Abstract
In order to investigate for a new effective and safe anticancer drug, we synthesized a novel series of quinazoline containing biologically active substituted-sulfonamide moiety at 3- position 4a–n. The structure of the newly prepared compounds was proved by microanalysis, IR, 1H-NMR, 13C-NMR and mass spectral data. All the synthesized compounds were evaluated for their in vitro cytotoxic activity in numerous cancer cell lines including A549, HepG-2, LoVo and MCF-7 and normal HUVEC cell line. The two most active compounds 4d and 4f were then tested for their apoptosis induction using DNA content and Annexin V-FITC/PI staining. Moreover, apoptosis initiation was also confirmed using RT-PCR and Western blot. To further understand the binding preferences of quinazoline sulfonamides, docking simulations were used. Among the fourteen new synthesized compounds, we found that compounds 4d and 4f exerted the strongest cytotoxicity against MCF-7 cells with an IC50 value of 2.5 and 5 μM, respectively. Flow cytometry data revealed the ability of compounds 4d and 4f to mediate apoptosis and arrest cell cycle growth at G1 phase. Furthermore, RT-PCR and Western blot results suggested that both 4d and 4f activates apoptotic cell death pathway in MCF-7 cells. Molecular docking assessments indicated that compounds 4d and 4f fit perfectly into Bcl2’s active site. Based on the biological properties, we conclude that both compounds 4d and 4f could be used as a new type of anticancer agent, which provides a scientific basis for further research into the treatment of cancer.
1. Introduction
Quinazoline and sulfonamide moieties have been identified as classes of chemotherapeutic agents with substantial therapeutic value against solid tumors [1,2,3,4]. Gefitinib (small-molecule inhibitor) being the original quinazoline molecule to be approved as an antitumor agent for treatment of Non-Small Cell Lung Cancer [5,6,7]. Due to the great performances of quinazoline compounds in antitumor applications, the enhancement of new quinazoline candidates as anticancer agents is a promising topic. In fact, several selective derivatives of quinazoline-containing drugs such as lapatinib, erlotinib, and vandetanib have been approved as anticancer drugs [8,9,10] (Figure 1). On the other hand, various biological activities have been attributed to sulfonamide-containing compounds, including antitumor, carbonic anhydrase inhibition, antimalarial, and antimicrobial activities [4,11]. In the new drug design, the development of hybrid derivatives through the combination of several pharmacophores may assign compounds with remarkable biological profiles [12]. Previously, various quinazoline sulfonamide derivatives were evaluated as anticancer agents against numerous cell lines, and many compounds had displayed a promising activity [3,13,14]. Apoptosis evasion is a remarkable feature of most cancer cells; therefore, designing drugs that induce apoptosis has emerged as a promising candidate for anti-cancer optimization [15]. In fact, several previous studies have demonstrated that quinazoline scaffolds had a remarkable antiproliferative capacity against a variety of cancer cells, highlighting their efficiency in the development of apoptosis inducers drugs. Moreover, numerous novel quinazoline-derived compounds which exert effective anticancer activity via apoptosis induction have been synthesized [10,16]. Lapatinib and erlotinib are among these quinazoline-containing drugs that have been demonstrated to exert cell cycle arrest and apoptosis initiation in various cancer cells [17,18]. Consequently, efforts have been in use to synthesize a hybrid of quinazoline–sulfonamide in expectation of a promising synergistic consequence on the inhibition of cancer cell propagation. Although, there are no literature reports on substituted quinazoline–sulfonamide hybrid synthesis and its anticancer activity on human breast cancer cell line and adenocarcinoma cell line. Hybridization has confirmed to be valuable for the synthesis of new antitumor agents and overcoming the problems of the traditionally used drugs [19]. Therefore, sulfonamides were included with quinazolin-4(3H)-ones to obtain more effective candidates. As a continuation of our previous efforts [13,20,21].
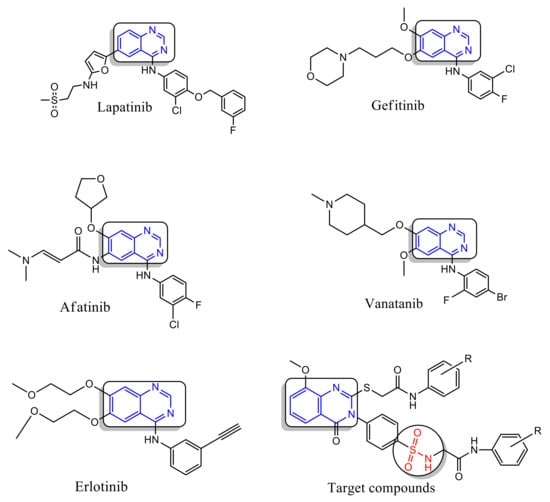
Figure 1.
Quinazoline-containing drugs.
In this respect, we designed novel quinazoline derivatives bearing substituted-sulfonamide moiety, these derivatives were subjected to in vitro cytotoxic evaluation against numerous cell lines including A549 (lung), LoVo (colon), HepG2 (liver) and MCF-7 (breast) cancer cells as well as HUVEC (normal) cell line. Moreover, the most active compounds were selected to explore its apoptotic effects against the most responsive cancer cells. Molecular docking was also performed to determine their binding mode and their ability to satisfy the pharmacophoric features required to induce the desired inhibition.
2. Results and Discussion
2.1. Chemistry
Scheme 1 shows the synthesis of quinazoline carrying biologically active substituted- benzenesulfonamide moieties 4a–n. The required strategic starting material 4-(2-mercapto-8-methoxy-4- oxoquinazolin-3(4H)-yl) benzenesulfonamide 3 was synthesized in quantitative yield by cyclocondensation of 2-amino-3-methoxybenzoic acid 1 with 4-isothiocyanatobenzenesulfonamide 2 [19], in refluxing 1,4-dioxan containing triethylamine (Scheme 1). IR spectrum of compound 3 exhibited a characteristic band for NH2, CH aromatic, C=O, C=N and SO2 groups. 1H-NMR spectrum revealed signals at 3.9 ppm assigned to OCH3 and 12.3 ppm attributed to SH proton. 13C-NMR showed signals at 55.7 ppm for OCH3 and 160.9 ppm due to C=O of quinazoline ring. The reaction of 3 with 2-chloro-N-arylacetamide derivatives was investigated. Thus, reaction of 3 with the excess of 2-chloro-N-substituted acetamide (2 mol) in dry acetone containing excess anhydrous K2CO3 (2 mol) gave the unexpected novel quinazoline incorporated substituted-sulfonamide moieties at 3-position 4a–n, rather than the expected quinazoline- containing free sulfonamide moiety at 3-position 5a–n [22] (Scheme 2). The formation of compounds 4a–n was explained in (Scheme 3). On the other hand, compounds 4a–n were obtained also in good yield via reaction of compounds 5a–n with 2-chloro-N-arylacetamide derivatives (1:1 mol) in presence of dry acetone containing (1 mol) anhydrous K2CO3. All the synthesized compounds 4a–n were established by elemental analysis, IR, 1H-NMR, 13C-NMR and mass spectral data. IR spectra of 4a–n displayed additional 2NH and 2C=O bands at their assigned regions. 1H-NMR spectra of 4a–n revealed signals at 3.7–3.9 ppm assigned to OCH3, two singlet signals, one at 3.9–4.1 ppm referring to the SCH2, 4.1–4.3 ppm due to N-CH2, and 7.5–8.0 ppm for aromatic protons (AB system), 8.0–8.2 ppm attributed for SO2NH proton and 8.8–10.5 ppm for 2NH protons. 13C-NMR of 4a–n displayed two signals peculiar to the SCH2, NCH2 and 3CO carbons. 1H-NMR spectrum of 4a showed singlet signals at 3.8 ppm for OCH3, 4.1 ppm for SCH2, 4.2 ppm for NCH2, 8.0 ppm for SO2NH and 10.3 ppm due to 2NH protons. 13C-NMR spectrum of compound 4a revealed signals at 31.6 ppm assigned to SCH2, 44.0 ppm attributed to NCH2, 56.7 ppm for OCH3, 161.4, 165.0, 165.6 ppm for 3C=O. 1H-NMR spectrum of 4b showed signals at 2.1 ppm for 2CH3 groups at the ortho positions of the phenyl ring, 3.8 ppm for OCH3, 4.1, 4.3 ppm for 2CH2, 8.1 ppm for SO2NH and 9.6 ppm due to 2NH protons. 13C-NMR spectrum of compound 4b exhibited signals at 18.1 ppm due to 2CH3 protons. 1H-NMR spectrum of 4c revealed signals at 2.3 ppm for 2CH3 group at the meta positions of the phenyl ring; 13C-NMR spectrum of compound 4c exhibited signals at 21.6 ppm due to 2CH3 protons. 1H-NMR spectrum of 4d revealed signals at 2.2 ppm for 2CH3 groups at the para positions of the phenyl ring. 13C-NMR spectrum of compound 4d showed singlet at 20.9 ppm for 2CH3 protons, 28.6, 44.0 ppm due to 2CH2 protons. 1H-NMR spectrum of 4e revealed triplets at 1.1 ppm attributed to the 2CH3 ethyl, and quartet at 2.5 ppm referring to the 2CH2 ethyl at the ortho positions of phenyl ring. 13C-NMR spectrum of compound 4e exhibited signals at 14.5 ppm for 2CH3 ethyl group, 24.0 ppm due to 2CH2 ethyl protons. 1H-NMR spectrum of 4f revealed a triplet at 1.1 ppm attributed to the 2CH3 ethyl, and quartet at 2.6 ppm referring to the 2CH2 ethyl at the meta positions. 13C-NMR spectrum of compound 4f exhibited signals at 15.9 ppm due to 2CH3 protons, 28.6 ppm for 2CH2 ethyl group. 1H-NMR spectrum of 4g revealed triplets at 1.1 ppm attributed to the 2CH3 ethyl, and quartet at 2.6 ppm referring to the 2CH2 ethyl at the para positions. 13C-NMR spectrum of compound 4g exhibited signals at 16.1 ppm due to 2CH3 protons, 28.0 ppm for 2CH2 ethyl groups. 1H-NMR spectrum of 4h revealed signals at 3.72, 3.73, 3.80 ppm for 3 OCH3 groups. 13C-NMR spectrum of compound 4h exhibited signals at 55.6 ppm due to 3 OCH3 protons. 1H-NMR spectrum of 4i revealed triplets at 1.3 ppm attributed to the 2CH3 ethoxy groups, and quartet at 4.0 ppm referring to the 2CH2 ethoxy groups at the para positions. 13C-NMR spectrum of compound 4i exhibited signals at 15.1 ppm for 2CH3 ethoxy groups, 63.5 ppm due to 2CH2 ethoxy groups. 1H-NMR spectrum of 4j revealed signals at 3.81, 3.84, 3.90 ppm for 5 OCH3 groups. 13C-NMR spectrum of 4j exhibited signals at 56.3 ppm due to 5 OCH3 protons. 1H-NMR spectrum of 4k revealed signals at 3.74, 3.77, 3.81, 3.91 ppm for 7 OCH3 groups. 13C-NMR spectrum of 4k exhibited signals at 55.3, 56.8, 62.7 ppm due to 7 OCH3 protons. IR spectra of compounds 4l–n showed bands of NO2 groups at their specified regions. 1H-NMR spectrum of 4l revealed singlet at 2.1 ppm and attributed to the 2CH3 at the ortho positions. 13C-NMR spectrum of 4l exhibited signals at 18.0 ppm for 2CH3 protons. 1H-NMR spectrum of 4m revealed singlet at 2.2 ppm attributed to the 2CH3 at the ortho positions. 13C-NMR spectrum of 4m exhibited signals at 18.0 ppm for 2CH3 protons. 1H-NMR spectrum of 4n exhibited singlet at 3.8 ppm attributed to the OCH3 moiety, 3.9, 4.2 ppm for 2CH2 protons. 13C-NMR spectrum of 4n showed signals at 28.6, 40.4 ppm assigned to 2CH2 protons, 57.3 ppm for OCH3 protons.
