
Chip-Based Spectrofluorimetric Determination of Iodine in a Multi-Syringe Flow Platform with and without In-Line Digestion—Application to Salt, Pharmaceuticals, and Algae Samples

 ,
,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Solutions
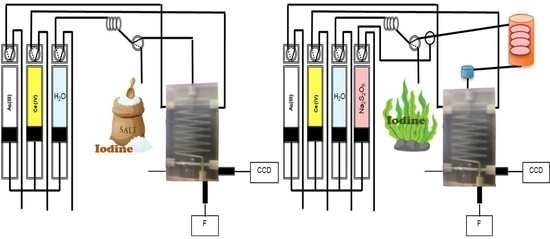
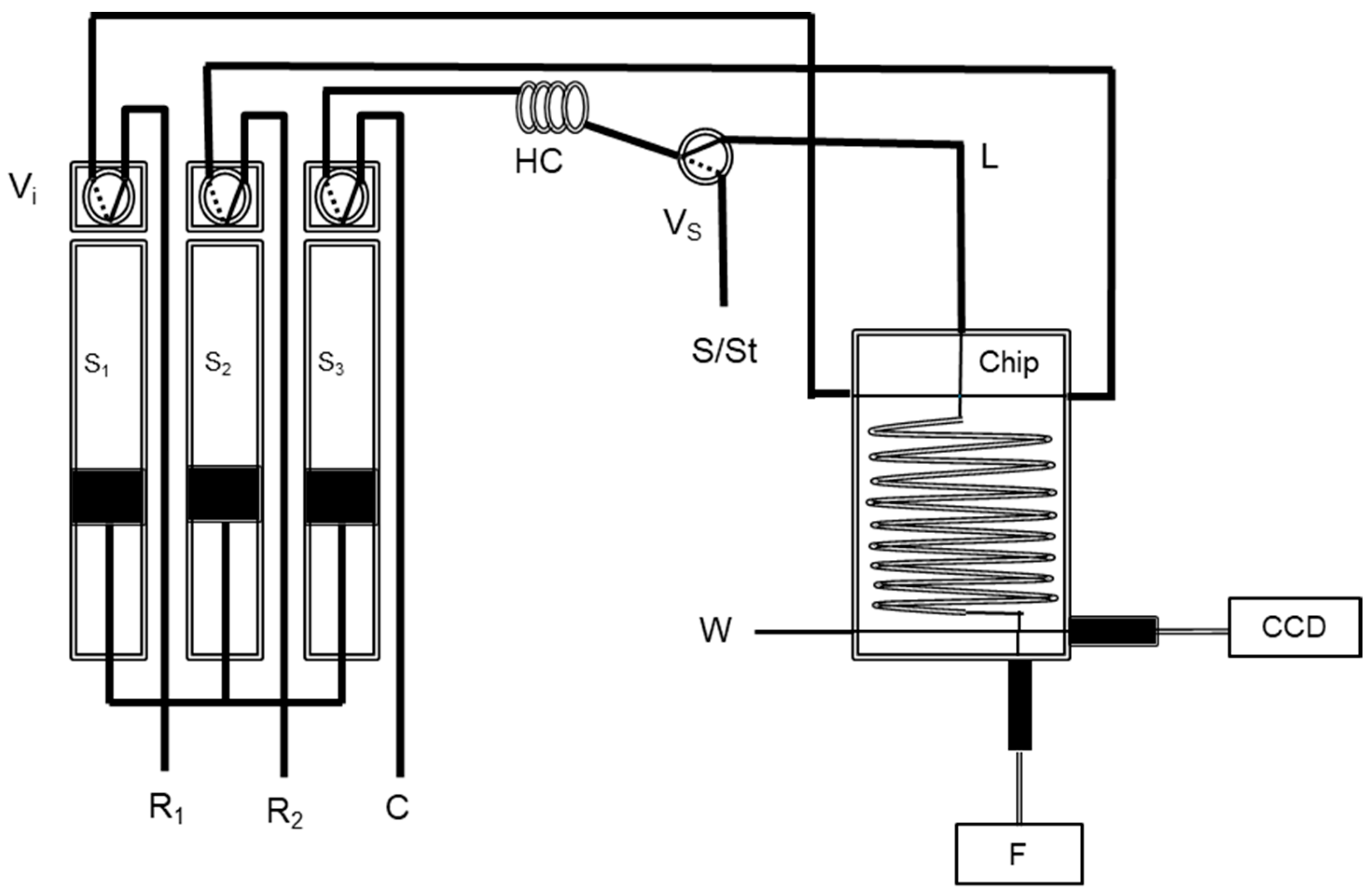
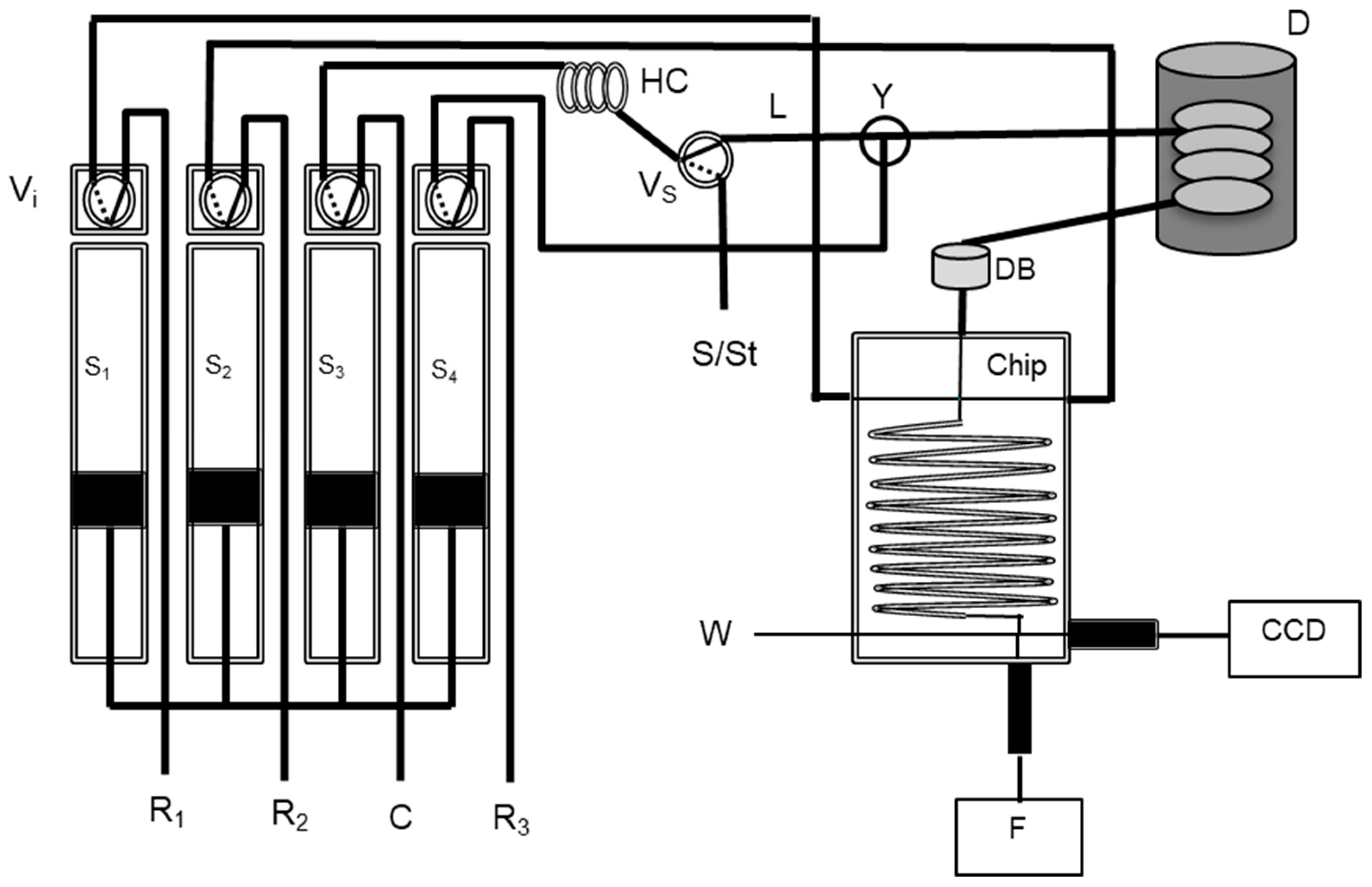
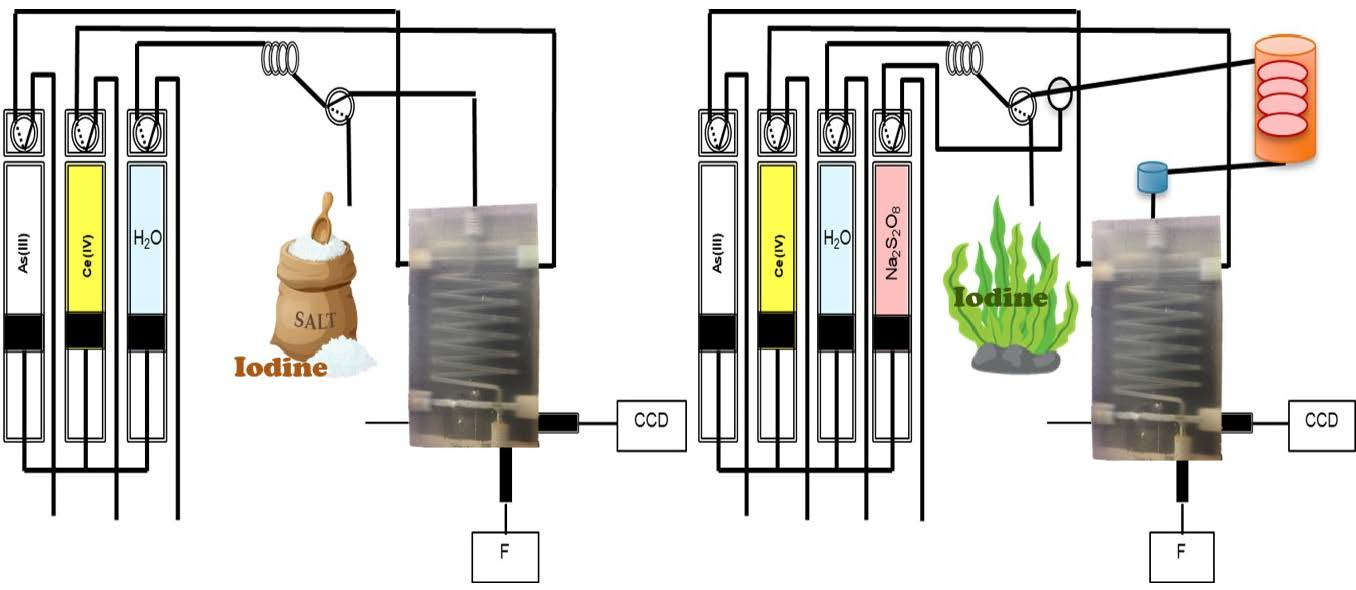
2.2. Chip-Based and Multi-Syringe Flow System Manifold and Procedure
2.2.1. Iodine Determination
2.2.2. Total Iodine Determination with In-Line UV Digestion
2.3. Sample Collection and Preparation
2.3.1. Salt Samples
2.3.2. Iodine Supplement Samples
2.4. Accuracy Assessment
2.4.1. Determination of Iodine
2.4.2. Determination of Total Iodine Using an In-Line UV Digestion
3. Results and Discussion
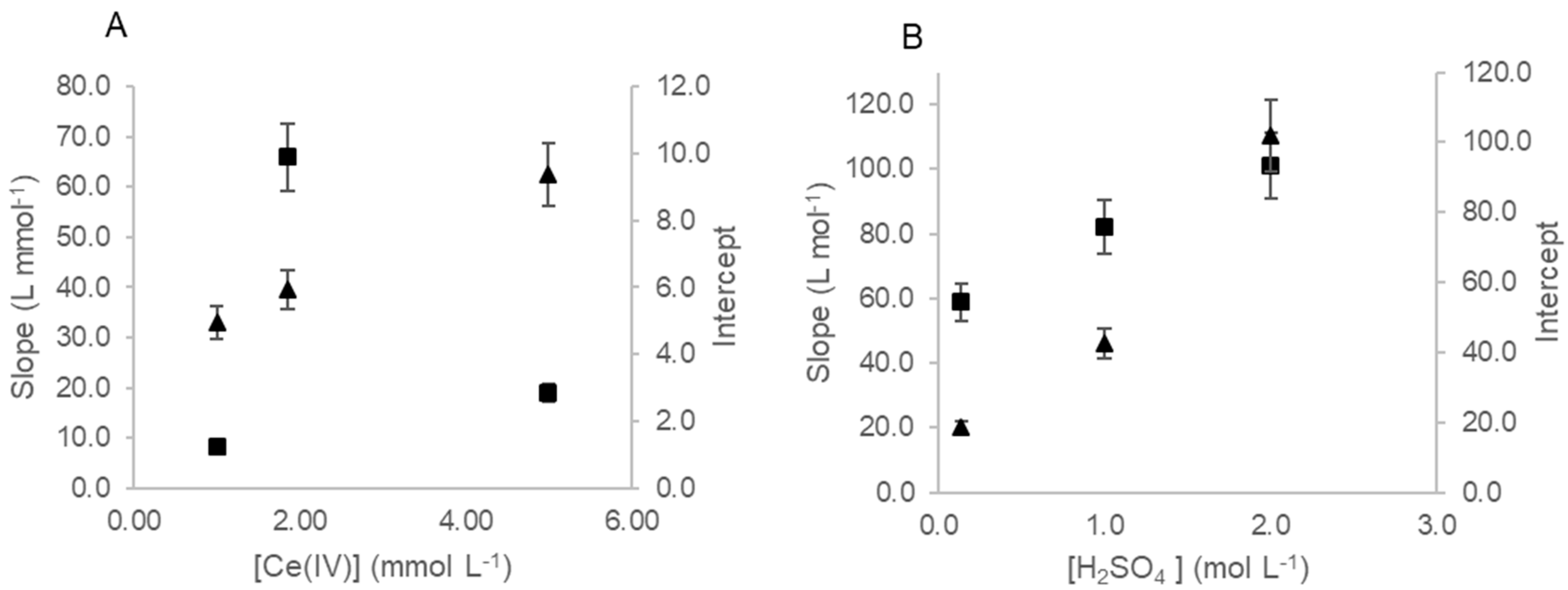
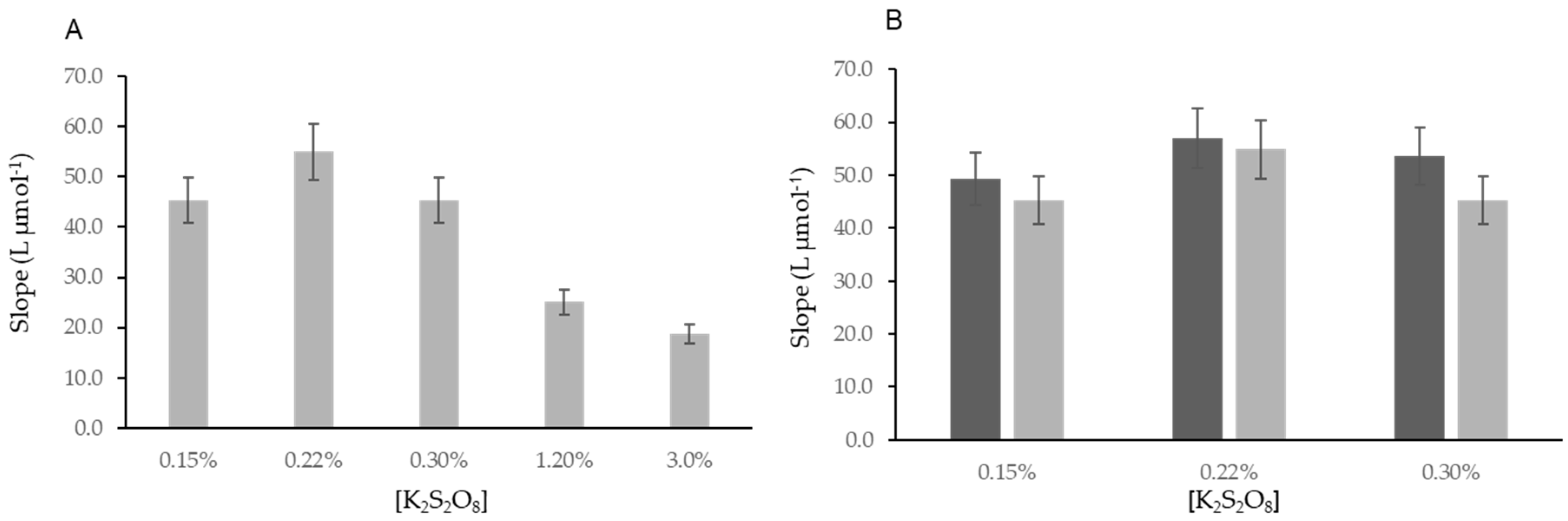
3.1. Study of Fluorometric Determination of Iodine
3.2. Study of the Fluorometric Determination of Total Iodine
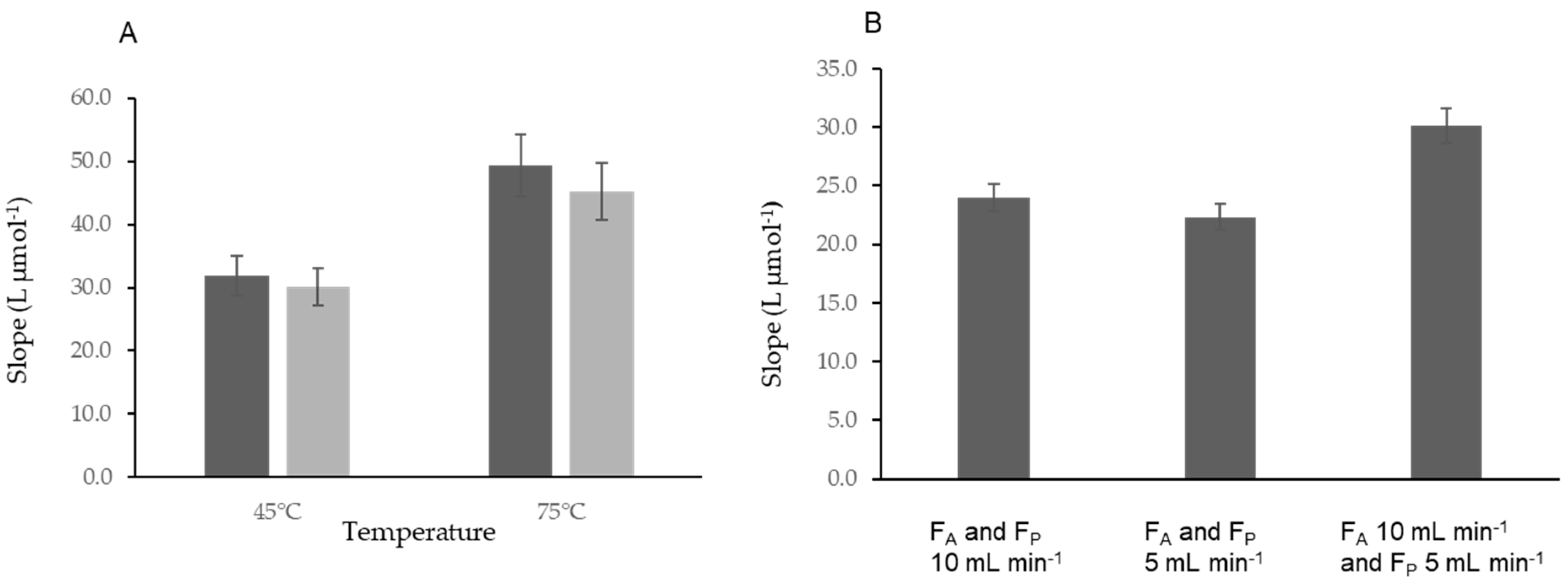
Study of the Temperature Influence
3.3. Study of the Flow Rate Influence Using the MS-Chip In-Line UV Digestion System
3.4. Interference Assessment
3.4.1. Interferences of the Sandell–Kolthoff Reaction
3.4.2. Other Potential Interfering Ions
3.5. Figures of Merit
3.6. Application to Iodine Containing Samples—Accuracy Assessment
3.6.1. Salt Samples
3.6.2. Supplement Iodine Samples
