90 (m, C16-H), 2.85–2.67 (m, C7-Hb), 2.15–2.03 (m, C6-H
2), 2.03–1.59 (m, C15-H
2 and C17-H
2), 1.48–1.39 (m, C12-H
3).
13C NMR (100 MHz, CDCl
3) δ: 167.16 (carboxyl C=O), 146.47 (C3), 128.62–127.27 (aromatic C), 125.29 (aromatic C), 110.72 (C10), 101.92 (β-C18), 5.83 (α-C18), 82.12 (C20), 79.77 (β-C19), 79.27 (C21), 76.06 (α-C19), 75.32 (benzyl CH2), 75.03 (C21), 74.77 (benzyl CH
2), 74.53 (benzyl CH
2), 73.94 (C20), 73.65 (C22) 73.13 (benzyl CH
2), 69.78 (C22), 69.31, 69.01 (C23), 32.64 (C15, C17), 26.04 (C6), 20.25 (C12), 18.93 (C7). FT-IR ν cm
−1: 3061.62, 3029.55 (w, aromatic C-H), 2920.54, 2861.87 (w, carboxylic C-OH), 1723.88 (m, carbonyl C=O), 1619 (m, C=C), 1490 (s, arene C-C), MS +ESI found 883.3593 (MH
+) C
53H
56FN
2O
9 expected 883.3970.
Nadifloxacin 2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranoside (6b)
![Molecules 27 01504 i014]()
Yield (1.75 g, 14%),
1H NMR (400 MHz, CDCl3) δ: 8.69 (s, C3-H), 8.00 (d,
J = 12.0 Hz, C10-H), 7.41–7.21 (m, benzyl H), 5.06–4.96 (m, α-C18-H, benzyl CH
2), 4.95–4.72 (m, benzyl CH
2), 4.71–4.44 (benzyl CH
2, β-C18-H, C5-H), 4.03 (at,
J = 9.5 Hz, α-C20-H), 3.94–3.87 (m, glucose H), 3.78–3.71 (m, C23-Ha), 3.71–3.44 (m, C23-Hb, α-C19-H, β-C19-H, glucose H), 3.38–3.16 (m, C7-Ha), 3.15–3.00 (m, C13-H
2 or C14-H
2), 2.93–2.75 (m, C7-Hb), 2.11–1.64 (m, C17-H
2, C15-H
2), 1.53 (d,
J = 6.5 Hz, C12-H
3), 1.52 (d,
J = 6.5 Hz, C12-H
3).
13C NMR (100 MHz, CDCl
3) δ: 177.14, 167.35 (carbonyl C=O), 110.77, 110.49 (C10), 101.88 (β-C18), 95.13 (α-C18), 84.52, (glucose C), 82.29 (β-C19), 82.04 (α-C20), 79.98, 77.88 (glucose C), 75.69, 75.25, 75.04, 74.95 (benzyl CH
2), 74.82 (β-glucose C), 73.50, 73.42, 73.33, 73.25 (benzyl CH2), 70.51 (glucose C), 68.62 (C23), 57.95 (C5), 33.12 (C17, C15), 30.97 (C6), 20.23 (C12), 18.83 (C7). FT-IR ν cm
−1: 3062.43, 3028.98 (w, aromatic C-H), 2900.68, 2865.08 (w, carboxylic C-OH), 1618.42 (m, C=C), 1452 (s, arene C-C), 1067.21, 1027.67 (s, C-O). MS + ESI found 883.3850 (MH
+) C
53H
56FN
2O
9 expected 883.3970.
Deprotection of per-O-benzylated nadifloxacin glycosides and resolution of anomers by enzyme hydrolysis (7a, 7b)–General method
De-O-benzylation of glycosides (6a) and (6b) was achieved using Pd/C (10 wt.%) in methanol under an atmosphere of hydrogen. After 24 h, the mixtures were filtered over Celite and concentrated in vacuo to afford anomeric mixture of nadifloxacin glycosides (7a α,β) and (7b α,β). These were then stirred in a suitable buffer, β-glycosidase (100 U) was added for 7a α,β: β-galactosidase from E. coli, for 7b α,β, β-glucosidase from almonds, and the suspensions were incubated at 37 °C and monitored by HPLC analysis for the decrease in the β-glycoside. Once the β-anomers had all been hydrolysed, the reaction mixtures were filtered, adjusted to pH 3.0 using formic acid and purified on C18 columns and eluted with 0.1% (v/v) formic acid in water (A) and acetonitrile (B) (70:30).