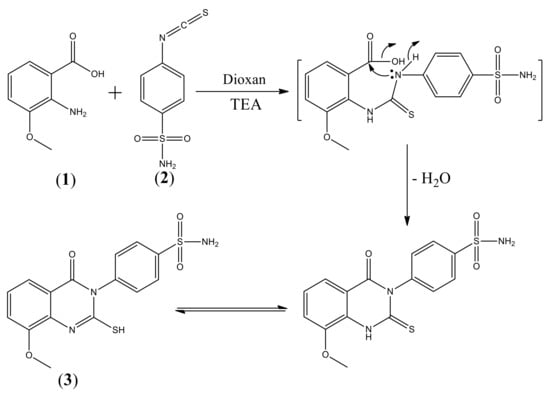
Scheme 1.
Synthesis of 4-(2-mercapto-8-methoxy-4-oxoquinazolin-3(4H)-yl)benzenesulfonamide 3.
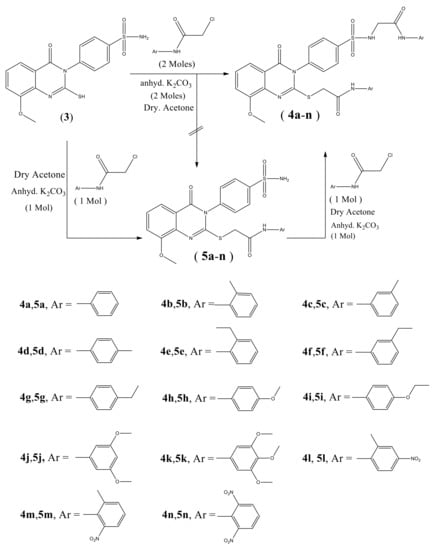
Scheme 2.
Synthesis of novel quinazolines-substituted sulfonamides 4a–n.
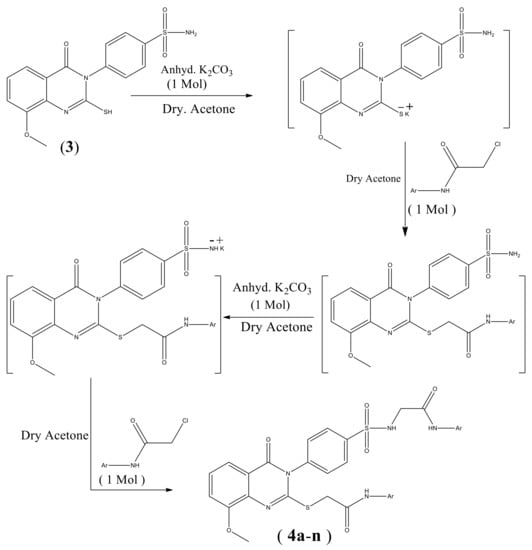
Scheme 3.
Formation of compounds 4a–n.
2.2. MTT Antiproliferative Evaluation
Compounds 4a–n were subjected to the cytotoxicity screening of the cell lines MCF-7 (breast), LoVo (colon), HepG2 (liver) and A549 (lung) carcinoma cells as well as HUVEC normal cell lines. The MTT assay was employed to assess cytotoxicity after 48 h of treatment with different concentrations of each compound and doxorubicin was used as a reference drug. The results were presented in Table 1 as growth inhibitory concentration (IC50 values). Our results demonstrate that six compounds (4a, 4b, 4d, 4f, 4h and 4i) out of fourteen exhibited promising antiproliferative activities. A remarkable antiproliferative activity was observed for compound 4d with an IC50 values of 2.5, 5.6, 6.87 and 9 µM against MCF-7, A549, LoVo and HepG2 cells, respectively. Additionally, compound 4f revealed a promising activity against all tested cell lines with an IC50 value of 5, 9.76, 10.14 and 11.7 µM against MCF-7, LoVo, A549 and HepG2 cells, respectively. Interestingly, compound 4f had a lower cytotoxic effect in terms of IC50 values against normal HUVEC cells (IC50 = 17.9). Structure activity relationship revealed that 4-tolyl 4d and 3-ethylphenyl 4f (IC50 value 2.5 and 5.0 µM) showed a demonstrated superior cytotoxic activity and are nearly as active as a reference drug doxorubicin (IC50 value 1.4 µM) against MCF-7 breast cancer cell line. It is obvious that replacement of 4-tolyl moiety 4d (IC50 value 2.5 µM) with 2-tolyl fragment 4b (IC50 value 36.2 µM) led to increase the antiproliferative activity against MCF-7 cell line, while incorporation of unsubstituted phenyl moiety as in compound 4a (IC50 value 14.2 µM) with a 4-tolyl fragment 4d (IC50 value 2.5 µM) exhibited intermediate activity. On the contrary, insertion of a methyl group at 3-position 4c abolishes the cytotoxicity, as opposed to compound 4d (IC50 value 2.5 µM) containing the methyl group in 4-position. On the other hand, the orientation of ethyl group significantly affected the cytotoxic activity of compounds 4e, 4f, and 4g where the presence of it at 3-position as in compound 4f enhanced the cytotoxic activity (IC50 value 5.0 µM) compared to compounds 4e and 4g carrying the ethyl group at 2-position and 4-position, respectively, which assigned no antiproliferative completely. Additionally, we observed that 4d and 3-ethylphenyl moiety 4f had a considerable potent cytotoxic activity compared to the corresponding unsubstituted phenyl 4a (IC50 value 14.2 µM) and 2-tolyl 4b (IC50 value 36.2 µM). Based on this result, compounds 4d and 4f were selected for further investigations.

Table 1.
Cytotoxic activity, expressed as IC50, of synthetic compounds 4a–n and doxorubicin against different cancer cell lines.
2.3. Compounds 4d and 4f Induces G1-Phase Cell Cycle Arrest on MCF-7 Cells
Because compounds 4d and 4f displayed the highest activity towards MCF-7 cells, we looked into their cellular effects with respect to cell cycle progression and cell death mode. Indeed, designing of new chemotherapeutic drugs targeting cell cycle progression and apoptosis is considered as an attractive strategy [23,24]. To explore the antiproliferative effect of compounds 4d and 4f, cell-cycle analysis was performed in MCF-7 cells were examined. The cells were treated with various concentrations of both compound for 24 h, stained with propidium iodide (PI) and analyzed by flow cytometry. As shown in (Figure 2), the incubation of MCF-7 cells with (1.25, 2.5, and 5 μM) of compound 4d for 24 h increased the percentage of cells at G1 (from 57.91 ± 0.61% in control) to 62.51 ± 0.14%, 64.95 ± 0.21% and 66.55 ± 0.78% in treated cells, respectively. At the same conditions, the percentage of cells in S and G2/M phases were obviously decreased. These results revealed that compound 4d arrested the cell cycle at the G1 stage. In the same manner, DNA content analyses also showed a statistically significant increase in the number of cells in the G1 phase of MCF-7 cells treated with compound 4f (increased to 61.65 ± 0.21% at 1.25 μM, 62.65 ± 0.35% at 2.5 μM and 65.15 ± 0.36% at 5 μM) when compared to control (57.9 ± 0.61%) (Figure 3). Collectivity, our data indicated that both compounds 4d and 4f inhibit MCF-7 cells proliferation by modulating cell cycle progress at G1 phase which is a critical stage in the cell cycle as well as a worth target for drug action [25].
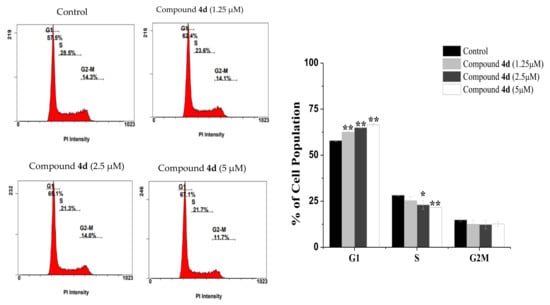
Figure 2.
The effects of compound 4d on cell cycle distribution. MCF-7 cells untreated (control) or treated with the indicated concentrations of compound 4d for 24 h followed by staining with PI dye. Representative histograms (upper) and graph bar (down) showed the variation in the percentage of cells in each phase of the cell cycle. Results are presented as the mean ± SD. Differences between treated and control groups were considered statistically significant at p < 0.05 (*) and p < 0.01 (**).