3.7. Recovery Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hess, S.Y. The impact of common micronutrient deficiencies on iodine and thyroid metabolism: The evidence from human studies. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Laurberg, P.; Cerqueira, C.; Ovesen, L.; Rasmussen, L.B.; Perrild, H.; Andersen, S.; Pedersen, I.B.; Carlé, A. Iodine intake as a determinant of thyroid disorders in populations. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Costeira, M.J.; Oliveira, P.; Santos, N.C.; Ares, S.; Saenz-Rico, B.; De Escobar, G.M.; Palha, J.A. Psychomotor development of children from an iodine-deficient region. J. Pediatr. 2011, 159, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Henjum, S.; Lilleengen, A.M.; Aakre, I.; Dudareva, A.; Gjengedal, E.L.F.; Meltzer, H.M.; Brantsæter, A.L. Suboptimal iodine concentration in breastmilk and inadequate iodine intake among lactating women in Norway. Nutrients 2017, 9, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnweidner-Holme, L.; Aakre, I.; Lilleengen, A.M.; Brantsæter, A.L.; Henjum, S. Knowledge about iodine in pregnant and lactating women in the Oslo area, Norway. Nutrients 2017, 9, 493. [Google Scholar] [CrossRef] [Green Version]
- Limbert, E.; Prazeres, S.; São Pedro, M.; Madureira, D.; Miranda, A.; Ribeiro, M.; Jacome de Castro, J.; Carrilho, F.; Oliveira, M.J.; Reguengo, H.; et al. Iodine intake in Portuguese pregnant women: Results of a countrywide study. Eur. J. Endocrinol. 2010, 163, 631–635. [Google Scholar] [CrossRef] [Green Version]
- Lopes, M.S.; de Castro, J.J.; Marcelino, M.; Oliveira, M.J.; Carrilho, F.; Limbert, E. Iodine and Thyroid: What a Clinic Should Know. Acta Med. Port. 2012, 25, 174–178. [Google Scholar] [CrossRef]
