
Activation of the Nrf2 Pathway as a Therapeutic Strategy for ALS Treatment



Abstract
:1. Introduction
2. Motoneurons Vulnerability to Oxidative Stress
3. Antioxidant Therapy in ALS
4. Nrf2 Involvement in ALS
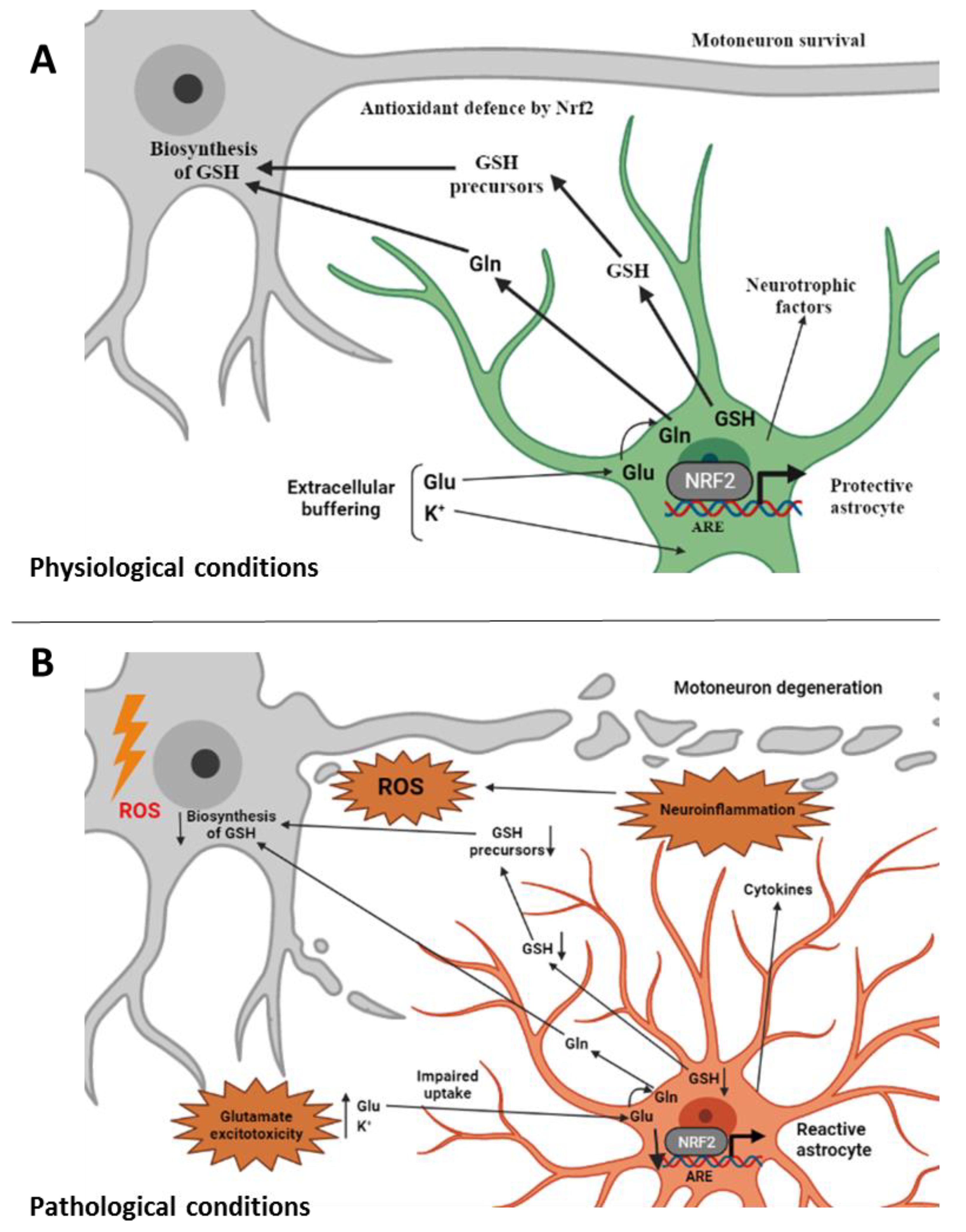
5. Role of Glial Cells in ALS
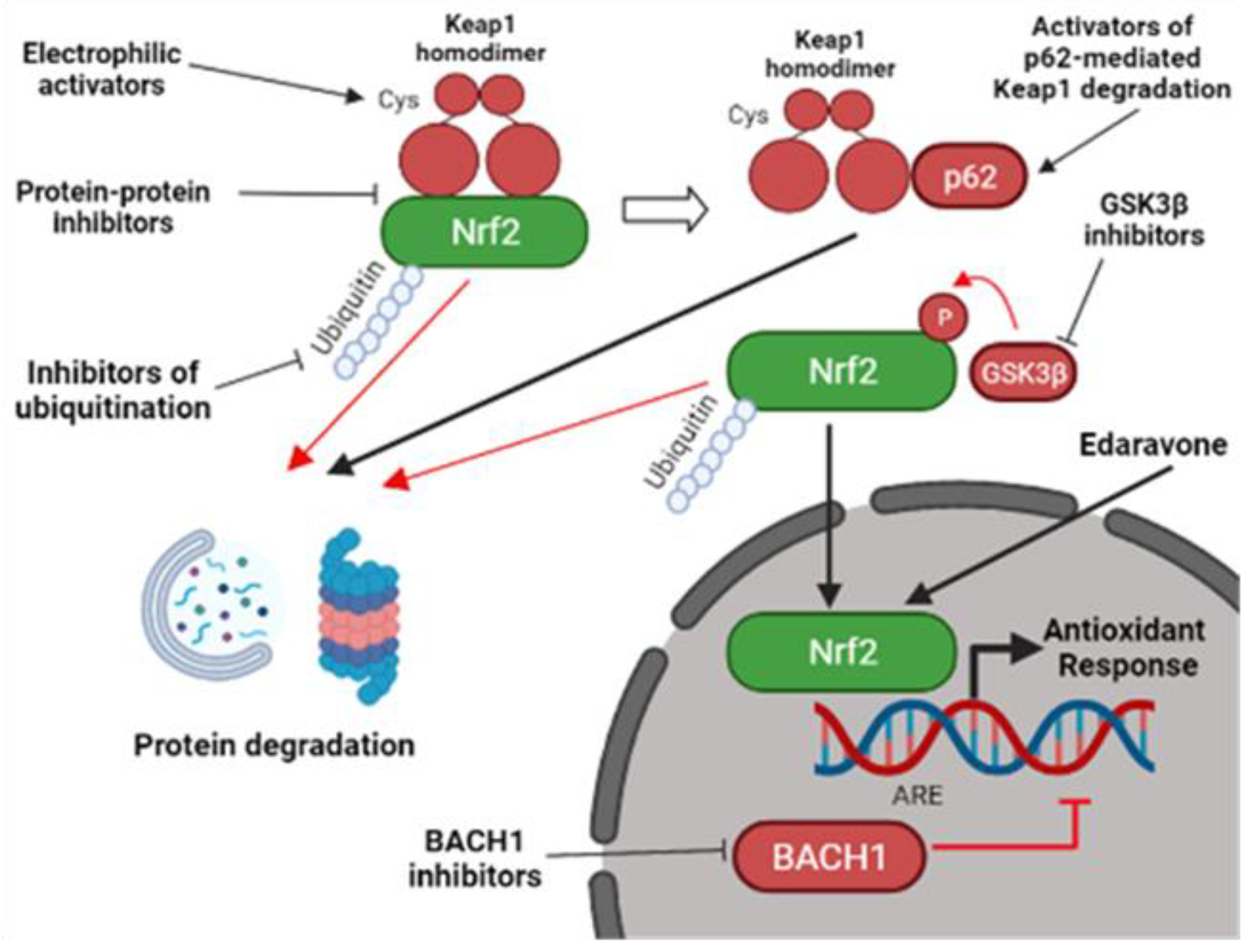
6. Nrf2-Targeted Pharmacological Approach
7. Electrophilic Inhibitors of Keap1
8. Protein–Protein Interaction (PPI) Inhibitors of the Keap1-Nrf2 Complex
9. Other Ways for Nrf2 Activation
10. Issues in Drug Discovery Related to ALS
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bono, S.; Feligioni, M.; Corbo, M. Impaired antioxidant KEAP1-NRF2 system in amyotrophic lateral sclerosis: NRF2 activation as a potential therapeutic strategy. Mol. Neurodegener. 2021, 16, 71. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Alsultan, A.A.; Waller, R.; Heath, P.R.; Kirby, J. The genetics of amyotrophic lateral sclerosis: Current insights. Degener Neurol Neuromuscul Dis. 2016, 6, 49–64. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Brites, D.; Vaz, A.R. Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front. Cell. Neurosci. 2014, 8, 117. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Talbot, K.; McDermott, C.J.; Hardiman, O.; Shefner, J.M.; Al-Chalabi, A.; Huynh, W.; Cudkowicz, M.; Talman, P.; et al. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2021, 17, 104–118. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calkins, M.J.; Johnson, D.A.; Townsend, J.A.; Vargas, M.R.; Dowell, J.A.; Williamson, T.P.; Kraft, A.D.; Lee, J.; Li, J.; Johnson, J.A. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid Redox Signal. 2009, 11, 497–508. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Antonucci, L.; Karin, M. 40th anniversary review article NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis 2020, 41, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Filograna, R.; Beltramini, M.; Bubacco, L.; Bisaglia, M. Anti-oxidants in parkinson’s disease therapy: A critical point of view. Curr. Neuropharmacol. 2016, 14, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddell, J.R. Are astrocytes the predominant cell type for activation of Nrf2 in aging and neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a therapeutic target in neurodegenerative diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef] [PubMed]
- Izrael, M.; Slutsky, S.G.; Revel, M. Rising stars: Astrocytes as a therapeutic target for ALS disease. Front. Neurosci. 2020, 14, 824. [Google Scholar] [CrossRef]
- Wang, T.; Tomas, D.; Perera, N.D.; Cuic, B.; Luikinga, S.; Viden, A.; Barton, S.K.; McLean, C.A.; Samson, A.L.; Southon, A.; et al. Ferroptosis mediates selective motor neuron death in amyotrophic lateral sclerosis. Cell Death Differ. in press. [CrossRef]
- Blasco, H.; Garcon, G.; Patin, F.; Veyrat-Durebex, C.; Boyer, J.; Devos, D.; Vourc’H, P.; Andres, C.R.; Corcia, P. Panel of oxidative stress and inflammatory biomarkers in ALS: A pilot study. Can. J. Neurol. Sci. 2017, 44, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, W.A.; Fu, W.; Keller, J.N.; Markesbery, W.R.; Appel, S.; Smith, R.G.; Kasarskis, E.; Mattson, M.P. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann. Neurol. 1998, 44, 819–824. [Google Scholar] [CrossRef]
- Weiduschat, N.; Mao, X.; Hupf, J.; Armstrong, N.; Kang, G.; Lange, D.J.; Mitsumoto, H.; Shungu, D.C. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci. Lett. 2014, 570, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Corp, P.; Henry, K.; Mattson, M.P.; Appel, S.H. Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann. Neurol. 1998, 44, 696–699. [Google Scholar] [CrossRef]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased lipid peroxidation in sera of ALS patients. A potential biomarker of disease burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef] [PubMed]