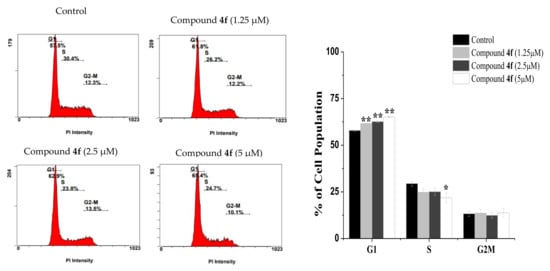
Figure 3.
Cell cycle distribution following 24 h exposure to different concentration of compound 4f. A statistically significant increase in the number of cells in G1 phase was observed in treated cells when compared to control cells. * p < 0.05, ** p < 0.01.
2.4. Apoptosis Quantification
In addition to cell cycle arrest, apoptosis is another important anticancer process that inhibits cancer cell proliferation [26]. Therefore, we further used a double labeling staining with Annexin V-FITC and PI to investigate apoptotic cell death. Phosphatidylserine exposure on the cell surface is a hallmark of early and late apoptotic cells, which can be easily identified with Annexin V-FITC [27]. As shown in (Figure 4), early apoptotic cells were increased along with the concentrations increment of compound 4d from 1.05 ± 0.07% in untreated MCF-7 cells to 44.45 ± 1.06% at 1.25 µM, (p < 0.05), 67.55 ± 1.34% at 2.5 µM (p < 0.01), and 83.75 ± 2.19% at 5 µM (p < 0.001). For compound 4f (Figure 5), it was noticed that most treated cells were in a late apoptotic stage which were increased in a dose-dependent manner (from 1.75 ± 0.07% in control to 15.4 ± 0.28%, 31.05 ± 1.06% and 52.4 ± 0.28% at 1.25, 2.5 and 10 µM, respectively). Interestingly, we noticed that most treated cells were in early apoptotic stage after 4d compound treatment while most cells populations were in a late apoptotic stage with compound 4f and these different effects of both compounds could be due to their difference attached functional groups (4-tolyl in 4d and 3-ethylphenyl in 4f) which may be targeting different sites for inducing apoptosis in different time periods which need further investigation. Overall, the findings obviously showed that both compounds effectively triggered apoptosis in MCF-7 cells.
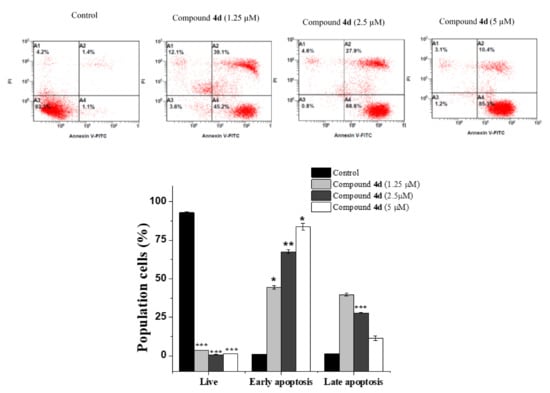
Figure 4.
Annexin V-FITC/PI data of untreated and treated MC-7 cells with compound 4d at the indicated concentrations for 24 h. Flow cytometric plot: A3 (live), A4 (early apoptosis), A2 (late apoptosis) and A1 (necrosis). The bar graph depicts the variation in the percentage of cells in different stages. Results are presented as the mean ± SD and differences between treated and control groups were considered statistically significant at * p < 0.05, ** p < 0.01, *** p < 0.001).
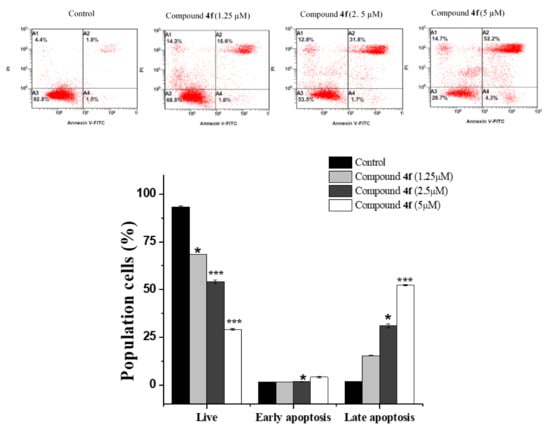
Figure 5.
Flow cytometry analysis of compound 4f for MCF-7 cells following 24 h incubation period at indicated concentrations. Dot plot of Annexin V-FITC/PI and bar quantification for evaluation of apoptosis. Values are presented as means ± SD. Significant differences from control group: * p < 0.05, and *** p < 0.001.
2.5. Effect of Compounds 4d and 4f on Apoptosis Regulatory Markers
We next used reverse transcription polymerase chain reaction (RT-PCR) and Western blot analysis to look at Bax (proapoptotic), Bcl-2 (antiapoptotic) and caspase 7 (executioner caspase) expression at gene and protein level as an additional marker of apoptosis occurrence [28]. In fact, apoptotic cell death is mostly directed by the levels of Bcl-2 family, whether they are pro-apoptotic or antiapoptotic and targeting these molecules as a therapeutic strategy is continuously under development [29]. As predicted, we observed an upregulation in the expression of Bax in MCF-7 treated cells to (1.7-, 2- and 2.4-fold) with compound 4d and to (1.2-, 1.4- and 1.6-fold) with compound 4f at (1.25, 2.5 and 5 µM) respectively (Figure 6). In contrast, Bcl-2 expression was downregulated into (0.88-, 0.68- and 0.49-fold) with 4d and into (0.92, 0.68 and 0.62) with 4f at indicated concentration in compare to DMSO-vehicle treated cells. Our results also showed that both 4d and 4f caused a significant increase in the expression of caspase 7 to (1.13-, 1.35- and 1.58-fold) relative to untreated MCF-7 cells (Figure 6). Consistently, Western blot analysis revealed that compounds 4d and 4f treatment markedly increased the expression of Bax whereas Bcl-2 was downregulated in a dose-dependent manner compared to that of control cells (Figure 7). This shifting in Bax to Bcl-2 ratio usually started caspases activation which plays a critical role in apoptosis execution [26,30]. Among all caspases, caspase 7 is considered as one of the most crucial executioner caspases that mediate cell death, and its expression strongly correlates with apoptosis stimulation [31,32]. Hence, we evaluated whether compounds 4d and 4f (1.25, 2.5 and 5 μM) promote the caspase 7 expression at protein level. We found that caspase 7 protein expression significantly increased after cell treatment with compound 4d (3-, 3.3- and 3.9-fold) and (1.14-, 1.2- and 1.5-fold) with compound 4f in comparison to the control, which suggested involvement of caspase 7 activation during compounds treatment. In line with our findings, several studies have demonstrated the apoptotic behavior of quinazoline derivatives through upregulating the proapoptotic genes and downregulating the antiapoptotic genes [33,34,35]. In particular, quinazoline-based anticancer drugs such as lapatinib and gefitinib were found to inhibit cancer cells growth through increasing the Bax and caspases gene levels and downregulating Bcl-2 levels, which is consistent with our finding in this study [36,37].
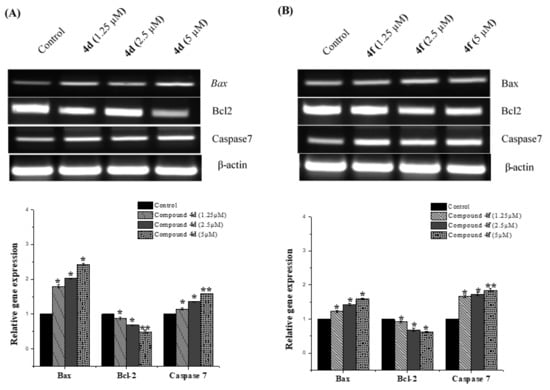
Figure 6.
RT-PCR analysis of apoptotic related genes. The RT-PCR expression profile of Bax, Bcl2 and caspase 7 genes were demonstrated in MCF-7 cells after treating them without and with different concentration of compounds 4d (A) and 4f (B) for 24 h. A dose dependent increase in the expression of Bax and caspase 7 with the increasing concentration of the compounds 4d and 4f (1.25, 2.5 and 5 µM) was documented while Bcl-2 was downregulated as compared to that of the control where the cells untreated. The levels of mRNA, normalized to that of the β-actin gene, were calculated (histograms). The results were expressed as the mean ± SD. * p < 0.05; ** p < 0.01 vs. normal control group.
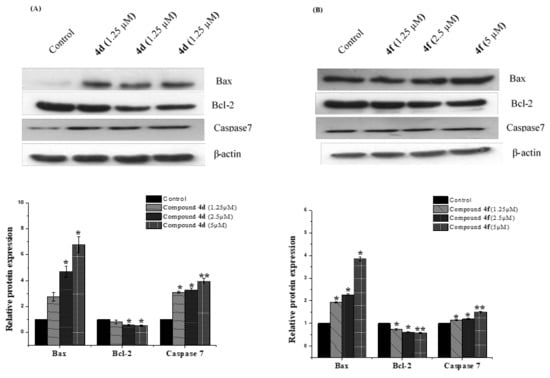
Figure 7.
Impact of compounds 4d and 4f at various on the expression of apoptosis related genes. MCF-7 cells were treated with vehicle or indicated concentrations (1.25, 2.5 and 5 μM), respectively, for 24 h time period. (A) After the treatment, Western blot analysis of Bax, Bcl-2 and caspase 7 was performed. Loading was normalized to the levels of β-actin (B) Band quantification of target proteins was performed using Image j software. Data shown are means ± SD (n = 3) (* p < 0.05, ** p < 0.01).
2.6. Molecular Docking
Overexpression of anti-apoptotic Bcl-2 protein has been linked to cancer progression and cell death avoidance. Hence, developing novel Bcl2 inhibitors is considered a promising field in designing chemotherapeutic agents [38]. The molecular docking procedure executed in this study was authenticated as reported by [39]. The X-ray bound ligand, i.e., 1-(2-{[(3S)-3-(aminomethyl)-3,4-dihydroisoquinolin-2(1H)-yl]carbonyl}phenyl)-4-chloro-5-methyl-N,N-diphenyl-1H-pyrazole-3-carboxamide (DRO), was extracted from the crystal structure and re-docked to Bcl-2. The poses of the bound and re-docked ligand were compared, and the root mean square deviation (RMSD) was calculated. The RMSD of the re-docked DRO was found to be 0.3842 Å (Figure S1). Because the calculated RMSD was within the acceptable limit (2.0 Å), we were confident in adopting the docking protocol to predict the binding of compounds 4d and 4f with Bcl-2.