- Lobato, C.B.; Machado, A.; Mesquita, R.B.R.; Lima, L.; Bordalo, A.A. Can non-fortified marine salt cover human needs for iodine? Int. J. Food Sci. Nutr. 2019, 70, 349–354. [Google Scholar] [CrossRef]
- Andersson, M.; Karumbunathan, V.; Zimmermann, M.B. Global iodine status in 2011 and trends over the past decade. J. Nutr. 2012, 142, 744–750. [Google Scholar] [CrossRef]
- Hess, S.Y.; Ouédraogo, C.T.; Young, R.R.; Bamba, I.F.; Stinca, S.; Zimmermann, M.B.; Wessells, K.R. Urinary iodine concentration identifies pregnant women as iodine deficient yet school-aged children as iodine sufficient in rural Niger. Public Health Nutr. 2017, 20, 1154–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, M.B.; Andersson, M. Prévalence du déficit iodé en Europe en 2010. Ann. Endocrinol. (Paris) 2011, 72, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Aquaron, R.; Delange, F.; Marchal, P.; Lognoné, V.; Ninane, L. Bioavailability of seaweed iodine in human beings. Cell Mol. Biol. (Noisy-Le-Grand) 2002, 48, 563–569. [Google Scholar]
- Yeh, T.S.; Hung, N.H.; Lin, T.C. Analysis of iodine content in seaweed by GC-ECD and estimation of iodine intake. J. Food Drug Anal. 2014, 22, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Shelor, C.P.; Dasgupta, P.K. Review of analytical methods for the quantification of iodine in complex matrices. Anal. Chim. Acta 2011, 702, 16–36. [Google Scholar] [CrossRef] [PubMed]
- Jooste, P.L.; Strydom, E. Methods for determination of iodine in urine and salt. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 77–88. [Google Scholar] [CrossRef]