- Weishaupt, J.H.; Bartels, C.; Polking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Bohn, M.; Huther, G.; Schneider, A.; et al. Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J. Pineal Res. 2006, 41, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Chang, Y.; Kong, Q.; Shan, X.; Tian, G.; Ilieva, H.; Cleveland, D.W.; Rothstein, J.D.; Borchelt, D.R.; Wong, P.C.; Lin, C.G. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE 2008, 3, e2849. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Grubman, A.; Acevedo, K.; Yang, S.; Ke, Y.D.; Moujalled, D.M.; Duncan, C.; Caragounis, A.; Perera, N.D.; Turner, B.J.; et al. TDP-43 mutations causing amyotrophic lateral sclerosis are associated with altered expression of RNA-binding protein hnRNP K and affect the Nrf2 antioxidant pathway. Hum. Mol. Genet. 2017, 26, 1732–1746. [Google Scholar] [CrossRef]
- Johnson, D.A.; Johnson, J.A. Nrf2—a therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 2015, 88, 253–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Decreased glutathione accelerates neurologiCal deficit and mitochondrial pathology in familial ALS-linked hSOD1G93A mice model. Neurobiol. Dis. 2011, 43, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Dantas, L.S.; Viviani, L.G.; Inague, A.; Piccirillo, E.; de Rezende, L.F.; Ronsein, G.E.; Augusto, O.; de Medeiros, M.H.; Amaral, A.T.; Miyamoto, S. Lipid-derived electrophiles induce covalent modification and aggregation of Cu, Zn-superoxide dismutase in a hydrophobicity-dependent manner. Free Radic. Biol. Med. 2020, 156, 157–167. [Google Scholar] [CrossRef]
- Li, C.; Chen, T.; Zhou, H.; Zhang, C.; Feng, Y.; Tang, F.; Hoi, M.P.; He, C.; Zheng, Y.; Lee, S.L. Schisantherin A Attenuates Neuroinflammation in Activated Microglia: Role of Nrf2 Activation Through ERK Phosphorylation. Cell Physiol Biochem 2018, 47, 1769–1784. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.M.; Mirkovic, J.; Stanisz, Z.A.; Patwari, F.S.; Yang, W.S. NSC-34 motor neuron-like cells are sensitized to ferroptosis upon differentiation. FEBS Open Bio 2019, 9, 582–593. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, E.J.; Mattson, M.P. How does hormesis impact biology, toxicology, and medicine ? NPJ Aging Mech Dis. 2017, 3, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, T.; Adachi-Tominari, K.; Sano, O.; Kamei, T.; Nogami, M.; Ogi, K.; Okano, H.; Yano, M. Involvement of ferroptosis in human motor neuron cell death. Biochem. Biophys. Res. Commun. 2021, 566, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Shin, J.H.; Gwag, B.J.; Choi, E.J. Iron accumulation promotes TACE-mediated TNF- α secretion and neurodegeneration in a mouse model of ALS. Neurobiol. Dis. 2015, 80, 63–69. [Google Scholar] [CrossRef]
- Moreau, C.; Danel, V.; Devedjian, J.C.; Grolez, G.; Timmerman, K.; Laloux, C.; Petrault, M.; Gouel, F.; Jonneaux, A.; Dutheil, M.; et al. Could conservative iron chelation lead to neuroprotection in amyotrophic lateral sclerosis ? Antioxid. Redox Signal. 2018, 29, 742–748. [Google Scholar] [CrossRef] [Green Version]
- Kasarskis, E.J.; Tandon, L.; Love, M.A.; Ehmann, W.D. Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: A preliminary study. J. Neurol. Sci. 1995, 130, 203–208. [Google Scholar] [CrossRef]
- Kuraszkiewicz, B.; Goszczyńska, H.; Podsiadły-Marczykowska, T.; Piotrkiewicz, M.; Andersen, P.; Gromicho, M.; Grosskreutz, J.; Kuźma-Kozakiewicz, M.; Petri, S.; Stubbendorf, B.; et al. Potential Preventive Strategies for Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 428. [Google Scholar] [CrossRef]
- Silva, J.M.; Nobre, M.S.C.; Albino, S.L.; Lócio, L.L.; Nascimento, A.P.S.; Scotti, L.; Scotti, M.T.; Oshiro-Junior, J.A.; Lima, M.C.A.; Mendonça-Junior, F.; et al. Secondary metabolites with antioxidant activities for the putative treatment of amyotrophic lateral sclerosis (ALS): “Experimental evidences”. Oxid. Med. Cell. Longev. 2020, 2020, 5642029. [Google Scholar] [CrossRef]