The results of molecular docking between ligands 4d and 4f and Bcl-2 were compared with that of DRO, and the results are given in (Table 2, Figure 8 and Figure 9). We found that both compounds 4d and 4f occupied a similar position at the Bcl-2 binding site as occupied by control ligand DRO (Figure 8A,B). The DRO-Bcl-2 complex was stabilized by a carbon interaction and an electrostatic interaction with the Asp70 residue (Figure 8C), as well as ten hydrophobic interactions with Phe63, Tyr67, Phe71, Met74, Val92, Leu96, and Ala108 (Table 2). The DRO-Bcl-2 complex was further stabilized by van der Waals’ interactions of DRO with the Arg88, Glu95, Gly104, Arg105, and Phe112 residues of Bcl-2. Analysis of the interactions between compound 4d and Bcl-2 revealed that the protein–ligand complex was stabilized by one conventional hydrogen bond with Glu95, and seven hydrophobic interactions with Tyr67, Phe71, Met74, Val92, Leu96, and Ala108 (Table 2). The compound 4d-Bcl-2 complex was further stabilized by van der Waals’ interactions of the compound with several other residues, including Phe63, Asp70, Gly104, and Arg105 (Figure 9A). Similarly, the compound 4f-Bcl-2 complex was stabilized by one conventional hydrogen bond with Tyr67, and eight hydrophobic interactions with Phe63, Tyr67, Met74, Glu95, Leu96, Arg105 and Ala108 (Table 2). Additionally, the Asp70, Phe71, Arg98, Asp99, Gly104, and Phe112 residues of Bcl-2 formed van der Waals’ interactions to stabilize the compound 4f-Bcl-2 complex (Figure 9B). In comparison, the binding energies and the corresponding binding affinities of DRO, compounds 4d and 4f towards Bcl-2, were estimated to be −10.2 kcal mol−1 and 3.03 × 107 M−1, −7.8 kcal mol−1 and 5.25 × 105 M−1, and −7.5 kcal mol−1 and 3.17 × 105 M−1, respectively (Table 1). It is interesting to note that Phe63, Tyr67, Asp70, Phe71, Met74, Val92, Glu95, Leu96, Gly104, Arg105, Ala108, and Phe112 residues of Bcl-2 showed interactions with both compounds 4d and DRO. Similarly, Phe63, Tyr67, Asp70, Phe71, Met74, Glu95, Leu96, Gly104, Arg105, Ala108, Phe112 residues of Bcl-2 showed interactions with compound 4f as well as the control ligand DRO.
Table 2.
Molecular docking of compound 4d and 4f with Bcl-2.
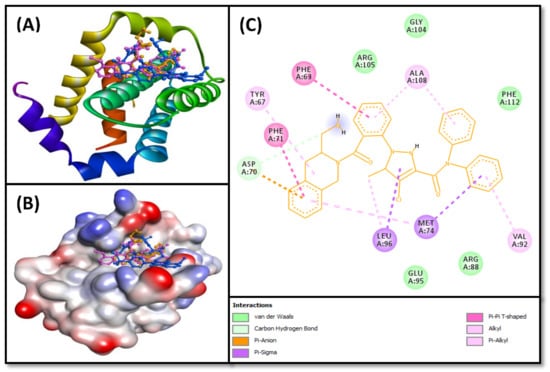
Figure 8.
(A) 2D cartoon representation of the binding of DRO (control ligand), compounds 4d and 4f at the binding site of the Bcl-2 protein, and (B) 3D representation of the binding of DRO (control ligand), compounds 4d and 4f at the binding site of the Bcl-2 protein. DRO, compounds 4d and 4f are represented by gold, pink, and blue, respectively. (C) Molecular docking and interaction between Bcl-2 protein and DRO (control ligand).
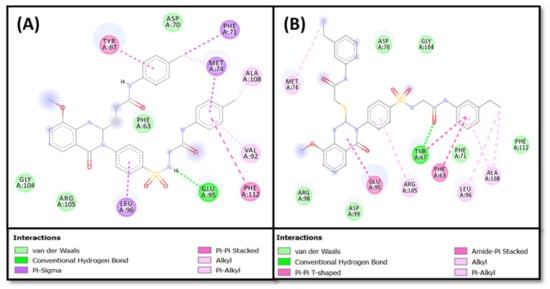
Figure 9.
Molecular docking and interaction of (A) compound 4d, and (B) compound 4f, with the Bcl-2 protein.
3. Experimental
3.1. Chemistry
Melting points (uncorrected) were determined in open capillary on a Gallen Kamp melting point apparatus (Sanyo Gallen Kamp, Southborough, UK). Precoated silica gel plates (Kieselgel 0.25 mm, 60 F254, Merck, Darmstadt, Germany) were used for thin-layer chromatography. A developing solvent system of chloroform/methanol (8:2) was used and the spots were detected by ultraviolet light. IR spectra (KBr disc) were recorded using an FT-IR spectrophotometer (Perkin Elmer, Norwalk, CT, USA). 1H-NMR spectra were scanned on an NMR spectrophotometer (Bruker AXS Inc., Flawil, Switzerland), operating at 500 MHz for 1H- and 125.76 MHz for 13C. Chemical shifts are expressed in δ-values (ppm) relative to TMS as an internal standard, using DMSO-d6 as a solvent. Elemental analyses were done on a model 2400 CHNSO analyser (Perkin Elmer, Norwalk, CT, USA). All the values were within ±0.4% of the theoretical values. All reagents used were of AR grads.
- 4-(2-Mercapto-8-methoxy-4- oxoquinazolin-3(4H)-yl) benzenesulfonamide (3)
A mixture of 2-amino-3-methoxybenzoic acid 1 (1.67 g, 0.01 mol) and 4-isothiocyanatobenzenesulfonamide 2 (2.14 g, 0.01 mol) in dioxan (50 mL) containing 3 drops of triethylamine, was refluxed for 8 h. The solid product formed while hot was collected by filtration and recrystallized from ethanol to give 3. 3: Yield, 89%; m.p. 342–344 °C. IR: 3321, 3262, 3181 (NH2), 3055 (arom.), 1691 (CO), 1620 (CN), 1381, 1156 (SO2). 1HNMR: 3.9 (s, 3H, OCH3), 7.0–7.7 [m, 5H, Ar-H + SO2NH2], 7.8, 8.0 (2d. 4H, AB system, Ar-H), 12.3 [s, 1H, SH]. 13CNMR: 55.7, 116.2, 118.6, 119.8, 120.6 (2), 127.0, 128.9 (2), 130.1, 134.6, 135.3, 150.4, 160.5, 160.9. MS m/z (%): 363 (M+) (14.54), 155 (100). Anal. Calcd. for C15H13N3O4S2 (363.41): C, 49.57; H, 3.61; N, 11.56. Found: C, 49.22; H, 3.86; N, 11.31. Quinazolin-3(4H)-yl)phenylsulfonamido)-N-phenylacetamide derivatives (4a-n).
3.2. General Procedure
3.2.1. Method A
A mixture of 3 (4.59 g, 0.01 mol) and 2-chloro-N-substitutedacetamide derivatives (0.02 mol) in dry acetone (50 mL) and anhydrous K2CO3 (2.56 g, 0.02 mol) was stirred at room temperature for 24 h., filtered and the solid product formed was recrystallized from ethanol to give 4a–n.
3.2.2. Method B
A mixture of 5a–n [20] (0.01 mol) and 2-chloro-N-substitutedacetamide derivatives (0.01 mol) in dry acetone (50 mL) and anhydrous K2CO3 (1. 38 g, 0.01 mol) was stirred at room temperature for 18 h, filtered and the solid product formed was recrystallized from ethanol to give 4a–n.
- 2-(4-(8-Methoxy-4-oxo-2-((2-oxo-2-(phenylamino)ethylthio)quinazolin-3(4H)-yl)phenylsul-fonamido)-N-phenylacetamide (4a).
5: Yield, 90%; m.p. 165–167 °C. IR: 3300, 3141 (NH), 3066 (arom.), 2965, 2844 (aliph.), 1700, 1693, 1665 (3CO), 1612 (CN), 1377, 1154 (SO2). 1HNMR: 3.8 [s, 3H, OCH3], 4.1 [s, 2H, SCH2], 4.2 [s, 2H, NCH2], 7.5, 7.6 (2d, 4H, AB system, Ar-H), 7.0- 7.3 [m, 13H, Ar-H), 8.0 (s, 1H, SO2NH], 10.3 [s, 2H, 2NH]. 13CNMR: 31.6, 44.0, 56.7, 116.2, 116.8, 119.8 (4), 120.6, 124.3 (2), 129.3 (2), 129.4 (4), 129.8 (2), 131.7 (2), 137.0, 138.9 (2), 145.7, 154.0, 158.8, 161.4, 165.0, 165.6. MS m/z (%): 629 (M+) (31.43), 231 (100). Anal. Calcd. for C31H27N5O6S2 (629.71): C, 59.13; H, 4.32; N, 11.12. Found: C, 59.45; H, 4.65; N, 10.83.
- 2-(4-(8-Methoxy-4-oxo-2-((2-oxo-2-(o-tolylamino)ethylthio)quinazolin-3(4H)-yl)phenylsul-fonamido)-N-(o-tolyl)acetamide (4b).