- Pino, S.; Fang, S.L.; Braverman, L.E. Ammonium persulfate: A safe alternative oxidizing reagent for measuring urinary iodine. Clin. Chem. 1996, 42, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, A.; Lima, L.; Mesquita, R.B.R.; Bordalo, A.A. Improvement of the Sandell-Kolthoff reaction method (ammonium persulfate digestion) for the determination of iodine in urine samples. Clin. Chem. Lab. Med. 2017, 55, E206–E208. [Google Scholar] [CrossRef] [Green Version]
- American Public Health Association; American Water Works Association; Water Environment Association. Standard Methods for the Examination of Water and Wastewater, 23th ed.; American Public Health Association: Denver, CO, USA, 2017; ISBN 978-087553-287-5. [Google Scholar]
- Zimmermann, M.B.; Jooste, P.L.; Pandav, C.S. Iodine-deficiency disorders. Lancet 2008, 372, 1251–1262. [Google Scholar] [CrossRef]
- Frizzarin, R.M.; Aguado, E.; Portugal, L.A.; Moreno, D.; Estela, J.M.; Rocha, F.R.P.; Cerda, V. A portable multi-syringe flow system for spectrofluorimetric determination of iodide in seawater. Talanta 2015, 144, 1155–1162. [Google Scholar] [CrossRef]
- Zagatto, E.A.G.G.; Arruda, M.A.Z.Z.; Jacintho, A.O.; Mattos, I.L. Compensation of the Schlieren effect in flow-injection analysis by using dual-wavelength spectrophotometry. Anal. Chim. Acta 1990, 234, 153–160. [Google Scholar] [CrossRef]
- Nacapricha, D.; Muangkaew, S.; Ratanawimarnwong, N.; Shiowatana, J.; Grudpan, K. Continuous and stopped flow injection for catalytic determination of total iodine in urine. Analyst 2001, 126, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Santos, I.C.; Mesquita, R.B.R.; Machado, A.; Bordalo, A.A.; Rangel, A.O.S.S. Sequential injection methodology for carbon speciation