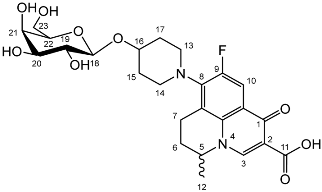
Nadifloxacin α-d-galactopyranoside (7a)
![Molecules 27 01504 i015]()
Yield (1.25 g, 33%), m.p. 143–150 °C,
1H NMR (400 MHz, DMSO-d6) δ: 8.94 (1H, s, C3-H), 7.83 (1H, d,
J = 12.0 Hz, C10-H), 4.99–4.76 (2H, m, C18-H and C5-H), 3.76–3.67 (3H, galactose C-H), 3.63–3.55 (2H, m, C19-H and galactose C-H), 3.54–3.40 (2H, m, C23-H
2), 3.47–3.17 (4H, m, C13-H
2 and C14-H
2), 3.03–2.83 (2H, m, C7-H
2), 2.21–196 (2H, m, C6-H
2), 2.00–1.54 (4H, m, C15-H
2 and C17-H
2), 1.41 (3H, d,
J= 5.0 Hz, C12-H
3).
13C NMR (100 MHz, DMSO-d6) δ: 166.08 (Carboxyl C=O), 147.25 (C3), 133.50 (Aromatic C), 108.87 (C10), 106.28 (Aromatic C), 97.64 (C18), 71.45, 69.55, 68.87, 68.24 (galactose C), 60.57 (C23), 57.06 (C5), 33.08 (C15 and C17), 24.76 (C7), 19.57 (C12), 18.51 (C6). FT-IR ν cm
−1: 3373.83 (m, alcohol OH), 2934.33 (w, carboxyl OH), 1708.29 (carboxylic or ester C=O), 1620.84 (aromatic C=C). MS + ESI found 523.2216 (MH
+) C
25H
32FN
2O
9 required 523.2092. HPLC analysis: retention time: 4.127 min, purity: 98.2%.
Nadifloxacin α-d-glucopyranoside (7b)
![Molecules 27 01504 i016]()
Yield (0.20 g, 27%), m.p. 162 °C,
1H NMR (400 MHz, DMSO-d6) δ: 9.01 (1H, s, C3-H), 7.90 (1H, d,
J = 12.0 Hz, C10-H), 5.03–4.83 (2H, m, C5-H and C18-H), 3.69 (1H, d,
J = 11.0 Hz, C23-Ha), 3.61–3.46 (3H, m, C20-H, C22-H, C23-Hb), 3.30–3.20 (1H, m, C19-H), 3.25–3.12 (1H, m, C6-Ha), 3.20–3.08 (1H, m, C21-H), 3.07–2.88 (1H, m, C6-Hb), 2.25–1.95 (2H, m, C7-H2), 2.07–1.60 (4H, m, C15-H
2 and C17-H
2), 1.48 (3H, d,
J= 7.0 Hz).
13C NMR (100 MHz, DMSO-d6) δ: 176.47 (carboxyl C=O), 166.08 (aromatic C), 147.40 (C3), 133.61 (aromatic C), 108.99 (C10), 106.44 (aromatic C), 97.28 (C18), 73.16 (C20), 73.05 (C22), 71.81 (C19), 70.37 (C21), 61.03 (C23), 57.07 (C5), 24.84 (C7), 19.57 (C12), 18.53 (C15 and C17). FT-IR ν cm
−1: 3359.86 (m, alcohol OH), 2928.52 (w, carboxyl OH), 1709.04 (carboxylic or ester C=O), 1621.75 (aromatic C=C). MS + ESI found 523.1920 (MH
+) C
25H
32FN
2O
9 required 523.2092. HPLC analysis: retention time: 5.028 min, purity 98.4%.
Determination of minimum inhibitory concentration
The glycosides (3a-e and 7a,b) and underivatised nadifloxacin (1) were individually dissolved in dimethyl sulfoxide, and amounts of the solutions were added to Tryptone Soya Agar (TSA) that had been autoclaved and then cooled to 50 °C to produce doubling concentrations of nadifloxacin (1) from 0.125–8 μg/mL and 1–128 μg/mL of the glycosides (3a-f and 7a,b). The agar was swirled to mix, and then plates were poured and dried. Specifically, a stock of nadifloxacin (1) was prepared in DMSO (1.6 mg/mL). Concentrations of 800, 400, 200, 100, 50 and 25 μg/mL were prepared by serial dilutions starting with addition of 0.5 mL of the 1.6 mg/mL solution into 0.5 mL of DMSO and so forth. An aliquot (100 μL) from each concentration was added individually to molten agar (20 mL) to give final agar concentrations of 8, 4, 2, 1, 0.5, 0.25 and 0.125 μg/mL. A stock of each nadifloxacin glycoside was prepared in DMSO (25.6 mg/mL). Concentrations of 12.8, 6.4, 3.2, 1.6, 0.8, 0.4 and 0.2 mg/mL were prepared by serial dilutions starting with addition of 0.5 mL of the 25.6 mg/mL solution into 0.5 mL of DMSO. A nadifloxacin glycoside concentration of 20 μg/mL was prepared by diluting 100 μL of concentration 0.2 mg/mL into 900 μL of DMSO. An aliquot (100 μL) from each concentration was added individually to molten agar (20 mL) to give final agar concentrations of 128, 64, 32, 16, 8, 4, 2, 1 and 0.1 μg/mL. A plate containing no nadifloxacin glycoside was used as control. Plates were cooled and dried before use. A plate containing no nadifloxacin was used as control. Plates were cooled and dried before use.