4b: Yield, 90%; m.p. 102–104 °C. IR: 3354, 3154 (NH), 3036 (arom.), 2972, 2873 (aliph.), 1698, 1668, 1660 (3CO), 1610 (CN), 1386, 1166 (SO2). 1HNMR: 2.1 [s, 3H, CH3], 3.8 [s, 3H, OCH3], 4.1 [s, 2H, SCH2], 4.3 [s, 2H, NCH2], 7.1, 7.7 [m, 11H, Ar-H), 7.9, 8.0 (2d, 4H, AB system, Ar-H), 8.1 (s, 1H, SO2NH], 9.6 [s, 2H, 2NH]. 13CNMR: 18.1 (2), 31.6, 43.6, 57.8, 116.6, 117.8, 121.0, 124.8 (2), 125.6 (2), 126.1 (2), 126.5, 129.6 (2), 130.8 (2), 132.4 (2), 132.6 (2), 136.0, 137.9, 138.2 (2), 139.8, 147.6, 155.6, 161.2, 165.6, 165.9. MS m/z (%): 657 (M+) (11.84), 188 (100). Anal. Calcd. for C33H31N5O6S2 (657.76): C, 60.26; H, 4.75; N, 10.65. Found: C, 60.60; H, 4.39; N, 10.36.
- 2-(4-(8-Methoxy-4-oxo-2-((2-oxo-2-(m-tolylamino)ethylthio)quinazolin-3(4H)-yl)phenylsul-fonamido)-N-(m-tolyl)acetamide (4c).
4c: Yield, 84%; m.p. 112–114 °C. IR: 3309, 3144 (NH), 3078 (arom.), 2954, 2863 (aliph.), 1692, 1677, 1661 (3CO), 1618 (CN), 1377, 1151 (SO2). 1HNMR: 2.3 [s, 6H, 2CH3], 3.9 [s, 3H, OCH3], 4.0 [s, 2H, SCH2], 4.2 [s, 2H, NCH2], 6.8–7.7 [m, 11H, Ar-H), 7.6, 8.0 (2d, 4H, AB system, Ar-H), 9.6 (s, 1H, SO2NH], 10.1 [s, 2H, 2NH]. 13CNMR: 21.6 (2), 26.7, 40.3, 56.7, 116.8, 117.1 (2), 117.4, 120.1 (2), 120.7, 124.5 (2), 124.8 (2), 127.4, 128.9 (2), 129.0 (2), 130.7, 138.2, 138.3 (2), 138.7 (2), 141.3, 155.6, 161.2, 165.0, 168.5, 171.3. MS m/z (%): 657 (M+) (3.76), 154 (100). Anal. Calcd. for C33H31N5O6S2 (657.76): C, 60.26; H, 4.75; N, 10.65. Found: C, 60.01; H, 4.43; N, 10.89.
- 2-(4-(8-Methoxy-4-oxo-2-((2-oxo-2-(p-tolylamino)ethylthio)quinazolin-3(4H)-yl)phenylsul-fonamido)-N-(p-tolyl)acetamide (4d).
4d: Yield, 84%; m.p. 160–162 °C. IR: 3353, 3132 (NH), 3064 (arom.), 2976, 2845 (aliph.), 1695, 1676, 1660 (3CO), 1620 (CN), 1387, 1155 (SO2). 1HNMR: 2.2 [s, 6H, 2CH3], 3.8 [s, 3H, OCH3], 4.0 [s, 2H, SCH2], 4.2 [s, 2H, NCH2], 7.1–7.4 [m, 3H, Ar-H), 7.6, 8.0 (2d, 12H, AB system, Ar-H), 8.1 (s, 1H, SO2NH], 10.2 [s, 2H, 2NH]. 13CNMR: 20.9 (2), 28.6, 44.0, 56.7, 118.0, 119.8, 120.8 (4), 120.9 (3), 129.6 (5), 129.7 (2), 129.9, 133.2 (2), 136.4, 138.0 (2), 139.2, 153.8, 158.6, 164.8, 165.2 (2). MS m/z (%): 657 (M+) (17.54), 224 (100). Anal. Calcd. for C33H31N5O6S2 (657.76): C, 60.26; H, 4.75; N, 10.65. Found: C, 60.50; H, 4.55; N, 10.44.
- N-(2-ethylphenyl)-2-((3-(4-(N-(2-((2-ethylphenyl)amino)-2-oxoethyl)sulfamoyl)-phe-nyl)- 8-methoxy-4-oxo-3,4-dihydroquinazolin-2yl) thio)acetamide (4e).
4e: Yield, 64%; m.p. 246–248 °C. IR: 3409, 3256 (NH), 3075 (arom.), 2944, 2856 (aliph.), 1694, 1683, 1665 (3CO), 1617 (CN), 1398, 1155 (SO2). 1HNMR: 1.0 (t, 6H, 2 CH3 ethyl), 2.5 (q, 4H, 2CH2 ethyl), 3.8 [s, 3H, OCH3], 3.9 [s, 2H, SCH2], 4.1 [s, 2H, NCH2], 7.1–7.7 [m, 11H, Ar-H), 7.9, 8.0 (2d, 4H, AB system, Ar-H), 8.1 (s, 1H, SO2NH], 9.6 [s, 2H, 2NH]. 13CNMR: 14.5 (2), 24.0 (2), 31.0, 40.3, 56.7, 116.3, 118.1, 120.9, 126.3 (2), 126.4 (2), 127.1 (2), 127.4 (2), 128.9 (2), 130.3 (2), 130.7 (2), 135.8 (2), 138.0 (2), 139.1 (2), 139.2, 153.7, 161.0, 166.4 (2). MS m/z (%): 685 (M+) (23.62), 288 (100). Anal. Calcd. for C35H35N5O6S2 (685.81): C, 61.30; H, 5.14; N, 10.21. Found: C, 61.00; H, 5.37; N, 10.46.
- N-(3-ethylphenyl)-2-((3-(4-(N-(2-((3-ethylphenyl)amino)-2-oxoethyl)sulfamoyl)-phe-nyl)-8-methoxy-4-oxo-3,4-dihydroquinazolin-2yl) thio)acetamide (4f).
4f: Yield, 57%; m.p. 239–241 °C. IR: 3321, 3123 (NH), 3084 (arom.), 2975, 2866 (aliph.), 1691, 1685, 1660 (3CO), 1619 (CN), 1376, 1151 (SO2). 1HNMR: 1.2 (t, 6H, 2CH3 ethyl), 2.6 (q, 4H, 2CH2), 3.8 [s, 3H, OCH3], 4,0 [s, 2H, SCH2], 4.2 [s, 2H, NCH2], 6.9–7.6 [m, 11H, Ar-H), 7.6, 8.0 (2d, 4H, AB system, Ar-H), 9.5 (s, 1H, SO2NH], 10,4 [s, 2H, 2NH]. 13CNMR: 15.9 (2), 28.6 (2), 28.7, 40.3, 56.7, 116.4, 117.0 (2), 117.2, 118.1 (2), 119.1, 120.9 (2), 123.8 (2), 127.0, 127.4 (2), 129.2 (2), 130.7, 138.0, 139.3 (2), 144.7, 145.9 (2), 153.7, 155.6, 160.9, 165.0, 166.0. MS m/z (%): 685 (M+) (10.74), 412 (100). Anal. Calcd. for C35H35N5O6S2 (685.81): C, 61.30; H, 5.14; N, 10.21. Found: C, 61.03; H, 4.87; N, 10.06.
- N-(4-ethylphenyl)-2-((3-(4-(N-(2-((4-ethylphenyl)amino)-2-oxoethyl)sulfamoyl)-phenyl)-8-methoxy-4-oxo-3,4-dihydroquinazolin-2yl)thio)acetamide (4g).
4g: Yield, 64%; m.p. 219–221 °C. IR: 3409, 3256 (NH), 3075 (arom.), 2944, 2856 (aliph.), 1694, 1683, 1665 (3CO), 1617 (CN), 1398, 1155 (SO2). 1HNMR: 1.1 (t, 6H, 2CH3 ethyl), 2.5 (q, 4H, 2CH2 ethyl), 3.8 [s, 3H, OCH3], 4.0 [s, 2H, SCH2], 4.2 [s, 2H, NCH2], 7.1–7.5 [m, 3H, Ar-H), 7.6, 8.0 (2d, 12H, AB system, Ar-H), 8.1 (s, 1H, SO2NH], 10.2 [s, 2H, 2NH]. 13CNMR: 16.1 (2, 28.0 (3), 44.0, 58.6, 115.6, 117.8, 119.9 (4), 120.1, 121.6 (2), 126.7 (4), 127.8, 128.5 (2), 129.8, 136.6 (2), 136.8, 139.7, 140.2 (2), 153.9, 161.2, 164.8, 165.6 (2). MS m/z (%): 685 (M+) (23.62), 288 (100). Anal. Calcd. for C35H35N5O6S2 (685.81): C, 61.30; H, 5.14; N, 10.21. Found: C, 61.66; H, 5.39; N, 10.04.
- 2-(4-(8-Methoxy-2-((2-((4-methoxyphenyl)amino)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)phenylsulfonamido)-N-(4-methoxy-phenyl)acetamide (4h).
4h: Yield, 71%; m.p. 117–119 °C. IR: 3350, 3231 (NH), 3054 (arom.), 2986, 2891 (aliph.), 1703, 1692, 1664 (3CO), 1611 (CN), 1364, 1152 (SO2). 1HNMR: 3.72, 3.73, 3.88 [3s, 9H, 3OCH3], 4.1 [s, 2H, SCH2], 4.2 [s, 2H, NCH2], 6.9, 7.5 (2d, 8H, AB system, Ar-H), 7.5–7.6 (m, 3H, Ar-H), 7.6, 8.0 (2d, 4H, AB system, Ar-H), 8.1 (s, 1H, SO2NH], 10.5 [s, 2H, 2NH]. 13CNMR: 31.3, 43.9, 55.6 (3), 114.4 (4), 116.8, 118.6, 121.4, 123.7 (2), 125.8 (4), 125.9, 128.6 (2), 129.1 (2), 132.0, 137.2, 137.6, 155.9, 156.0 (2), 162.6, 164.6, 165.2 (2). MS m/z (%): 689 (M+) (9.86), 312 (100). Anal. Calcd. for C33H31N5O8S2 (689.76): C, 57.46; H, 4.53; N, 10.15. Found: C, 57.13; H, 4.23; N, 10.35.
- N-(4-Ethoxyphenyl)-2-((3-(4-N-(2((4-ethoxyphenyl)amino)-2-oxoethyl)sulfamoyl)-phenyl-8-methoxy-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (4i).