in bathing waters. Anal. Chim. Acta 2013, 778, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Zhang, X. Determination of iodide, iodate and organo-iodine in waters with a new total organic iodine measurement approach. Water Res. 2013, 47, 6660–6669. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, X. Total organic iodine measurement: A new approach with UPLC/ESI-MS for off-line iodide separation/detection. Water Res. 2013, 47, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, K.; Namba, H.; Nomura, T.; Yokoyama, N.; Yamashita, S.; Izumi, M.; Nagataki, S. Automated Measurement of Urinary Iodine with Use of Ultraviolet-Irradiation. Clin. Chem. 1995, 41, 581–585. [Google Scholar] [CrossRef]
- Ford, H.C.; Johnson, L.A. Ascorbic acid interferes with an automated urinary iodide determination based on the ceric-arsenious acid reaction. Clin. Chem. 1991, 37, 759. [Google Scholar] [CrossRef] [PubMed]
- Amorim, K.; Lage-Yusty, M.A.; López-Hernández, J. Changes in bioactive compounds content and antioxidant activity of seaweed after cooking processing. CyTA-J. Food 2012, 10, 321–324. [Google Scholar] [CrossRef] [Green Version]
- Umar, S.; Iqbal, M. Nitrate accumulation in plants, factors affecting the process, and human health implications. A review. Agron. Sustain. Dev. 2007, 27, 45–57. [Google Scholar] [CrossRef]
- Mikkelsen, R. A Closer Look at Phosphorus Uptake by Plants; International Plant Nutrition Institute (IPNI): Canada, 2013; Available online: http://www.ipni.net/publication/insights-na.nsf/0/324D239E9065F43385257CD800765F01/$FILE/INSIGHTS-NA-2013-07.pdf (accessed on 30 January 2022).