- Orrell, R.W.; Lane, R.J.M.; Ross, M. A systematic review of antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease. Amyotroph. Lateral Scler. 2008, 9, 195–211. [Google Scholar] [CrossRef]
- Nieves, J.W.; Gennings, C.; Factor-Litvak, P.; Hupf, J.; Singleton, J.; Sharf, V.; Oskarsson, B.; Fernandes Filho, J.A.; Sorenson, E.J.; D’Amico, E.; et al. Association between dietary intake and function in amyotrophic lateral sclerosis. JAMA Neurol. 2016, 73, 1425–1432. [Google Scholar] [CrossRef] [Green Version]
- Petri, S.; Sonja, K.; Kiaei, M. Nrf2/ARE signaling pathway: Key mediator in oxidative stress and potential therapeutic target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef]
- Ahmadi, M.; Agah, E.; Nafissi, S.; Jaafari, M.R.; Harirchian, M.H.; Sarraf, P.; Faghihi-Kashani, S.; Hosseini, S.J.; Ghoreishi, A.; Aghamollaii, V.; et al. Safety and efficacy of nanocurcumin as add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A pilot randomized clinical trial. Neurotherapeutics 2018, 15, 430–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louwerse, E.S.; Weverling, G.J.; Bossuyt, P.M.; Meyjes, F.E.; de Jong, J.M. Randomized, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch. Neurol. 1995, 52, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Cucatto, A.; Terreni, A.A.; Schiffer, D. Reduced glutathione in amyotrophic lateral sclerosis: An open, crossover, randomized trial. Ital. J. Neurol. Sci. 1998, 19, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Y.; Rayner, S.L.; Chung, R.; Shi, B.Y.; Liang, X.J. Advances in nanotechnology-based strategies for the treatments of amyotrophic lateral sclerosis. Mater. Today Bio 2020, 6, 100055. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Porras, G.; Arribas-Blázquez, M.; Maestro, I.; Borrego-Hernández, D.; Boya, P.; Cerdán, S.; García-Redondo, A.; Martínez, A.; Martin-Requero, Á. Molecular alterations in sporadic and sod1-als immortalized lymphocytes: Towards a personalized therapy. Int. J. Mol. Sci. 2021, 22, 3007. [Google Scholar] [CrossRef] [PubMed]
- Pall, M.L.; Levine, S. Nrf2, a master regulator of detoxification and also antioxidant, anti-inflammatory and other cytoprotective mechanisms, is raised by health promoting factors. Sheng Li Xue Bao 2015, 67, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.V.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pehar, M.; Vargas, M.R.; Robinson, K.M.; Cassina, P.; Díaz-Amarilla, P.J.; Hagen, T.M.; Radi, R.; Barbeito, L.; Beckman, J.S. Mitochondrial superoxide production and nuclear factor erythroid 2-related factor 2 activation in p75 neurotrophin receptor-induced motor neuron apoptosis. J. Neurosci. 2007, 27, 7777–7785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanou, A.; Higginbottom, A.; Valori, C.F.; Wyles, M.; Ning, K.; Shaw, P.; Azzouz, M. Viral delivery of antioxidant genes as a therapeutic strategy in experimental models of amyotrophic lateral sclerosis. Mol. Ther. 2013, 21, 1486–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardo, G.; Iennaco, R.; Fusi, N.; Heath, P.R.; Marino, M.; Trolese, M.C.; Ferraiuolo, L.; Lawrence, N.; Shaw, P.J.; Bendotti, C. Transcriptomic indices of fast and slow disease progression in two mouse models of amyotrophic lateral sclerosis. Brain 2013, 136, 3305–3332. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhang, Y.; Wen, D.; Duan, W.; An, T.; Shi, P.; Wang, J.; Li, Z.; Chen, X.; Li, C. The modest impact of transcription factor Nrf2 on the course of disease in an ALS animal model. Lab. Investig. 2013, 93, 825–833. [Google Scholar] [CrossRef]
- Vargas, M.R.; Burton, N.C.; Kutzke, J.; Gan, L.; Johnson, D.A.; Schäfer, M.; Werner, S.; Johnson, J.A. Absence of Nrf2 or its selective overexpression in neurons and muscle does not affect survival in ALS- linked mutant hSOD1 mouse models. PLoS ONE 2013, 8, e56625. [Google Scholar] [CrossRef]