Cultures of bacteria were grown overnight in Nutrient Broth 2 (NB2) at 37 °C from beads stored at −80 °C. Cultures were then diluted decimally in Maximum Recovery Diluent (MRD) to approximately 106 CFU/mL, and 300 µL of each organism added to the wells of a multipoint inoculator (“Oxoid Cathra Replicator”, Thermofisher Scientific). Pins of the inoculator then dispensed amounts of bacterial suspension onto the surface of the agar plates. Plates were incubated at 37 °C and inspected after 18 h for growth to obtain cultures in the log phase, wherein cells were actively dividing. The lowest amount of antibacterial agent that totally inhibited growth was recorded as the MIC for that organism.



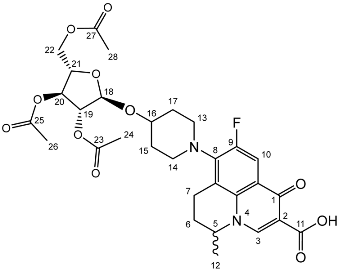

 Yield (14.8 g, 69%), m.p. 102–104 °C, isolated as a mixture of anomers, α:β 1:1.59, 1H NMR (400 MHz, CDCl3) δ: 7.38–7.22 (m, benzyl C-H), 6.38 (d, J = 3.5 Hz, α-C1-H), 5.57 (d, J = 8.0 Hz, β-C1-H), 4.95 (d, J = 11.5 Hz, α-benzyl CH2), 4.94 (d, J = 11.5 Hz, β-benzyl CH2), 4.84 (d, J = 11.5 Hz, β-benzyl CH2), 4.82 (d, J = 11.5 Hz, α-benzyl CH2), 4.74 (d, J = 11.5 Hz, α-benzyl CH2), 4.75–4.69 (m, benzyl CH2), 4.62 (d, J = 11.5 Hz, β-benzyl CH2), 4.57 (d, J = 11.5 Hz, α-benzyl CH2), 4.46 (d, J = 11.5 Hz, α-benzyl CH2), 4.43 (d, J = 11.5 Hz, β-benzyl CH2), 4.39 (d, J = 11.5 Hz, α-benzyl CH2), 4.38 (d, J = 11.5 Hz, β-benzyl CH2), 4.16 (dd, J = 10.0, 3.5 Hz, α-C2-H), 4.06–3.99 (m, C5-H), 3.94 (m, β-C2-H), 3.89 (dd, J = 10.0, 2.5 Hz, α-C3-H), 3.72–3.66 (m, β-C4-H), 2.11 (s, α-C8-H3), 2.03 (s, β-C8-H3). 13C NMR (100 MHz, DMSO-d6) δ: 169.63, 169.41 (carboxyl C=O), 138.64, 138.52, 138.42, 138.22, 138.06, 137.80, 137.77 (benzyl ipso C), 128.63–127.36 (benzyl CH), 94.30 (β-C1), 90.81 (α-C1), 82.42 (α-C5), 78.61 (α-C3), 78.19 (β-C2), 75.42 (α-C2), 75.35, 74.95, 74.72 (benzyl CH2), 74.08 (β-C4), 73.61, 73.54, 73.40, 73.40 (benzyl CH2), 73.04 (β-C5), 72.89 (benzyl CH2), 68.41 (α-C6), 67.94 (β-C6), 21.24 (α-C8), 21.06 (β-C8). FT-IR ν cm−1: 3059.70, 3026.72 (w, aromatic C-H), 1747.65 (m, ester C=O), 1081.32, 1045.51, 1024.29 (s, ether C-O). MS + ESI found 621.3333 (M + K+) C36H38KO7 required 621.2249.