- Kurashova, I.; Halevy, I.; Kamyshny, A. Kinetics of Decomposition of Thiocyanate in Natural Aquatic Systems. Environ. Sci. Technol. 2018, 52, 1234–1243. [Google Scholar] [CrossRef]
- Currie, L.A. Nomenclature in Evaluation of Analytical Methods Including Detection and Quantification Capabilities (IUPAC Recommendations 1995). Int. Union Pure Appl. Chem. 1995, 67, 1699–1723. [Google Scholar] [CrossRef]
- International Union of Pure and Applied Chemistry Nomenclature, symbols, units and their usage in spectrochemical analysis-ii. data interpretation. Pure Appl. Chem. 1976, 45, 99–103. [CrossRef]
- Miller, J.N.; Miller, J.C. Statistics and Chemometrics for Analytical Chemistry, 6th ed.; Pearson Education Limited: Harlow, UK, 2010. [Google Scholar]
- Burns, D.; Danzer, K.; Townshend, A. Use of the Terms “Recovery” and “Apparent Recovery” in Analytical Procedures. Int. Union Pure Appl. Chem. 2002, 74, 2201–2205. [Google Scholar] [CrossRef]
- Choengchan, N.; Lukkanakul, K.; Ratanawimarnwong, N.; Waiyawat, W.; Wilairat, P.; Nacapricha, D. Use of pseudo-first order kinetics in flow injection for determination of trace inorganic iodine. Anal. Chim. Acta 2003, 499, 115–122. [Google Scholar] [CrossRef]
- Abouhiat, F.Z.; Henríquez, C.; Horstkotte, B.; El Yousfi, F.; Cerdà, V. A miniaturized analyzer for the catalytic determination of iodide in seawater and pharmaceutical samples. Talanta 2013, 108, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Inpota, P.; Strzelak, K.; Koncki, R.; Sripumkhai, W.; Jeamsaksiri, W.; Ratanawimarnwong, N.; Wilairat, P.; Choengchan, N.; Chantiwas, R.; Nacapricha, D. Microfluidic Analysis with Front-Face Fluorometric Detection for the Determination of Total Inorganic Iodine in Drinking Water. Anal. Sci. 2018, 34, 161–167. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Urinary iodine concentrations for determining iodine status in populations. In Vitamin and Mineral Nutrition Information System; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Garry, P.J.; Lashley, D.W.; Owen, G.M. Automated Measurement of Urinary Iodine. Clin. Chem. 1973, 19, 950–953. [Google Scholar] [CrossRef]
- Burguera, J.L.; Brunetto, M.R.; Contreras, Y.; Burguera, M.; Gallignani, M.; Carrero, P. Head-space flow injection for the on-line determination of iodide in urine samples with chemiluminescence detection. Talanta 1996, 43, 839–850. [Google Scholar] [CrossRef]
- Yaqoob, M.; Atiq-Ur-Rehman; Waseem, A.; Nabi, A. Determination of iodide using flow injection with acidic potassium permanganate chemiluminescence detection. Luminescence 2006, 21, 221–225. [Google Scholar] [CrossRef]
- Machado, A.; Mesquita, R.B.R.; Oliveira, S.; Bordalo, A.A. Development of a robust, fast screening method for the potentiometric determination of iodide in urine and salt samples. Talanta 2017, 167, 688–694. [Google Scholar] [CrossRef]
- Yaping, Z.; Dongxing, Y.; Jixiang, C.; Tianshiu, L.; Huiqin, C. Spectrophotometric determination of urinary iodine by flow-injection analysis with on-line catalytic digestion. Clin. Chem. 1996, 42, 2021–2027. [Google Scholar] [CrossRef]
- Waseem, A.; Yaqoob, M.; Nabi, A. Flow-injection method for the determination of iodide/iodine using Ru(bpy)33+-NADH chemiluminescence detection. Luminescence 2008, 23, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.C.; Rocha, F.R.P. Liquid-liquid microextraction in a multicommuted flow system for direct spectrophotometric determination of iodine value in biodiesel. Anal. Chim. Acta 2014, 829, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.V.; Ermolenko, Y.V.; Seffar, L. Flow-injection determination of elemental iodine by polyvinyl alcohol. J. Anal. Chem. 2004, 59, 688–693. [Google Scholar] [CrossRef]
- Kuznetsov, V.V.; Ermolenko, Y.V.; Seffar, L. Amylose and amylopectin as reagents for the flow-injection determination of elemental iodine. J. Anal. Chem. 2007, 62, 479–485. [Google Scholar] [CrossRef]
- Trojánek, A.; Papoff, P. Pneumatoamperometric flow-injection determination of iodide. Anal. Chim. Acta 1991, 247, 73–77. [Google Scholar] [CrossRef]
- Motomizu, S.; Yoden, T. Porous membrane permeation of halogens and its application to the determination of halide ions and residual chlorine by flow-injection analysis. Anal. Chim. Acta 1992, 261, 461–469. [Google Scholar] [CrossRef]
- Williamson, J.P.; Emmert, G.L. A flow injection analysis system for monitoring silver (I) ion and iodine residuals in recycled water from recovery systems used for spaceflight. Anal. Chim. Acta 2013, 792, 72–78. [Google Scholar] [CrossRef]
- Jakmunee, J.; Grudpan, K. Flow injection amperometry for the determination of iodate in iodized table salt. Anal. Chim. Acta 2001, 438, 299–304. [Google Scholar] [CrossRef]
- De Nogueira, A.R.A.; Mockiuti, F.; De Souza, G.B.; Primavesi, O. Flow Injection Spectrophotometric Catalytic Determination of Iodine in Milk. Anal. Sci. 1998, 14, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Hassan, S.S.M.; Marzouk, S.A.M. Sequential flow-injection potentiometric determination of iodide and iodine in povidone iodine pharmaceuticals. Electroanalysis 1993, 5, 855–861. [Google Scholar] [CrossRef]