- Hadano, S.; Mitsui, S.; Pan, L.; Otomo, A.; Kubo, M.; Sato, K.; Ono, S.; Onodera, W.; Abe, K.; Chen, X.; et al. Functional links between SQSTM1 and ALS2 in the pathogenesis of ALS: Cumulative impact on the protection against mutant SOD1-mediated motor dysfunction in mice. Hum. Mol. Genet. 2016, 25, 3321–3340. [Google Scholar] [CrossRef] [Green Version]
- Van Harten, A.C.M.; Phatnani, H.; Przedborski, S. Non-cell-autonomous pathogenic mechanisms in amyotrophic lateral sclerosis. Trends Neurosci. 2021, 44, 658–668. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited ALS. Nat. Neurosci. 2011, 11, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [Green Version]
- Kim, K. Glutathione in the nervous system as a potential therapeutic target to control the development and progression of amyotrophic lateral sclerosis. Antioxidants 2021, 10, 1011. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Pehar, M.; Cassina, P.; Beckman, J.S.; Barbeito, L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75 NTR -dependent motor neuron apoptosis. J. Neurochem. 2006, 97, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Andronesi, O.C.; Nicholson, K.; Jafari-Khouzani, K.; Bogner, W.; Wang, J.; Chan, J.; Macklin, E.A.; Levine-Weinberg, M.; Breen, C.; Schwarzschild, M.A.; et al. Imaging neurochemistry and brain structure tracks clinical decline and mechanisms of ALS in patients. Front. Neurol. 2020, 11, 1575. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K. Glutathione in the brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef]
- Mimoto, T.; Miyazaki, K.; Morimoto, N.; Kurata, T.; Satoh, K.; Ikeda, Y.; Abe, K. Impaired antioxydative Keap1/Nrf2 system and the downstream stress protein responses in the motor neuron of ALS model mice. Brain Res. 2012, 1446, 109–118. [Google Scholar] [CrossRef]
- Kraft, A.D.; Resch, J.M.; Johnson, D.A.; Johnson, J.A. Activation of the Nrf2-ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp. Neurol. 2007, 207, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, M.R.; Pehar, M.; Cassina, P.; Martínez-Palma, L.; Thompson, J.A.; Beckman, J.S.; Barbeito, L. Fibroblast growth factor-1 induces heme oxygenase-1 via nuclear factor erythroid 2-related factor 2 (Nrf2) in spinal cord astrocytes. J. Biol. Chem. 2005, 280, 25571–25579. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, P.; Rodriguez-Muela, N.; Klim, J.R.; de Boer, A.S.; Agrawal, S.; Sandoe, J.; Lopes, C.S.; Ogliari, K.S.; Williams, L.A.; Shear, M.; et al. Reactive astrocytes promote ALS-like degeneration and intracellular protein aggregation in human motor neurons by disrupting autophagy through TGF-β1. Stem Cell Rep. 2017, 9, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.N.; Gochenaur, L.; Singh, A.; Grant, R.; Patel, K.; Watkins, S.; Wu, J.Y.; Pandey, U.B. Traumatic injury induces stress granule formation and enhances motor dysfunctions in ALS/FTD models. Hum. Mol. Genet. 2018, 27, 1366–1381. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Buttari, B.; Profumo, E.; Tucci, P.; Saso, L. A perspective on Nrf2 signaling pathway for neuroinflammation: A potential therapeutic target in alzheimer’s and parkinson’s diseases. Front. Cell. Neurosci. 2022, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Boas, S.M.; Joyce, K.L.; Cowell, R.M. The NRF2-dependent transcriptional regulation of antioxidant defense pathways: Relevance for cell type-specific vulnerability to neurodegeneration and therapeutic intervention. Antioxidants 2021, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Traiffort, E.; Morisset-lopez, S.; Moussaed, M.; Zahaf, A. Defective oligodendroglial lineage and demyelination in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2021, 22, 3426. [Google Scholar] [CrossRef] [PubMed]