4i: Yield, 66%; m.p. > 350 °C. IR: 3343, 3222 (NH), 3077 (arom.), 2965, 2836 (aliph.), 1698, 1685, 1666 (3CO), 1611 (CN), 1367, 1155 (SO2). 1HNMR: 1.3 (t, 6H, 2CH3 ethyl), 3.9 [s, 3H, OCH3], 4.0 (q, 4H, 2CH2 ethyl), 4.1 [s, 2H, SCH2], 4.2 [s, 2H, NCH2], 6.8, 7.4 (2d, 8H, AB system, Ar-H), 7.6–7.8 (m, 3H, Ar-H), 7.9, 8.0 (2d, 4H, AB system, Ar-H), 8.1 (s, 1H, SO2NH], 10.1 [s, 2H, 2NH]. 13CNMR: 15.1 (2), 31.3, 43.9, 57.2, 63.5 (2), 114.9 (4), 116.0, 118.6, 121.3, 121.4 (2), 126.2 (4), 126.3, 129.7 (2), 131.9 (2), 132.0, 138.9, 139.0, 155.1, 157.4 (2), 161.6, 164.5, 165.1 (2). MS m/z (%): 717 (M+) (51.82), 365 (100). Anal. Calcd. for C35H35N5O8S2 (717.81): C, 58.56; H, 4.91; N, 9.76. Found: C, 58.21; H, 4.65; N, 9.45.
- N-(3,5-Dimethoxyphenyl)-2-((3-(4-N-(2((3,5-dimethoxyphenyl)amino)-2-oxoethyl)-sulfa-moyl)phenyl-8-methoxy-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (4j).
4j: Yield, 65%; m.p. 239–241 °C. IR: 3384, 3198 (NH), 3045 (arom.), 2978, 2865 (aliph.), 1700, 1681, 1662 (3CO), 1612 (CN), 1356, 1154 (SO2). 1HNMR: 3.81, 3.84, 3.90 [3s, 15H, 5OCH3], 4.0 [s, 2H, SCH2], 4.1 [s, 2H, NCH2], 6.2–7.6 [m, 9H, Ar-H), 7.8, 8.0 (2d, 4H, AB system, Ar-H), 8.2 (s, 1H, SO2NH], 10.1 [s, 2H, 2NH]. 13CNMR: 27.6, 41.8, 56.3 (5), 97.2 (2), 101.0 (4), 114.0, 117.2, 120.3, 120.9 (2), 127.4, 128.3 (2), 131.9, 134.6, 139.7, 140.6 (2), 151.8, 158.6, 159.0 (4), 161.2, 167.4, 168.2, MS m/z (%): 749 (M+) (1.85), 147 (100). Anal. Calcd. for C35H35N5O10S2 (749.81): C, 56.06; H, 4.70; N, 9.34. Found: C, 56.29; H, 4.45; N, 9.66.
- 2-(4-(8-Methoxy-4-oxo-2-((2-oxo-2-(3,4,5 trimethoxyphenyl)amino)ethyl)thio)quinazolin-3(4H)-yl)phenylsulfonamido)-N-(3,4,5-trimethoxyphenyl)acetamide (4k).
4k: Yield, 78%; m.p. 260–262 °C. IR: 3377, 3154 (NH), 3043 (arom.), 2933, 2875 (aliph.), 1690, 1672, 1665 (3CO), 1623 (CN), 1376, 1151 (SO2). 1HNMR: 3.74, 3.77, 3.81, 3.91 [4s, 21H, 7OCH3], 4.0 [s, 2H, SCH2], 4.1 [s, 2H, NCH2], 6.9–7.7 [m, 7H,), 7.8, 8.0 (2d, 4H, AB system, Ar-H), 8.3 (s, 1H, SO2NH], 10.2 [s, 2H, 2NH]. 13CNMR: 28.3, 42.7, 55.3, 56.8 (4), 62.7 (2), 97.0 (4), 114.6, 120.6, 120.8, 121.2 (2), 129.8, 130.5 (2), 131.7, 133.0, 134.2, 136.4 (2), 137.1, 139.6, 151.8, 155.1 (4), 158.3, 161.0, 168.1, 168.9. MS m/z (%): 809 (M+) (3.42), 364 (100). Anal. Calcd. for C37H39N5O12S2 (809.86): C, 54.87; H, 4.85; N, 8.65. Found: C, 54. 49; H, 4.58; N, 8.28.
- 2-(4-(8-Methoxy-2-((2-((2-methyl-4-nitrophenyl)amino)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)phenylsulfonamido)-N-(2-methyl-4-nitrophenyl)acetamide (4l).
4l: Yield, 61%; m.p. 127–129 °C. IR: 3325, 3245 (NH), 3076 (arom.), 2976, 2843 (aliph.), 1694, 1682, 1660 (3CO), 1608 (CN), 1387, 1161 (SO2). 1HNMR: 2.1 (s, 6H, 2CH3), 3.7 [s, 3H, OCH3], 3.9 [s, 2H, SCH2], 4.2 [s, 2H, NCH2], 7.4–7.8 (m, 9H, Ar-H), 7.9, 8.0 (2d, 4H, AB system, Ar-H), 8.1 (s, 1H, SO2NH], 9.9 [s, 2H, 2NH]. 13CNMR: 18.0 (2), 28.6, 40.3, 56.5, 112.2 (2), 116.3, 118.0, 120.9 (2), 122.2, 125.8 (2), 127.1 (2), 127.4 (2), 130.7 (2), 130.9, 138.0, 139.1 (2), 143.2 (2), 145.9 (2), 153.7, 161.0, 166.9 (2), 172.0. MS m/z (%): 747 (M+) (16.66), 384 (100). Anal. Calcd. for C33H29N7O10S2 (747.75): C, 53.01; H, 3.91; N, 13.11. Found: C, 53.33; H, 3.62; N, 13.43.
- 2-(4-(8-Methoxy-2-((2-((2-methyl-6-nitrophenyl)amino)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)phenylsulfonamido)-N-(2-methyl-6-nitrophenyl)acetamide (4m).
4m: Yield, 85%; m.p. 272–274 °C. IR: 3340, 3212 (NH), 3055 (arom.), 2954, 2865 (aliph.), 1695, 1684, 1661 (3CO), 1618 (CN), 1377, 1151 (SO2). 1HNMR: 2.2 (s, 6H, 2CH3), 3.8 [s, 3H, OCH3], 3.9 [s, 2H, SCH2], 4.1 [s, 2H, NCH2], 7.4–7.5 (m, 9H, Ar-H), 7.6, 8.0 (2d, 4H, AB system, Ar-H), 8.1 (s, 1H, SO2NH], 10.2 [s, 2H, 2NH]. 13CNMR: 18.0 (2), 31.6, 40.4, 56.7, 115.3, 118.1, 120.9, 122.6 (2), 123.0 (2), 126.7 (2), 127.1 (2), 127.4, 127.5 (2), 128.8 (2), 130.3, 135.4 (2), 137.7, 137.9 (2), 139.0, 153.7, 161.0, 162.5, 166.4 (2). MS m/z (%): 747 (M+) (28.14), 341 (100). Anal. Calcd. for C33H29N7O10S2 (747.75): C, 53.01; H, 3.91; N, 13.11. Found: C, 52.76; H, 4.22; N, 13.35.
- N-(2,4-dinitrophenyl)-2-((3-(4-N-(2((2,4-dinitrophenyl)amino)-2-oxoethyl)sulfamoyl)phen-yl-8-methoxy-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (4n).
4n: Yield, 76%; m.p. 248–250 °C. IR: 3342, 3200 (NH), 3093 (arom.), 2984, 2876 (aliph.), 1696, 1681, 1669 (3CO), 1608 (CN), 1398, 1166 (SO2). 1HNMR: 3.8 [s, 3H, OCH3], 3.9 [s, 2H, SCH2], 4.2 [s, 2H, NCH2], 7.1–7.6 (m, 9H, Ar-H), 7.7, 7.9 (2d, 4H, AB system, Ar-H), 8.1 (s, 1H, SO2NH], 8.8 [s, 2H, 2NH]. 13CNMR: 28.6, 40.4, 57.3, 115.1, 120.2, 123.9 (2), 124.0, 125.6 (2), 126.9 (2), 129.1, 129.8 (2), 130.6 (2), 135.5, 136.7, 138.0 (2), 138.7, 138.9, 143.1 (2), 147.3, 150.3, 156.4, 161.0, 163.5, 164.6. MS m/z (%): 809 (M+) (16.42), 294 (100). Anal. Calcd. for C31H23N9O14S2 (809.70): C, 45.98; H, 2.86; N, 15.57. Found: C, 45.64; H, 2.56; N, 15.25.
3.3. Biological Evaluations
3.3.1. MTT Cell Viability Assay
Cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay as stated previously [40]. In brief, A549 (lung), HepG2 (liver), LoVo (colon), MCF-7 (breast) cancer cells as well as normal cells (HUVEC) were seeded in 96-well plates (105 per well) and compounds were added in a different final concentration. All cells were treated for 48 h, and then 10μL of MTT reagent (5 mg/mL) was added to each well and further incubated for 4 h. Thereafter, 0.1 mL of acidified isopropanol was added to solubilize formazan product and the developed color was measured at 570 nm in Microplate ELISA Reader (BioTek, Elx-800, Winooski, VT, USA). The IC50 values (concentration required for 50% inhibition of cell viability) were calculated using dose response curve. Cell viability was calculated as = (Ab Treated/Ab Control) × 100.
The A549, HepG2, LoVo and MCF-7 cancer cell lines were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ) (Braunschweig, Germany). For Human umbilical vein endothelial cells (HUVEC cells), it was purchased from American Type Culture Collection (16549; Cat no. PCS-100-010; ATTCC, Manassas, VA, USA).
3.3.2. Cell Cycle Analysis
Cell cycle distribution was evaluated by PI staining as previously described [41]. Briefly, MCF-7 cells were seeded and incubated for a period of 24 h to allow for attachment. Subsequently, cells were exposed to different concentrations of compounds 4d and 4f. After 24 h, cells were harvested and fixed in 70% ethanol. Thereafter, cells were washed with PBS, resuspended in 1 mL of PBS containing PI (50 μg/mL), RNase A (0.5 mg/mL), sodium citrate (0.1%) and Triton X-100 (0.1%), then incubated for 15 min in the dark at room temperature. Flow cytometry was carried out by a FACS Scan Flow Cytometer (Cytomics FC 500; Beckman Coulter, Brea, CA, USA). The data were analyzed using CXP software V. 3.0.