Yield (14.8 g, 69%), m.p. 102–104 °C, isolated as a mixture of anomers, α:β 1:1.59, 1H NMR (400 MHz, CDCl3) δ: 7.38–7.22 (m, benzyl C-H), 6.38 (d, J = 3.5 Hz, α-C1-H), 5.57 (d, J = 8.0 Hz, β-C1-H), 4.95 (d, J = 11.5 Hz, α-benzyl CH2), 4.94 (d, J = 11.5 Hz, β-benzyl CH2), 4.84 (d, J = 11.5 Hz, β-benzyl CH2), 4.82 (d, J = 11.5 Hz, α-benzyl CH2), 4.74 (d, J = 11.5 Hz, α-benzyl CH2), 4.75–4.69 (m, benzyl CH2), 4.62 (d, J = 11.5 Hz, β-benzyl CH2), 4.57 (d, J = 11.5 Hz, α-benzyl CH2), 4.46 (d, J = 11.5 Hz, α-benzyl CH2), 4.43 (d, J = 11.5 Hz, β-benzyl CH2), 4.39 (d, J = 11.5 Hz, α-benzyl CH2), 4.38 (d, J = 11.5 Hz, β-benzyl CH2), 4.16 (dd, J = 10.0, 3.5 Hz, α-C2-H), 4.06–3.99 (m, C5-H), 3.94 (m, β-C2-H), 3.89 (dd, J = 10.0, 2.5 Hz, α-C3-H), 3.72–3.66 (m, β-C4-H), 2.11 (s, α-C8-H3), 2.03 (s, β-C8-H3). 13C NMR (100 MHz, DMSO-d6) δ: 169.63, 169.41 (carboxyl C=O), 138.64, 138.52, 138.42, 138.22, 138.06, 137.80, 137.77 (benzyl ipso C), 128.63–127.36 (benzyl CH), 94.30 (β-C1), 90.81 (α-C1), 82.42 (α-C5), 78.61 (α-C3), 78.19 (β-C2), 75.42 (α-C2), 75.35, 74.95, 74.72 (benzyl CH2), 74.08 (β-C4), 73.61, 73.54, 73.40, 73.40 (benzyl CH2), 73.04 (β-C5), 72.89 (benzyl CH2), 68.41 (α-C6), 67.94 (β-C6), 21.24 (α-C8), 21.06 (β-C8). FT-IR ν cm−1: 3059.70, 3026.72 (w, aromatic C-H), 1747.65 (m, ester C=O), 1081.32, 1045.51, 1024.29 (s, ether C-O). MS + ESI found 621.3333 (M + K+) C36H38KO7 required 621.2249.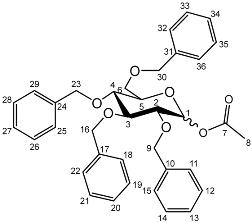 Yield (18.0 g, 83%). Isolated as a mixture of anomers, α:β 1:1.23, 1H NMR (400 MHz, CDCl3) δ: 7.38–7.22 (m, benzyl C-H), 7.16–7.10 (m, benzyl C-H), 6.35 (d, J = 3.5 Hz, α-C1-H), 5.60 (d, J = 8.0 Hz, β-C1-H), 4.99–4.44 (m, benzyl CH2), 3.99–3.90 (m, glucose H), 3.89–3.83 (m, glucose H), 3.79–3.53 (m, α-, β-C2-H and α-, β-C6-H2), 2.13 (s, α-C8-H3), 2.05 (s, β-C8-H3). 13C NMR (100 MHz, DMSO-d6) δ: 169.47, 169.31 (C7), 138.64, 138.11, 138.02, 137.89, 137.82, 137.60 (aromatic C), 128.58–127.59 (aromatic C-H), 94.05 (α-C1), 84.82, 81.68, 81.07, 78.88, 77.22, 76.95 (glucose C), 75.72 (benzyl CH2), 75.40 (glucose C), 75.30, 75.04, 73.55, 73.22 (benzyl CH2), 72.83 (glucose C), 68.09 (C6), 21.10 (C8). FT-IR ν cm−1: 3088.08, 3062.95, 3030.18 (w, aromatic C-H), 1755.44 (m, ester C=O), 1072.79, 1027.24 (s, ether C-O). MS-ESI found 621.5834 (M + K+) KC36H38O7 required 621.2249.
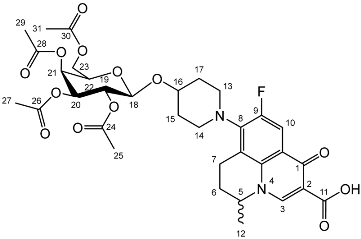
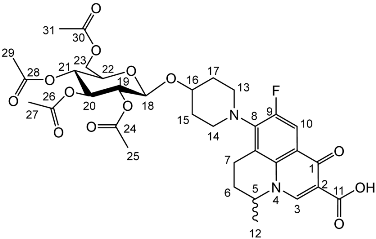