- Alhindi, A.; Boehm, I.; Chaytow, H. Small junction, big problems: Neuromuscular junction pathology in mouse models of amyotrophic lateral sclerosis (ALS). J. Anat. in press. [CrossRef]
- Verma, S.; Khurana, S.; Vats, A.; Sahu, B.; Ganguly, N.K.; Chakraborti, P.; Gourie-Devi, M.; Taneja, V. Neuromuscular junction dysfunction in amyotrophic lateral sclerosis. Mol. Neurobiol. in press. [CrossRef]
- Tang, W.; Xiangfang, C.; Liu, H.; Lv, Q.; Zou, J.; Shi, Y.; Liu, Z. Expression of Nrf2 Promotes schwann cell-mediated sciatic nerve recovery in diabetic peripheral neuropathy. Cell. Physiol. Biochem. 2018, 46, 1879–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Huan, Y.; Li, C.; Cao, H.; Sun, S.; Lei, L.; Liu, Q.; Liu, S.; Ji, W.; Liu, H.; et al. Diphenyl diselenide alleviates diabetic peripheral neuropathy in rats with streptozotocin-induced diabetes by modulating oxidative stress. Biochem. Pharmacol. 2020, 182, 114221. [Google Scholar] [CrossRef]
- Tatsumi, Y.; Kato, A.; Sango, K.; Himeno, T.; Kondo, M.; Kato, Y.; Kamiya, H.; Nakamura, J.; Kato, K. Omega-3 polyunsaturated fatty acids exert anti-oxidant effects through the nuclear factor in immortalized mouse Schwann cells. J. Diabetes Investig. 2019, 10, 602–612. [Google Scholar] [CrossRef]
- Jeong, J.Y.; Cha, H.; Choi, E.O.; Kim, C.H.; Kim, G.; Yoo, Y.H.; Hwang, H.; Park, H.T.; Yoon, H.M.; Choi, Y.H. Activation of the Nrf2/HO-1 signaling pathway contributes to the protective effects of baicalein against oxidative stress-induced DNA damage and apoptosis in HEI193 Schwann cells. Int. J. Med. Sci. 2019, 16, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Hou, B.; He, P.; Ma, P.; Yang, X.; Yang, X.; Zhang, L.; Qiang, G.; Li, W.; Du, G. Neuroprotective effect of salvianolic acid a against diabetic peripheral neuropathy through modulation of Nrf2. Oxidative Med. Cell. Longev. 2020, 2020, 1–22. [Google Scholar] [CrossRef]
- Jiménez-Villegas, J.; Ferraiuolo, L.; Mead, R.J.; Shaw, P.J.; Cuadrado, A.; Rojo, A.I. NRF2 as a therapeutic opportunity to impact in the molecular roadmap of ALS. Free Radic. Biol. Med. 2021, 173, 125–141. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Ohta, Y.; Nomura, E.; Shang, J.; Feng, T.; Huang, Y.; Liu, X.; Shi, X.; Nakano, Y.; Hishikawa, N.; Sato, K.; et al. Enhanced oxidative stress and the treatment by edaravone in mice model of amyotrophic lateral sclerosis. J. Neurosci. Res. 2019, 97, 607–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Teng, C.; Wu, F.; Ge, L.; Xiao, J.; Zhang, H.; Chen, D. Edaravone attenuates traumatic brain injury through anti-inflammatory and anti-oxidative modulation. Exp. Ther. Med. 2019, 18, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Cores, Á.; Piquero, M.; Villacampa, M.; León, R.; Menéndez, J.C. NRF2 regulation processes as a source of potential drug targets against neurodegenerative diseases. Biomolecules 2020, 10, 904. [Google Scholar] [CrossRef] [PubMed]
- Hushpulian, D.M.; Ammal Kaidery, N.; Ahuja, M.; Poloznikov, A.A.; Sharma, S.M.; Gazaryan, I.G.; Thomas, B. Challenges and limitations of targeting the keap1-Nrf2 pathway for neurotherapeutics: Bach1 de-repression to the rescue. Front. Aging Neurosci. 2021, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.J.; Kiyoung, K. Effects of the edaravone, a drug approved for the treatment of amyotrophic lateral sclerosis, on mitochondrial function and neuroprotection. Antioxidants 2022, 11, 195. [Google Scholar] [CrossRef]