- Nacapricha, D.; Uraisin, K.; Ratanawimarnwong, N.; Grudpan, K. Simple and selective method for determination of iodide in pharmaceutical products by flow injection analysis using the iodine-starch reaction. Anal. Bioanal. Chem. 2004, 378, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Srivorakul, T.; Varanusupakul, P.; Alahmad, W. Development of a Sample Treatment Method for a Flow Injection Determination of Iodine in Eggs: A Comparison Study. Anal. Sci. 2020, 36, 491–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomaidis, N.S.; Georgiou, C.A. Direct parallel flow injection multichannel spectrophotometric determination of olive oil iodine value. Anal. Chim. Acta 2000, 405, 239–245. [Google Scholar] [CrossRef]
- Ratanawimarnwong, N.; Amornthammarong, N.; Choengchan, N.; Chaisuwan, P.; Amatatongchai, M.; Wilairat, P.; McKelvie, I.D.; Nacapricha, D. Determination of iodide by detection of iodine using gas-diffusion flow injection and chemiluminescence. Talanta 2005, 65, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Zabala, J.; Carrión, N.; Murillo, M.; Quintana, M.; Chirinos, J.; Seijas, N.; Duarte, L.; Brätter, P. Determination of normal human intrathyroidal iodine in Caracas population. J. Trace Elem. Med. Biol. 2009, 23, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Choengchan, N.; Uraisin, K.; Choden, K.; Veerasai, W.; Grudpan, K.; Nacapricha, D. Simple flow injection system for colorimetric determination of iodate in iodized salt. Talanta 2002, 58, 1195–1201. [Google Scholar] [CrossRef]
- Håkedal, J.T.; Egeberg, P.K. Determination of Iodide in Brines by Membrane Permeation Flow Injection Analysis. Analyst 1997, 122, 1235–1238. [Google Scholar] [CrossRef]
- Oguma, K.; Kitada, K.; Kuroda, R. Microchemical determination of lodate and iodide in sea waters by flow injection analysis. Microchim. Acta 1993, 110, 71–77. [Google Scholar] [CrossRef]
- Monks, C.D.; Nacapricha, D.; Taylor, C.G. Determination of iodide ion in impregnated charcoals by flow injection. Analyst 1993, 118, 623–626. [Google Scholar] [CrossRef]
- Fujiwara, T.; Mohammadzai, I.U.; Kojima, M.; Kumamaru, T. An improved method for the flow-injection determination of iodine using the luminol chemiluminescence reaction in a reversed micellar medium of cetyltrimethylammonium chloride in 1-hexanol-cyclohexane. Anal. Sci. 2006, 22, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Yonehara, N.; Kozono, S.; Sakamoto, H. Flow Injection-Spectrophotometric Determination of Trace Amounts of Iodide by Its Catalytic Effect on the 4, 4′-Bis(dimethylamino)-diphenylmethane- Chioramine T Reaction. Anal. Sci. 1991, 7, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Davey, D.E.; Mulcahy, D.E.; O’Connell, G.R. Potentiometric flow-injection determination of iodide and iodine. Talanta 1990, 37, 313–316. [Google Scholar] [CrossRef]
- Nacapricha, D.; Sangkarn, P.; Karuwan, C.; Mantim, T.; Waiyawat, W.; Wilairat, P.; Cardwell, T.; McKelvie, I.D.; Ratanawimarnwong, N. Pervaporation-flow injection with chemiluminescence detection for determination of iodide in multivitamin tablets. Talanta 2007, 72, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Li, J.; Zhao, X. Ion Exchange-Flow Injection Spectrometric Simultaneous Determination of Traces of Bromide and Iodide in some Chinese Standard Samples. Geostand. Newsl. 1995, 19, 215–220. [Google Scholar] [CrossRef]
- Nellaiappan, S.; Kumar, A.S. Selective flow injection analysis of iodate in iodized table salts by riboflavin immobilized multiwalled carbon nanotubes chemically modified electrode. Electrochim. Acta 2013, 109, 59–66. [Google Scholar] [CrossRef]
- Nikolic, S.D.; Mutic, J.J.; Lolic, A.D.; Manojlovic, D.D. Sensitive flow-injection amperometric detection of iodide using Mn3+ and As3+. Anal. Sci. 2005, 21, 525–529. [Google Scholar] [CrossRef] [Green Version]
- Ensafi, A.A.; Dehaghi, G.B. Flow-injection simultaneous determination of iodate and periodate by spectrophotometric and spectrofluorometric detection. Anal. Sci. 2000, 16, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Tesfaldet, Z.O.; Van Staden, J.F.; Stefan, R.I. Sequential injection spectrophotometric determination of trace amounts of iodide by its catalytic effect on the 4,4′-methylenebis(N,N-dimethylaniline)-chloramine-T reaction. Talanta 2004, 64, 1213–1219. [Google Scholar] [CrossRef]
- Santos, I.C.; Mesquita, R.B.R.; Bordalo, A.A.; Rangel, A. Iodine speciation in coastal and inland bathing waters and seaweeds extracts using a sequential injection standard addition flow-batch method. Talanta 2015, 133, 7–14. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Active Devices | Action | Flow Rate | Description | |
|---|---|---|---|---|---|
| Preparation Steps | 1 | S1, S2, S3 | Aspirate 5.00 mL | 10 mL min−1 | Filling of syringes with reagents and carrier |
| 2 | S1, S2, S3 | Dispense 2.15 mL | 5 mL min−1 | Enabling the syringes to have capacity for sample aspiration | |
| 3 | S3, VS | Aspirate 0.150 mL | 10 mL min−1 | Sample aspiration for channel preparation | |
| 4 | S3 | Dispense 3.00 mL | 5 mL min−1 | Cleaning the analytical path | |
| 5 | S1, S2, S3 | Aspirate 1.50 mL | 10 mL min−1 | Filling of syringes with reagents and carrier | |
| Loop for iodine determination | 6 | S3, VS | Aspirate 0.35 mL | 10 mL min−1 | Sample aspiration |
| 7 | S3, | Dispense 0.05 mL | 5 mL min−1 | Sample injection | |
| 8 | S1, S2, S3 | Dispense 0.30 mL | 5 mL min−1 | Reagents and sample injection into the chip | |