Yield (18.0 g, 83%). Isolated as a mixture of anomers, α:β 1:1.23, 1H NMR (400 MHz, CDCl3) δ: 7.38–7.22 (m, benzyl C-H), 7.16–7.10 (m, benzyl C-H), 6.35 (d, J = 3.5 Hz, α-C1-H), 5.60 (d, J = 8.0 Hz, β-C1-H), 4.99–4.44 (m, benzyl CH2), 3.99–3.90 (m, glucose H), 3.89–3.83 (m, glucose H), 3.79–3.53 (m, α-, β-C2-H and α-, β-C6-H2), 2.13 (s, α-C8-H3), 2.05 (s, β-C8-H3). 13C NMR (100 MHz, DMSO-d6) δ: 169.47, 169.31 (C7), 138.64, 138.11, 138.02, 137.89, 137.82, 137.60 (aromatic C), 128.58–127.59 (aromatic C-H), 94.05 (α-C1), 84.82, 81.68, 81.07, 78.88, 77.22, 76.95 (glucose C), 75.72 (benzyl CH2), 75.40 (glucose C), 75.30, 75.04, 73.55, 73.22 (benzyl CH2), 72.83 (glucose C), 68.09 (C6), 21.10 (C8). FT-IR ν cm−1: 3088.08, 3062.95, 3030.18 (w, aromatic C-H), 1755.44 (m, ester C=O), 1072.79, 1027.24 (s, ether C-O). MS-ESI found 621.5834 (M + K+) KC36H38O7 required 621.2249.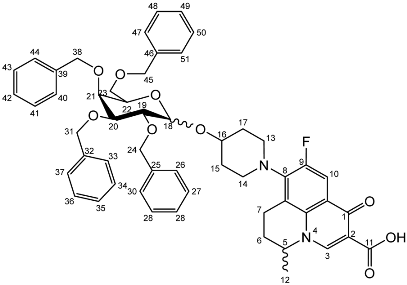 Yield (6.83 g, 56%). m.p. 61–65 °C, isolated as a mixture of anomers, α:β 3:5, 1H NMR (400 MHz, CDCl3) δ: 8.63 (1H, s, C3-H), 7.93 (1H, d, J = 12.0 Hz, C10-H), 7.36–7.15 (m, benzyl C-H), 4.98–4.94 (m, α-C18-H), 4.94–4.84 (m, benzyl CH2), 4.83–4.46 (m, benzyl CH2, C5-H), 4.44 (d, J = 8.5 Hz, β-C18-H), 4.41–4.30 (m, benzyl CH2), 4.03–3.95 (m, α-C19-H and C22-H), 3.95–3.88 (C21-H), 3.82 (d, J = 9.5 Hz, C22-H), 3.77 (d, J = 9.0 Hz, β-C19-H), 3.52–3.44 (m, C20-H and C23-H2), 3.21 (d, J = 17.0 Hz, C7-Ha), 3.03–2.90 (m, C16-H), 2.85–2.67 (m, C7-Hb), 2.15–2.03 (m, C6-H2), 2.03–1.59 (m, C15-H2 and C17-H2), 1.48–1.39 (m, C12-H3). 13C NMR (100 MHz, CDCl3) δ: 167.16 (carboxyl C=O), 146.47 (C3), 128.62–127.27 (aromatic C), 125.29 (aromatic C), 110.72 (C10), 101.92 (β-C18), 5.83 (α-C18), 82.12 (C20), 79.77 (β-C19), 79.27 (C21), 76.06 (α-C19), 75.32 (benzyl CH2), 75.03 (C21), 74.77 (benzyl CH2), 74.53 (benzyl CH2), 73.94 (C20), 73.65 (C22) 73.13 (benzyl CH2), 69.78 (C22), 69.31, 69.01 (C23), 32.64 (C15, C17), 26.04 (C6), 20.25 (C12), 18.93 (C7). FT-IR ν cm−1: 3061.62, 3029.55 (w, aromatic C-H), 2920.54, 2861.87 (w, carboxylic C-OH), 1723.88 (m, carbonyl C=O), 1619 (m, C=C), 1490 (s, arene C-C), MS +ESI found 883.3593 (MH+) C53H56FN2O9 expected 883.3970.
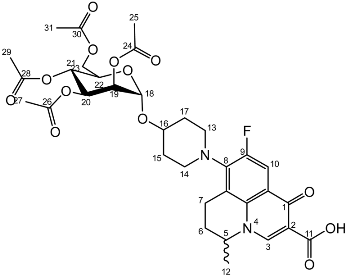
Yield (6.83 g, 56%). m.p. 61–65 °C, isolated as a mixture of anomers, α:β 3:5, 1H NMR (400 MHz, CDCl3) δ: 8.63 (1H, s, C3-H), 7.93 (1H, d, J = 12.0 Hz, C10-H), 7.36–7.15 (m, benzyl C-H), 4.98–4.94 (m, α-C18-H), 4.94–4.84 (m, benzyl CH2), 4.83–4.46 (m, benzyl CH2, C5-H), 4.44 (d, J = 8.5 Hz, β-C18-H), 4.41–4.30 (m, benzyl CH2), 4.03–3.95 (m, α-C19-H and C22-H), 3.95–3.88 (C21-H), 3.82 (d, J = 9.5 Hz, C22-H), 3.77 (d, J = 9.0 Hz, β-C19-H), 3.52–3.44 (m, C20-H and C23-H2), 3.21 (d, J = 17.0 Hz, C7-Ha), 3.03–2.90 (m, C16-H), 2.85–2.67 (m, C7-Hb), 2.15–2.03 (m, C6-H2), 2.03–1.59 (m, C15-H2 and C17-H2), 1.48–1.39 (m, C12-H3). 