- Lee, S.; Hu, L. Nrf2 activation through the inhibition of Keap1–Nrf2 protein–protein interaction. Med. Chem. Res. 2020, 29, 846–867. [Google Scholar] [CrossRef]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Furusawa, Y.; et al. Characterizations of three major cysteine sensors of Keap1 in stress response. Mol. Cell. Biol. 2015, 36, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Tian, W.; Tao, S.; Tillotson, J.; Wijeratne, E.M.K.; Gunatilaka, A.A.L.; Zhang, D.D.; Chapman, E. Non-covalent NRF2 activation confers greater cellular protection than covalent activation. Cell Chem. Biol. 2019, 26, 1427–1435. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Activation of Nrf2 by natural bioactive compounds: A promising approach for stroke? Int. J. Mol. Sci. 2020, 21, 4875. [Google Scholar] [CrossRef]
- Said Ahmed, M.; Hung, W.Y.; Zu, J.S.; Hockberger, P.; Siddique, T. Increased reactive oxygen species in familial amyotrophic lateral sclerosis with mutations in SOD1. J. Neurol. Sci. 2000, 176, 88–94. [Google Scholar] [CrossRef]
- Bhatia, N.K.; Srivastava, A.; Katyal, N.; Jain, N.; Khan, M.A.I.; Kundu, B.; Deep, S. Curcumin binds to the pre-fibrillar aggregates of Cu/Zn superoxide dismutase (SOD1) and alters its amyloidogenic pathway resulting in reduced cytotoxicity. Biochim. Biophys. Acta-Proteins Proteom. 2015, 1854, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Xu, L.; Wu, L.; Wang, X.; Duan, W.; Li, H.; Li, C. Curcumin abolishes mutant TDP-43 induced excitability in a motoneuron-like cellular model of ALS. Neuroscience 2014, 272, 141–153. [Google Scholar] [CrossRef]
- Laudati, G.; Mascolo, L.; Guida, N.; Sirabella, R.; Pizzorusso, V.; Bruzzaniti, S.; Serani, A.; Di Renzo, G.; Canzoniero, L.M.T.; Formisano, L. Resveratrol treatment reduces the vulnerability of SH-SY5Y cells and cortical neurons overexpressing SOD1-G93A to Thimerosal toxicity through SIRT1/DREAM/PDYN pathway. Neurotoxicology 2019, 71, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Wang, S.Y.; Wang, X.D.; Jiang, H.Q.; Yang, Y.Q.; Wang, Y.; Cheng, J.L.; Zhang, C.T.; Liang, W.W.; Feng, H.L. Fisetin exerts antioxidant and neuroprotective effects in multiple mutant hSOD1 models of amyotrophic lateral sclerosis by activating ERK. Neuroscience 2018, 379, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Markert, C.D.; Kim, E.; Gifondorwa, D.J.; Childers, M.K.; Milligan, C.E. A single-dose resveratrol treatment in a mouse model of amyotrophic lateral sclerosis. J. Med. Food 2010, 13, 1081–1085. [Google Scholar] [CrossRef]
- Vucic, S.; Henderson, R.D.; Mathers, S.; Needham, M.; Schultz, D.; Kiernan, M.C.; TEALS study group. Safety and efficacy of dimethyl fumarate in ALS: Randomised controlled study. Ann. Clin. Transl. Neurol. 2021, 8, 1991–1999. [Google Scholar] [CrossRef]
- Dolati, S.; Ahmadi, M.; Aghebti-Maleki, L.; Nikmaram, A.; Marofi, F.; Rikhtegar, R.; Ayromlou, H.; Yousefi, M. Nanocurcumin is a potential novel therapy for multiple sclerosis by influencing inflammatory mediators. Pharmacol. Rep. 2018, 70, 1158–1167. [Google Scholar] [CrossRef]
- Park, H.R.; Yang, E.J. Oxidative stress as a therapeutic target in amyotrophic lateral sclerosis: Opportunities and limitations. Diagnostics 2021, 11, 1546. [Google Scholar] [CrossRef]
- Wobst, H.J.; Mack, K.L.; Brown, D.G.; Brandon, N.J.; Shorter, J. The clinical trial landscape in amyotrophic lateral sclerosis—Past, present, and future. Med. Res. Rev. 2020, 40, 1352–1384. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.T.; Pallesen, J.S.; Solbak, S.M.; Narayanan, D.; Baig, A.; Zang, J.; Aguayo-Orozco, A.; Carmona, R.M.C.; Garcia, A.D.; Bach, A. A comparative assessment study of known small-molecule Keap1-Nrf2 protein-protein interaction inhibitors: Chemical synthesis, binding properties, and cellular activity. J. Med. Chem. 2019, 62, 8028–8052. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.X.; Garneau-Tsodikova, S. What are the drugs of the future? Medchemcomm 2018, 9, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Mizunoe, Y.; Kobayashi, M.; Sudo, Y.; Watanabe, S.; Yasukawa, H.; Natori, D.; Hoshino, A.; Negishi, A.; Okita, N.; Komatsu, M.; et al. Trehalose protects against oxidative stress by regulating the Keap1–Nrf2 and autophagy pathways. Redox Biol. 2018, 15, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Zuo, R.; Wang, Y.; Li, J.; Wu, J.; Wang, W.; Li, B.; Sun, C.; Wang, Z.; Shi, C.; Zhou, Y.; et al. Rapamycin induced autophagy inhibits inflammation-mediated endplate degeneration by enhancing Nrf2/Keap1 signaling of cartilage endplate stem cells. Stem Cells 2019, 37, 828–840. [Google Scholar] [CrossRef]