3.3.3. Annexin V-FITC/PI Apoptosis Detection
Apoptotic cells were detected using Annexin V-FITC/PI staining protocol (BioLegend, San Diego, CA, USA) according to the manufacturer’s recommendations. Briefly, MCF-7 cells were incubated for 24 h with the different doses of promising compounds 4d and 4f. The cells were then harvested, washed twice with cold PBS and re-suspended with 1× Annexin V binding buffer (100 µL). Thereafter, cells were transferred into flow cytometry tube and stained with 5 μL of Annexin V- FITC and 5 μL PI for 15 min at room temperature in the dark. The cells were processed for apoptosis using a FACS Scan Flow Cytometer (Cytomics FC 500; Beckman Coulter, Brea, CA, USA) and analyzed using CXP software V. 3.0.
3.3.4. Analysis of Apoptotic Genes by RT-PCR
The MCF-7 cells (0.1 × 106) in 12 well plates were treated with different concentrations of compounds 4d and 4f for 24 h and untreated cells were used as a control. After 24 h of treatment, cell culture media was removed and the cells were lysed with Trizol (Invitrogen, Waltham, MA, USA), and total RNA was extracted from all the samples [42]. The RNA was estimated by Nano-drop and 1 µg of RNA was used in the cDNA synthesis. The cDNA was synthesized from RNA by using a cDNA synthesis kit (Invitrogen, Thermo Fisher Scientific, lnc., Waltham, MA, USA) according to the manufacturer’s instruction. After Reverse transcription at 42 °C for 60 min, RT-PCR was performed using 5× Firepol Master Mix ready to load (Solis BioDyne) according to manufacturer’s instruction. RT-PCR was used to analyze the gene expression level of P53, Bcl2, Bax and caspase 7 using specific sets of primers for each gene [22], while β-actin was utilized as a housekeeping gene. The program was run as follows, initial denaturation at 95 °C for 5 min; denaturation at 95 °C for 30 s; annealing at 55 °C for 45 s; elongation at 72 °C for 45 s (30 Cycles) and final extension at 72 °C for 10 min. The RT-PCR products were electrophoresed on a 1.2% agarose gel containing ethidium bromide and the gel was imaged on a Licor machine.
3.3.5. Western Blotting to See the Effect of 4d and 4f Compounds
MCF-7 cells were plated in six well plates at the density of 0.5 × 106 and kept for adherence for overnight. On the next day, complete media was replaced with the fresh one and treated the cells with compounds 4d and 4f for 24 h at various concentrations. After 24 h of treatment, cells were harvested and washed twice with 1× PBS before adding the lysis buffer (20 mM Tris pH 7.5, 150 mM Nacl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X100, 1 μg/mL Leupeptin and 100 µM phenylmethylsulfonyl fluoride—PMSF) and kept it for 1 h on ice, with intermittent vortex mixing every 10 min. The samples were centrifuged at 13,000 rpm at 4 °C for 15 min and then the supernatants were transferred into new tubes. Total protein was measured by using Bio-Rad Bradford kit and then 35 µg of the total protein was run on a 12% SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked with 5% BSA in TBST (150 mM Nacl, 20 mM Tris base and 0.1% Tween 20) for 2 h at room temperature and then washed three times with TBST buffer. Then the membrane was incubated with the desired antibodies (p53, Bcl-2, Bax, caspase7 and β-actin) (1:200, Santa Cruz Biotecnology, Inc., Dallas, TX, USA) and kept on a rocker at 4 °C overnight. On the next day, the membrane was washed three times with TBST buffer and then incubated with horseradish peroxidase-conjugated secondary antibody (1:3000) for 1 h at room temperature. After incubation, membranes were washed three times with TBST buffer and then were ready for immunodetection. For immunodetection, membranes were incubated with ECL Western blotting detecting reagents (Amersaham Pharmacia Biotech Inc., Zurich, Switzerland) and then bands were obtained by exposing to X-ray films (Amersham).
3.3.6. Molecular Docking
Molecular docking between potential compounds 4d and 4f with the target protein Bcl-2 was performed as reported earlier [43]. The X-ray crystal structure Bcl-2 bound with DRO (2.10 Å resolution) was used to retrieve 3D coordinates of the target protein [44]. The protein was processed to remove non-essential water molecules and other heterogeneous atoms. Hydrogen atoms were added, bond orders were defined, and rotatable bonds were assigned, as reported by [45]. Prior to molecular docking, hydrogen bonds were redefined, and the energy of the protein was minimized using MMFF (Merck Molecular Force Field) force field. The 2D structures of compounds 4d and 4f were drawn using ChemDraw, and their energies were minimized using Universal Forcefield (UFF). Molecular docking between Bcl-2 and ligands (4d and 4f) along with the control ligand (DRO) was performed inside a grid box of 27 × 30 × 25 Å, placed at 39 × 28 × −12 Å, keeping 0.375 Å spacing between the grid points, using Autodock 4.2 (Scripps Research, San Diego, CA, USA), as reported by Rabbani and coworkers [46]. Lamarckian Genetic Algorithm (LGA) and Solis and Wets local search methods were used to perform molecular docking. Initial torsions, positions, and orientations of ligands were set arbitrarily. A maximum of 2,500,000 calculations was for each docking run. The population size, translational step, quaternion and torsion were set to 150, 0.2 Å, 5 and 5, respectively. The docked protein-ligand complex with the lowest binding energy was selected for molecular interaction analysis in Discovery Studio (BIOVIA, San Diego, CA, USA). The binding affinity (Kd) and binding energy (ΔG) of compounds 4d and 4f towards Bcl-2 were evaluated using the following relation [47]
where, R and T are the universal gas constant (1.987 kcal mol−1 K−1) and temperature (298 K), respectively.
3.3.7. Statistical Analysis
OriginPro 8.5 software was used to conduct the statistical analysis. The mean and standard deviation of the data were displayed. Student’s t-test was used to examine differences, and p < 0.05 was regarded statistically significant. * p < 0.05, ** p < 0.01, *** p < 0.001.
4. Conclusions
Synthesizing a novel quinazoline incorporating substituted sulfonamide moieties in order to obtain anticancer activity with lower toxicity has attracted attention on the beneficial effect of this group to overcome one of the most serious problems in cancer chemotherapy, which is the lower efficacy and higher toxicity. Therefore, we aimed in this study to synthesize a novel series of quinazolinone derivatives carrying biologically active substituted-sulfonamide moieties at 3-position to achieve new compounds with the anticancer effect. Herein, a novel unexpected series of quinazoline incorporating substituted sulfonamide moieties was synthesized and evaluated in vitro for their anticancer activity. Some compounds were found to exhibit high cytotoxicity in the MCF-7 breast cancer cell line. Compounds 4d and 4f were appeared as the most effective derivatives against MCF-7 cells. Flow cytometry data showed that compounds 4d and 4f successfully triggered apoptosis in MCF-7 cells. Furthermore, changes in the expression of apoptosis-related markers at the gene and protein level were also indicative of apoptotic activity. Molecular docking of compounds 4d and 4f also suggested that they possess good binding affinity for Bcl-2. Overall, screening of the tested compounds could offer an encouraging framework in this field, which may lead to develop more potent, selective, clinically useful analogues anticancer agents. Furthermore, the existing compounds are currently under investigations for their in vivo anti-cancer activity in different transplantable mouse tumors model. A series of toxicity studies are also underway to determine the safety of these existing compounds.
Supplementary Materials
The following are available online, Figure S1: Validation of docking protocol by redocking the ligand present in the crystal structure and comparing RMSD between the docking pose and crystal structure pose.
Author Contributions
Conceptualization, A.S.A. and M.M.G.; methodology, F.A.N., M.Z.A. and S.M.A.; resources, A.A.A.-M., supervision, A.S.A. and M.M.G.; original draft preparation, M.M.G. and F.A.N.; writing, review, and editing A.S.A., M.Z.A. and S.M.A. All authors have read and agreed to the published version of the manuscript.
Funding
This project was funded by the National Plan for Science, Technology, and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia (Award number: 13-MED 997-02).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
Authors are thankful to the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia, Award number (13-MED 997-02) for funding this research work.
Conflicts of Interest
The authors declare that they have no competing interest.