13C NMR (100 MHz, CDCl3) δ: 167.16 (carboxyl C=O), 146.47 (C3), 128.62–127.27 (aromatic C), 125.29 (aromatic C), 110.72 (C10), 101.92 (β-C18), 5.83 (α-C18), 82.12 (C20), 79.77 (β-C19), 79.27 (C21), 76.06 (α-C19), 75.32 (benzyl CH2), 75.03 (C21), 74.77 (benzyl CH2), 74.53 (benzyl CH2), 73.94 (C20), 73.65 (C22) 73.13 (benzyl CH2), 69.78 (C22), 69.31, 69.01 (C23), 32.64 (C15, C17), 26.04 (C6), 20.25 (C12), 18.93 (C7). FT-IR ν cm−1: 3061.62, 3029.55 (w, aromatic C-H), 2920.54, 2861.87 (w, carboxylic C-OH), 1723.88 (m, carbonyl C=O), 1619 (m, C=C), 1490 (s, arene C-C), MS +ESI found 883.3593 (MH+) C53H56FN2O9 expected 883.3970.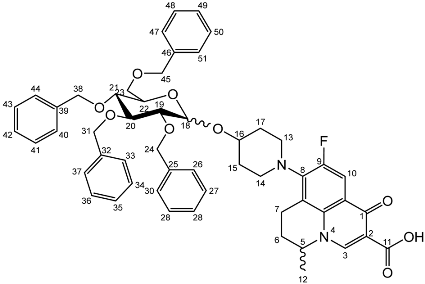 Yield (1.75 g, 14%), 1H NMR (400 MHz, CDCl3) δ: 8.69 (s, C3-H), 8.00 (d, J = 12.0 Hz, C10-H), 7.41–7.21 (m, benzyl H), 5.06–4.96 (m, α-C18-H, benzyl CH2), 4.95–4.72 (m, benzyl CH2), 4.71–4.44 (benzyl CH2, β-C18-H, C5-H), 4.03 (at, J = 9.5 Hz, α-C20-H), 3.94–3.87 (m, glucose H), 3.78–3.71 (m, C23-Ha), 3.71–3.44 (m, C23-Hb, α-C19-H, β-C19-H, glucose H), 3.38–3.16 (m, C7-Ha), 3.15–3.00 (m, C13-H2 or C14-H2), 2.93–2.75 (m, C7-Hb), 2.11–1.64 (m, C17-H2, C15-H2), 1.53 (d, J = 6.5 Hz, C12-H3), 1.52 (d, J = 6.5 Hz, C12-H3).13C NMR (100 MHz, CDCl3) δ: 177.14, 167.35 (carbonyl C=O), 110.77, 110.49 (C10), 101.88 (β-C18), 95.13 (α-C18), 84.52, (glucose C), 82.29 (β-C19), 82.04 (α-C20), 79.98, 77.88 (glucose C), 75.69, 75.25, 75.04, 74.95 (benzyl CH2), 74.82 (β-glucose C), 73.50, 73.42, 73.33, 73.25 (benzyl CH2), 70.51 (glucose C), 68.62 (C23), 57.95 (C5), 33.12 (C17, C15), 30.97 (C6), 20.23 (C12), 18.83 (C7). FT-IR ν cm−1: 3062.43, 3028.98 (w, aromatic C-H), 2900.68, 2865.08 (w, carboxylic C-OH), 1618.42 (m, C=C), 1452 (s, arene C-C), 1067.21, 1027.67 (s, C-O). MS + ESI found 883.3850 (MH+) C53H56FN2O9 expected 883.3970.
Yield (1.75 g, 14%), 1H NMR (400 MHz, CDCl3) δ: 8.69 (s, C3-H), 8.00 (d, J = 12.0 Hz, C10-H), 7.41–7.21 (m, benzyl H), 5.06–4.96 (m, α-C18-H, benzyl CH2), 4.95–4.72 (m, benzyl CH2), 4.71–4.44 (benzyl CH2, β-C18-H, C5-H), 4.03 (at, J = 9.5 Hz, α-C20-H), 3.94–3.87 (m, glucose H), 3.78–3.71 (m, C23-Ha), 3.71–3.44 (m, C23-Hb, α-C19-H, β-C19-H, glucose H), 3.38–3.16 (m, C7-Ha), 3.15–3.00 (m, C13-H2 or C14-H2), 2.93–2.75 (m, C7-Hb), 2.11–1.64 (m, C17-H2, C15-H2), 1.53 (d, J = 6.5 Hz, C12-H3), 1.52 (d, J = 6.5 Hz, C12-H3).13C NMR (100 MHz, CDCl3) δ: 177.14, 167.35 (carbonyl C=O), 110.77, 110.49 (C10), 101.88 (β-C18), 95.13 (α-C18), 84.52, (glucose C), 82.29 (β-C19), 82.04 (α-C20), 79.98, 77.88 (glucose C), 75.69, 75.25, 75.04, 74.95 (benzyl CH2), 74.82 (β-glucose C), 73.50, 73.42, 73.33, 73.25 (benzyl CH2), 70.51 (glucose C), 68.62 (C23), 57.95 (C5), 33.12 (C17, C15), 30.97 (C6), 20.23 (C12), 18.83 (C7). FT-IR ν cm−1: 3062.43, 3028.98 (w, aromatic C-H), 2900.68, 2865.08 (w, carboxylic C-OH), 1618.42 (m, C=C), 1452 (s, arene C-C), 1067.21, 1027.67 (s, C-O). MS + ESI found 883.3850 (MH+) C53H56FN2O9 expected 883.3970.