- Al-Sarraj, S.; King, A.; Troakes, C.; Smith, B.; Maekawa, S.; Bodi, I.; Rogelj, B.; Al-Chalabi, A.; Hortobágyi, T.; Shaw, C.E. P62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011, 122, 691–702. [Google Scholar] [CrossRef]
- Mitsui, S.; Otomo, A.; Nozaki, M.; Ono, S.; Sato, K.; Shirakawa, R.; Adachi, H.; Aoki, M.; Sobue, G.; Shang, H.F.; et al. Systemic overexpression of SQSTM1/p62 accelerates disease onset in a SOD1H46R-expressing ALS mouse model. Mol. Brain 2018, 11, 30. [Google Scholar] [CrossRef]
- Choi, H.J.; Cha, S.J.; Lee, J.W.; Kim, H.J.; Kim, K. Recent advances on the role of gsk3β in the pathogenesis of amyotrophic lateral sclerosis. Brain Sci. 2020, 10, 675. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, Y.; Wang, H.; Zhao, L.; Ma, Z.; Li, T.; Liu, J.; Sun, M.; Jian, Y.; Yao, L.; et al. Edaravone reduces Aβ-induced oxidative damage in SH-SY5Y cells by activating the Nrf2/ARE signaling pathway. Life Sci. 2019, 221, 259–266. [Google Scholar] [CrossRef]
- Kato, H.; Sato, H.; Okuda, M.; Wu, J.; Koyama, S.; Izumi, Y.; Waku, T.; Iino, M.; Aoki, M.; Arawaka, S.; et al. Therapeutic effect of a novel curcumin derivative GT863 on a mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2021, in press. [Google Scholar] [CrossRef]
- Novak, V.; Rogelj, B.; Župunski, V. Therapeutic potential of polyphenols in amyotrophic lateral sclerosis and frontotemporal dementia. Antioxidants 2021, 10, 1328. [Google Scholar] [CrossRef]
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; Zundert, B.; Hetz, C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013, 9, 1308–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Guo, Y.; Wang, X.; Yu, X.; Duan, W.; Hong, K.; Wang, J.; Han, H.; Li, C. Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience 2015, 298, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Lattante, S.; de Calbiac, H.; Le Ber, I.; Brice, A.; Ciura, S.; Kabashi, E. Sqstm1 knock-down causes a locomotor phenotype ameliorated by rapamycin in a zebrafish model of ALS/FTLD. Hum. Mol. Genet. 2015, 24, 1682–1690. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Lin, M.; Shen, C.J. Rapamycin alleviates pathogenesis of a new Drosophila model of ALS-TDP. J. Neurogenet. 2015, 29, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Martínez-González, L.; Gonzalo-Consuegra, C.; Gómez-Almería, M.; Porras, G.; de Lago, E.; Martín-Requero, Á.; Martínez, A. Tideglusib, a Non-ATP competitive inhibitor of GSK-3 β as a drug candidate for the treatment of amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2021, 22, 8975. [Google Scholar] [CrossRef]
- Aggarwal, S.P.; Zinman, L.; Simpson, E.; Mckinley, J.; Jackson, K.E.; Pinto, H.; Kaufman, P.; Conwit, R.A.; Schoenfeld, D.; Shefner, J.; et al. Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010, 9, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zhang, H.; Davies, K.J.A.; Forman, H.J. Aging-related decline in the induction of Nrf2-regulated antioxidant genes in human bronchial epithelial cells. Redox Biol. 2018, 14, 35–40. [Google Scholar] [CrossRef]
- Morrice, J.R.; Gregory-Evans, C.Y.; Shaw, C.A. Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural Regen. Res. 2018, 13, 2050–2054. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Markers of Oxidative Stress | Experimental Model | Ref. |
|---|---|---|
| - MDA accumulation (lipid peroxidation) | SOD1G93A mouse model | [17] |