| 9 | S3 | Dispense 0.90 mL | 0.4 mL min−1 | Transport through the chip and signal acquisition (CCD) | |
| 10 | S3 | Dispense 0.60 mL | 5 mL min−1 | Cleaning the analytical path | |
| 11 | S1, S2, S3 | Aspirate 1.50 mL | 10 mL min−1 | Refilling of syringes with reagents and carrier |
| Step | Active Devices | Action | Flow Rate | Description | |
|---|---|---|---|---|---|
| Preparation Steps | 1 | S1, S2, S3, S4 | Aspirate 5.00 mL | 10 mL min−1 | Filling of syringes with reagents and carrier |
| 2 | S1, S2, S3, S4 | Dispense 2.35 mL | 5 mL min−1 | Enabling the syringes to have capacity for sample aspiration | |
| 3 | S3, VS | Aspirate 0.150 mL | 10 mL min−1 | Sample aspiration for channel preparation | |
| 4 | S3 | Dispense 3.00 mL | 5 mL min−1 | Cleaning the analytical path | |
| 5 | S1, S2, S3, S4 | Aspirate 3.32 mL | 10 mL min−1 | Filling of syringes with reagents and carrier | |
| Loop for Total Iodine Determination | 6 | S3, VS | Aspirate 0.150 mL | 10 mL min−1 | Sample aspiration |
| 7 | S3, S4 | Dispense 0.150 mL | 5 mL min−1 | Sample injection and oxidant reagent propeled through Y confluence for mixing | |
| 8 | S3 | Dispense 1.02 mL | Propeling the sample and oxidant reagent mixture through the UV digestor | ||
| 9 | S1, S2, S3 | Dispense 0.300 mL | Reagents and sample injection into the chip | ||
| 10 | S3 | Dispense 0.900 mL | 0.4 mL min−1 | Transport through the chip manifold and signal acquisition (CCD) | |
| 11 | S3 | Dispense 1.10 mL | 5 mL min−1 | Cleaning the analytical path | |
| 12 | S1, S2, S3, S4 | Aspirate 3.32 mL | 10 mL min−1 | Refilling of syringes with reagents and carrier |
| Sample ID | Amount of Sample (Dissolved in 100 mL in MQW) | Dilution Factor | |
|---|---|---|---|
| Pharmaceuticals | #Pharm 1 | 1/10 of pill | - |
| #Pharm 2 | |||
| #Pharm 3 | |||
| #Pharm 4 | Pill | 10× | |
| #Pharm 5 | |||
| Algae Pills | #Algae 1 | Pill | 10× |
| #Algae 2 | 1/10 of pill | - | |
| #Algae 3 |
| Interferent | Concentration (mg L−1) | Interference (%) |
|---|---|---|
| SCN− | 5 | 3 |
| 22 | 9 | |
| C6H8O6 | 6.6 | 6 |
| 13.2 | 4 | |
| 26.5 | 13 |
| Potential Interferent | Reference Concentration in Algae (mg Kg−1) | Expected Concentration in Tested Sample (mg L−1) | Tested Concentration (mg L−1) | Interference (%) |
|---|---|---|---|---|
| NO3− | 4500 ** | 45 | 50 | −4 |
| 150 | −10 | |||
| 500 | −20 | |||
| NO2− | 40.9 ** | 0.41 | 40 | −7 |
| 50 | −11 | |||
| PO43− | 5400 | 54 | 10,000 | −2 |
| 15,000 | −11 | |||
| CN− * | 0.3 | 0.003 | 2.2 | 3 |
| 9.9 | 9 | |||
| C6H8O6 * | 118.8 | 1.2 | 13.2 | 4 |
| 26.5 | 13 |
| Dynamic Range (µmol L−1) | Typical Calibration Curve a A = S × µmol L−1 I + b | LOD (µmol L−1) | LOQ (µmol L−1) | One Determination (h−1) | Analysis Rate (h−1) b | Effluent Production (mL) b | Reagent Consumption (µmol) c | |
|---|---|---|---|---|---|---|---|---|
| MS-Chip | 0.20–4.0 | A = 91.17 ± 1.24 × [I] + 65.87 ± 9.326 R2 = 0.9985 ± 0.0016 | 0.025 | 0.199 | 0.049 | 1.18 | 71 | (Ce(IV)) 0.555 (As(III)) 30.0 (K2S2O8) 1.65 d |
| MS-Chip w/ in-line UV digestion | 0.23–4.0 | A = 42.18 ± 3.524 × [I] + 177.7 ± 15.86 R2 = 0.9963 ± 0.0010 | 0.028 | 0.231 | 0.058 | 1.40 | 110 |
| Sample ID | Expected Value [I−] (µmol L−1) | MS-Chip Method [I−] ± SD (µmol L−1) | RD (%) |
|---|---|---|---|
| #Pharm 1 | 0.81 | 0.83 ± 0.02 | 1.5 |
| #Pharm 2 | 1.76 | 1.78 ± 0.01 | 1.5 |
| #Pharm 3 | 2.52 | 2.61 ± 0.20 | 3.6 |
| #Pharm 4 | 1.58 | 1.60 ± 0.01 | 1.3 |
| #Pharm 5 | 2.36 | 2.40 ± 0.03 | 1.5 |
| #Algae 1 | 1.58 | 1.66 * ± 0.20 | 5.2 |
| Sample ID | Initial | Added | Found | Recovery (%) | ||
|---|---|---|---|---|---|---|
| [I] ± SD (µmol L−1) | RSD (%) | [I] (µmol L−1) | [I] ± SD (µmol L−1) | RSD (%) | ||
| #Algae 2 | 1.74 ± 0.01 | 0.6 | 1.58 | 3.33 ± 0.03 | 0.9 | 100 |
| #Algae 3 | 2.13 ± 0.05 | 2.3 | 0.79 | 2.97 ± 0.17 | 5.7 | 106 |
| 1.58 | 3.96 ± 0.15 | 3.8 | 116 | |||
| #Algae 4 | 0.27 ± 0.02 | 7.4 | 0.79 | 0.97 ± 0.12 | 12 | 89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda, J.L.A.; Mesquita, R.B.R.; Palacio, E.; Estela, J.M.; Cerdà, V.; Rangel, A.O.S.S. Chip-Based Spectrofluorimetric Determination of Iodine in a Multi-Syringe Flow Platform with and without In-Line Digestion—Application to Salt, Pharmaceuticals, and Algae Samples. Molecules 2022, 27, 1325. https://doi.org/10.3390/molecules27041325
Miranda JLA, Mesquita RBR, Palacio E, Estela JM, Cerdà V, Rangel AOSS. Chip-Based Spectrofluorimetric Determination of Iodine in a Multi-Syringe Flow Platform with and without In-Line Digestion—Application to Salt, Pharmaceuticals, and Algae Samples. Molecules. 2022; 27(4):1325. https://doi.org/10.3390/molecules27041325
Chicago/Turabian StyleMiranda, Joana L. A., Raquel B. R. Mesquita, Edwin Palacio, José M. Estela, Víctor Cerdà, and António O. S. S. Rangel. 2022. "Chip-Based Spectrofluorimetric Determination of Iodine in a Multi-Syringe Flow Platform with and without In-Line Digestion—Application to Salt, Pharmaceuticals, and Algae Samples" Molecules 27, no. 4: 1325. https://doi.org/10.3390/molecules27041325
APA StyleMiranda, J. L. A., Mesquita, R. B. R., Palacio, E., Estela, J. M., Cerdà, V., & Rangel, A. O. S. S. (2022). Chip-Based Spectrofluorimetric Determination of Iodine in a Multi-Syringe Flow Platform with and without In-Line Digestion—Application to Salt, Pharmaceuticals, and Algae Samples. Molecules, 27(4), 1325. https://doi.org/10.3390/molecules27041325