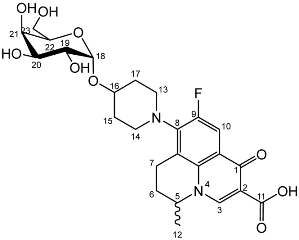 Yield (1.25 g, 33%), m.p. 143–150 °C, 1H NMR (400 MHz, DMSO-d6) δ: 8.94 (1H, s, C3-H), 7.83 (1H, d, J = 12.0 Hz, C10-H), 4.99–4.76 (2H, m, C18-H and C5-H), 3.76–3.67 (3H, galactose C-H), 3.63–3.55 (2H, m, C19-H and galactose C-H), 3.54–3.40 (2H, m, C23-H2), 3.47–3.17 (4H, m, C13-H2 and C14-H2), 3.03–2.83 (2H, m, C7-H2), 2.21–196 (2H, m, C6-H2), 2.00–1.54 (4H, m, C15-H2 and C17-H2), 1.41 (3H, d, J= 5.0 Hz, C12-H3). 13C NMR (100 MHz, DMSO-d6) δ: 166.08 (Carboxyl C=O), 147.25 (C3), 133.50 (Aromatic C), 108.87 (C10), 106.28 (Aromatic C), 97.64 (C18), 71.45, 69.55, 68.87, 68.24 (galactose C), 60.57 (C23), 57.06 (C5), 33.08 (C15 and C17), 24.76 (C7), 19.57 (C12), 18.51 (C6). FT-IR ν cm−1: 3373.83 (m, alcohol OH), 2934.33 (w, carboxyl OH), 1708.29 (carboxylic or ester C=O), 1620.84 (aromatic C=C). MS + ESI found 523.2216 (MH+) C25H32FN2O9 required 523.2092. HPLC analysis: retention time: 4.127 min, purity: 98.2%.
Yield (1.25 g, 33%), m.p. 143–150 °C, 1H NMR (400 MHz, DMSO-d6) δ: 8.94 (1H, s, C3-H), 7.83 (1H, d, J = 12.0 Hz, C10-H), 4.99–4.76 (2H, m, C18-H and C5-H), 3.76–3.67 (3H, galactose C-H), 3.63–3.55 (2H, m, C19-H and galactose C-H), 3.54–3.40 (2H, m, C23-H2), 3.47–3.17 (4H, m, C13-H2 and C14-H2), 3.03–2.83 (2H, m, C7-H2), 2.21–196 (2H, m, C6-H2), 2.00–1.54 (4H, m, C15-H2 and C17-H2), 1.41 (3H, d, J= 5.0 Hz, C12-H3). 13C NMR (100 MHz, DMSO-d6) δ: 166.08 (Carboxyl C=O), 147.25 (C3), 133.50 (Aromatic C), 108.87 (C10), 106.28 (Aromatic C), 97.64 (C18), 71.45, 69.55, 68.87, 68.24 (galactose C), 60.57 (C23), 57.06 (C5), 33.08 (C15 and C17), 24.76 (C7), 19.57 (C12), 18.51 (C6). FT-IR ν cm−1: 3373.83 (m, alcohol OH), 2934.33 (w, carboxyl OH), 1708.29 (carboxylic or ester C=O), 1620.84 (aromatic C=C). MS + ESI found 523.2216 (MH+) C25H32FN2O9 required 523.2092. HPLC analysis: retention time: 4.127 min, purity: 98.2%.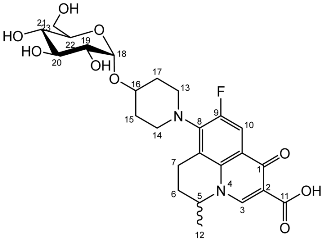 Yield (0.20 g, 27%), m.p. 162 °C, 1H NMR (400 MHz, DMSO-d6) δ: 9.01 (1H, s, C3-H), 7.90 (1H, d, J = 12.0 Hz, C10-H), 5.03–4.83 (2H, m, C5-H and C18-H), 3.69 (1H, d, J = 11.0 Hz, C23-Ha), 3.61–3.46 (3H, m, C20-H, C22-H, C23-Hb), 3.30–3.20 (1H, m, C19-H), 3.25–3.12 (1H, m, C6-Ha), 3.20–3.08 (1H, m, C21-H), 3.07–2.88 (1H, m, C6-Hb), 2.25–1.95 (2H, m, C7-H2), 2.07–1.60 (4H, m, C15-H2 and C17-H2), 1.48 (3H, d, J= 7.0 Hz). 13C NMR (100 MHz, DMSO-d6) δ: 176.47 (carboxyl C=O), 166.08 (aromatic C), 147.40 (C3), 133.61 (aromatic C), 108.99 (C10), 106.44 (aromatic C), 97.28 (C18), 73.16 (C20), 73.05 (C22), 71.81 (C19), 70.37 (C21), 61.03 (C23), 57.07 (C5), 24.84 (C7), 19.57 (C12), 18.53 (C15 and C17). FT-IR ν cm−1: 3359.86 (m, alcohol OH), 2928.52 (w, carboxyl OH), 1709.04 (carboxylic or ester C=O), 1621.75 (aromatic C=C). MS + ESI found 523.1920 (MH+) C25H32FN2O9 required 523.2092. HPLC analysis: retention time: 5.028 min, purity 98.4%.