| - Increase levels of MDA (lipid peroxidation) - Increased levels of 8-OHdG (oxidative DNA damage) - Increased GSSG/GSH ratio (oxidative stress). | Blood samples of SALS patients | [18] |
| - Increased levels of 4-HNE (lipid peroxidation) | Post-mortem specimens of lumbar spinal cord and/or occipital cortex | [19] |
| - Decreased GSH levels (oxidative stress) | Motor cortex of ALS patients | [20] |
| - Increased levels of urinary 8-OhdG and IsoP (lipid peroxidation) | Urine of SALS patients | [21] |
| - Increased HNE level (lipid peroxidation) | CSF of SALS patients | [22] |
| - Increased HNE level (lipid peroxidation) | Serum and CSF of SALS patients | [23] |
| - Increased protein carbonyl levels | Serum of SALS patients | [24] |
| - Increased levels of adducts of HNEH and CRAL (lipid peroxidation) - Increased levels of CML and pentosidine (protein glycoxidation) | Postmorten spinal cords of SALS patients | [25] |
| - Increase 8-OHG levels (DNA oxidation) | Postmortem brain and spinal cord tissues of ALS patients and SOD1G93A mouse model | [26] |
| Mechanism of Nrf2 Activation | Compound | Model and trial | Outcome | Ref. |
|---|---|---|---|---|
| Electrophilic inhibition of Keap1-mediated Nrf2 degradation | Curcumin derivatives | SOD1H46R mouse model | Improved motor function | [110] |
| Clinical trial NCT04499963 (ongoing) | ||||
| Clinical trial as add-on therapy to riluzole (completed) | Increased probability of survival without changes in motor function. | [43] | ||
| Resveratrol | SOD1G93A mouse model | Conflicting preclinical results | [111] | |
| Combined treatment of resveratrol and curcumin | Clinical trial NCT04654689 (ongoing) | |||
| CC100 | Clinical trial NCT03049046 (completed) | Short-term treatment is safe and tolerable. | [101] | |
| DMF | Clinical trial as add-on to riluzole therapy ACTRN12618000534280 (completed phase II) | No improvement of survival and respiratory function | [81,98] | |
| p62-mediated Keap1 degradation | Trehalose | SOD1G86R mouse model | Increased survival and attenuated disease progress in mouse models. | [112] |
| SOD1G93A mouse model | Postponed disease onset, slowed down disease progress, no changes in survival. | [113] | ||
| Clinical trial NCT05136885 (ongoing) | ||||
| Rapamycin | p62 knockdown zebrafish model | Imrpoved motor function | [114] | |
| TDP-43 Drosophila model | Partially improved survival and motor function | [115] | ||
| Clinical trial NCT03359538 (ongoing) | ||||
| Inhibition of GSK3β-promoted Nrf2 degradation by phosphorylation | Tideglusib | TDP-43 transgenic mice | Reduced TDP-43 phosphorylation in the spinal cord of TDP-43 transgenic mice. | [116] |
| Clinical trial NCT05105958 (ongoing). | ||||
| Lithium | Clinical studies (completed) | Pilot study NCT00818389 showed slowing ALS progression. | [108] | |
| Combined treatment of lithium and riluzole NCT00818389 did not show any effect | [117] | |||
| Unknown | Edaravone | FDA-approved drug in USA Canada and some other countries | Reduced level of oxidative stress and imrpoved motor and respiratory function. | [87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arslanbaeva, L.; Bisaglia, M. Activation of the Nrf2 Pathway as a Therapeutic Strategy for ALS Treatment. Molecules 2022, 27, 1471. https://doi.org/10.3390/molecules27051471
Arslanbaeva L, Bisaglia M. Activation of the Nrf2 Pathway as a Therapeutic Strategy for ALS Treatment. Molecules. 2022; 27(5):1471. https://doi.org/10.3390/molecules27051471
Chicago/Turabian StyleArslanbaeva, Liaisan, and Marco Bisaglia. 2022. "Activation of the Nrf2 Pathway as a Therapeutic Strategy for ALS Treatment" Molecules 27, no. 5: 1471. https://doi.org/10.3390/molecules27051471
APA StyleArslanbaeva, L., & Bisaglia, M. (2022). Activation of the Nrf2 Pathway as a Therapeutic Strategy for ALS Treatment. Molecules, 27(5), 1471. https://doi.org/10.3390/molecules27051471