Yield (0.20 g, 27%), m.p. 162 °C, 1H NMR (400 MHz, DMSO-d6) δ: 9.01 (1H, s, C3-H), 7.90 (1H, d, J = 12.0 Hz, C10-H), 5.03–4.83 (2H, m, C5-H and C18-H), 3.69 (1H, d, J = 11.0 Hz, C23-Ha), 3.61–3.46 (3H, m, C20-H, C22-H, C23-Hb), 3.30–3.20 (1H, m, C19-H), 3.25–3.12 (1H, m, C6-Ha), 3.20–3.08 (1H, m, C21-H), 3.07–2.88 (1H, m, C6-Hb), 2.25–1.95 (2H, m, C7-H2), 2.07–1.60 (4H, m, C15-H2 and C17-H2), 1.48 (3H, d, J= 7.0 Hz). 13C NMR (100 MHz, DMSO-d6) δ: 176.47 (carboxyl C=O), 166.08 (aromatic C), 147.40 (C3), 133.61 (aromatic C), 108.99 (C10), 106.44 (aromatic C), 97.28 (C18), 73.16 (C20), 73.05 (C22), 71.81 (C19), 70.37 (C21), 61.03 (C23), 57.07 (C5), 24.84 (C7), 19.57 (C12), 18.53 (C15 and C17). FT-IR ν cm−1: 3359.86 (m, alcohol OH), 2928.52 (w, carboxyl OH), 1709.04 (carboxylic or ester C=O), 1621.75 (aromatic C=C). MS + ESI found 523.1920 (MH+) C25H32FN2O9 required 523.2092. HPLC analysis: retention time: 5.028 min, purity 98.4%.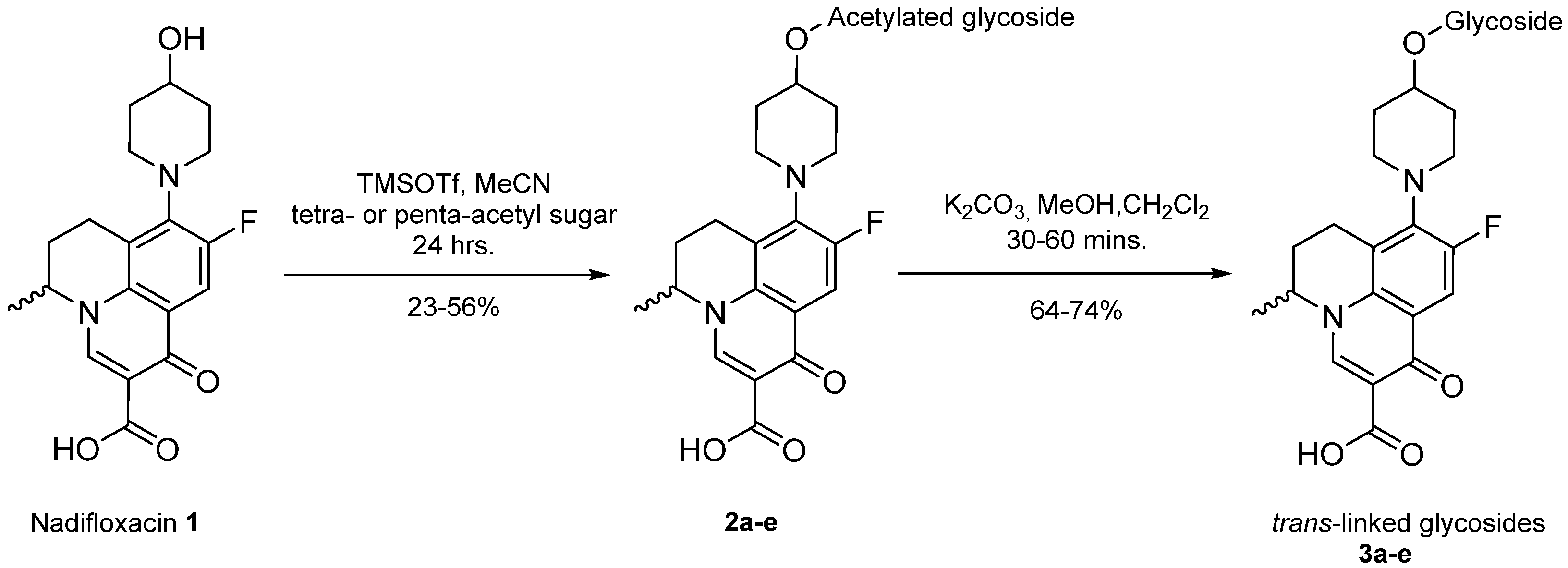

{kind=link}
{kind=link}
{kind=link}