References
- Ghorab, M.M.; Ragab, F.A.; Heiba, H.I.; Bayomi, A.A. Novel quinazoline derivatives bearing a sulfapyridine moiety as anticancer and radiosensitizing agents. J. Heterocycl. Chem. 2014, 51, E255–E262. [Google Scholar] [CrossRef]
- Yong, J.; Lu, C.; Wu, X. Synthesis and Biological Evaluation of Quinazoline Derivatives as Potential Anticancer Agents (II). Anticancer Agents Med. Chem. 2015, 15, 1326–1332. [Google Scholar] [CrossRef] [PubMed]
- Poudapally, S.; Battu, S.; Velatooru, L.R.; Bethu, M.S.; Janapala, V.R.; Sharma, S.; Sen, S.; Pottabathini, N.; Iska, V.B.R.; Katangoor, V. Synthesis and biological evaluation of novel quinazoline-sulfonamides as anti-cancer agents. Bioorg. Med. Chem. Lett. 2017, 27, 1923–1928. [Google Scholar] [CrossRef] [PubMed]
- Alagarsamy, V.; Chitra, K.; Saravanan, G.; Solomon, V.R.; Sulthana, M.T.; Narendhar, B. An overview of quinazolines: Pharmacological significance and recent developments. Eur. J. Med. Chem. 2018, 151, 628–685. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; De Marinis, F.; Di Maio, M.; Cortinovis, D.; Cappuzzo, F.; Mok, T. Gefitinib as first-line treatment for patients with advanced non-small-cell lung cancer with activating Epidermal Growth Factor Receptor mutation: Implications for clinical practice and open issues. Lung Cancer 2011, 72, 3–8. [Google Scholar] [CrossRef]
- Bronte, G.; Rolfo, C.; Giovannetti, E.; Cicero, G.; Pauwels, P.; Passiglia, F.; Castiglia, M.; Rizzo, S.; Vullo, F.L.; Fiorentino, E.; et al. Are erlotinib and gefitinib interchangeable, opposite or complementary for non-small cell lung cancer treatment? Biological, pharmacological and clinical aspects. Crit. Rev. Oncol. Hematol. 2014, 89, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-H.; Lee, Y.-C.; Whang-Peng, J.; Chen, Y.-M. Critical Appraisal of Gefitinib in the Treatment of Non-Small Cell Lung Cancer. J. Cancer Res. Pract. 2015, 2, 179–194. [Google Scholar] [CrossRef]
- Ismail, R.S.M.; Ismail, N.S.M.; Abuserii, S.; Abou El Ella, D.A. Recent advances in 4-aminoquinazoline based scaffold derivatives targeting EGFR kinases as anticancer agents. Future J. Pharm. Sci. 2016, 2, 9–19. [Google Scholar] [CrossRef]
- Bhatia, P.; Sharma, V.; Alam, O.; Manaithiya, A.; Alam, P.; Kahksha; Alam, M.T.; Imran, M. Novel quinazoline-based EGFR kinase inhibitors: A review focussing on SAR and molecular docking studies (2015–2019). Eur. J. Med. Chem. 2020, 204, 112640. [Google Scholar] [CrossRef]
- Bansal, R.; Malhotra, A. Therapeutic progression of quinazolines as targeted chemotherapeutic agents. Eur. J. Med. Chem. 2021, 211, 113016. [Google Scholar] [CrossRef]
- Supuran, C.T.; Casini, A.; Mastrolorenzo, A.; Scozzafava, A. COX-2 selective inhibitors, carbonic anhydrase inhibition and anticancer properties of sulfonamides belonging to this class of pharmacological agents. Mini Rev. Med. Chem. 2004, 4, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Solomon, V.R.; Hu, C.; Lee, H. Hybrid pharmacophore design and synthesis of isatin-benzothiazole analogs for their anti-breast cancer activity. Bioorg. Med. Chem. 2009, 17, 7585–7592. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Alsaid, M.S.; Al-Dosari, M.S.; El-Gazzar, M.G.; Parvez, M.K. Design, Synthesis and Anticancer Evaluation of Novel Quinazoline-Sulfonamide Hybrids. Molecules 2016, 21, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkahtani, H.M.; Abdalla, A.N.; Obaidullah, A.J.; Alanazi, M.M.; Almehizia, A.A.; Alanazi, M.G.; Ahmed, A.Y.; Alwassil, O.I.; Darwish, H.W.; Abdel-Aziz, A.A.; et al. Synthesis, cytotoxic evaluation, and molecular docking studies of novel quinazoline derivatives with benzenesulfonamide and anilide tails: Dual inhibitors of EGFR/HER2. Bioorg. Chem. 2020, 95, 103461. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Mehndiratta, S.; Sapra, S.; Singh, G.; Singh, M.; Nepali, K. Quinazolines as Apoptosis Inducers and Inhibitors: A Review of Patent Literature. Recent Pat. Anticancer Drug Discov. 2016, 11, 2–66. [Google Scholar] [CrossRef]
- Nahta, R.; Yuan, L.X.; Du, Y.; Esteva, F.J. Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: Effects on insulin-like growth factor I signaling. Mol. Cancer 2007, 6, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Ling, Y.H.; Lin, R.; Perez-Soler, R. Erlotinib induces mitochondrial-mediated apoptosis in human H3255 non-small-cell lung cancer cells with epidermal growth factor receptorL858R mutation through mitochondrial oxidative phosphorylation-dependent activation of BAX and BAK. Mol. Pharm. 2008, 74, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.H.; Addla, D.; Lv, J.S.; Zhou, C.H. Heterocyclic Naphthalimides as New Skeleton Structure of Compounds with Increasingly Expanding Relational Medicinal Applications. Curr. Top. Med. Chem. 2016, 16, 3303–3364. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Alsaid, M.S.; Soliman, A.M.; Ragab, F.A. VEGFR-2 inhibitors and apoptosis inducers: Synthesis and molecular design of new benzo[g]quinazolin bearing benzenesulfonamide moiety. J. Enzym. Inhib. Med. Chem. 2017, 32, 893–907. [Google Scholar] [CrossRef] [Green Version]
- Ghorab, M.M.; Alsaid, M.S.; Soliman, A.M.; Al-Mishari, A.A. Benzo[g]quinazolin-based scaffold derivatives as dual EGFR/HER2 inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.S.; Ghorab, M.M.; Nasr, F.A.; Ahmed, M.Z.; Al-Mishari, A.A.; Attia, S.M. Novel sulfonamide-bearing methoxyquinazolinone derivatives as anticancer and apoptosis inducers: Synthesis, biological evaluation and in silico studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Alimbetov, D.; Askarova, S.; Umbayev, B.; Davis, T.; Kipling, D. Pharmacological Targeting of Cell Cycle, Apoptotic and Cell Adhesion Signaling Pathways Implicated in Chemoresistance of Cancer Cells. Int. J. Mol. Sci 2018, 19, 1690. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liu, Y.; Ma, X.; Hu, H. The Influence of Cell Cycle Regulation on Chemotherapy. Int. J. Mol. Sci. 2021, 22, 6923. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Wang, X.; Luo, W.; Zhan, Y.; Zhang, Y. Brucine, an effective natural compound derived from nux-vomica, induces G1 phase arrest and apoptosis in LoVo cells. Food Chem. Toxicol. 2013, 58, 332–339. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlodkowic, D.; Skommer, J.; Darzynkiewicz, Z. Flow cytometry-based apoptosis detection. Methods Mol. Biol. 2009, 559, 19–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [Green Version]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [Green Version]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Lamkanfi, M.; Kanneganti, T.-D. Caspase-7: A protease involved in apoptosis and inflammation. Int J. Biochem. Cell Biol. 2010, 42, 21–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Zahedifard, M.; Faraj, F.L.; Paydar, M.; Looi, C.Y.; Hasandarvish, P.; Hajrezaie, M.; Kamalidehghan, B.; Majid, N.A.; Khalifa, S.A.; Ali, H.M.; et al. Synthesis of Apoptotic New Quinazolinone-Based Compound and Identification of its Underlying Mitochondrial Signalling Pathway in Breast Cancer Cells. Curr. Pharm. Des. 2015, 21, 3417–3426. [Google Scholar] [CrossRef] [PubMed]
- Nowar, R.M.; EE, A.O.; Abou-Seri, S.M.; El Moghazy, S.M.; Abou El Ella, D.A. Design, synthesis and biological evaluation of some novel quinazolinone derivatives as potent apoptotic inducers. Future Med. Chem. 2018, 10, 1191–1205. [Google Scholar] [CrossRef] [PubMed]
- ElZahabi, H.S.A.; Nafie, M.S.; Osman, D.; Elghazawy, N.H.; Soliman, D.H.; El-Helby, A.A.H.; Arafa, R.K. Design, synthesis and evaluation of new quinazolin-4-one derivatives as apoptotic enhancers and autophagy inhibitors with potent antitumor activity. Eur. J. Med. Chem. 2021, 222, 113609. [Google Scholar] [CrossRef] [PubMed]
- Ariyama, H.; Qin, B.; Baba, E.; Tanaka, R.; Mitsugi, K.; Harada, M.; Nakano, S. Gefitinib, a selective EGFR tyrosine kinase inhibitor, induces apoptosis through activation of Bax in human gallbladder adenocarcinoma cells. J. Cell Biochem. 2006, 97, 724–734. [Google Scholar] [CrossRef]
- Liu, L.; Zhong, L.; Zhao, Y.; Chen, M.; Yao, S.; Li, L.; Xiao, C.; Shan, Z.; Gan, L.; Xu, T.; et al. Effects of lapatinib on cell proliferation and apoptosis in NB4 cells. Oncol. Lett. 2018, 15, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Krishna, S.; Kumar, S.B.; Murthy, T.P.K.; Murahari, M. Structure-based design approach of potential BCL-2 inhibitors for cancer chemotherapy. Comput. Biol. Med. 2021, 134, 104455. [Google Scholar] [CrossRef]
- AlAjmi, M.F.; Rehman, M.T.; Hussain, A.; Rather, G.M. Pharmacoinformatics approach for the identification of Polo-like kinase-1 inhibitors from natural sources as anti-cancer agents. Int. J. Biol. Macromol. 2018, 116, 173–181. [Google Scholar] [CrossRef]
- Nasr, F.A.; Shahat, A.A.; Alqahtani, A.S.; Ahmed, M.Z.; Qamar, W.; Al-Mishari, A.A.; Almoqbil, A.N. Centaurea bruguierana inhibits cell proliferation, causes cell cycle arrest, and induces apoptosis in human MCF-7 breast carcinoma cells. Mol. Biol. Rep. 2020, 47, 6043–6051. [Google Scholar] [CrossRef]
- Alqahtani, A.S.; Nasr, F.A.; Noman, O.M.; Farooq, M.; Alhawassi, T.; Qamar, W.; El-Gamal, A. Cytotoxic Evaluation and Anti-Angiogenic Effects of Two Furano-Sesquiterpenoids from Commiphora myrrh Resin. Molecules 2020, 25, 1318. [Google Scholar] [CrossRef] [Green Version]
- Rio, D.C.; Ares, M.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5439. [Google Scholar] [CrossRef]
- Al-Shabib, N.A.; Khan, J.M.; Malik, A.; Alsenaidy, M.A.; Rehman, M.T.; AlAjmi, M.F.; Alsenaidy, A.M.; Husain, F.M.; Khan, R.H. Molecular insight into binding behavior of polyphenol (rutin) with beta lactoglobulin: Spectroscopic, molecular docking and MD simulation studies. J. Mol. Liq. 2018, 269, 511–520. [Google Scholar] [CrossRef]
- Porter, J.; Payne, A.; De Candole, B.; Ford, D.; Hutchinson, B.; Trevitt, G.; Turner, J.; Edwards, C.; Watkins, C.; Whitcombe, I.; et al. Tetrahydroisoquinoline amide substituted phenyl pyrazoles as selective Bcl-2 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.T.; Shamsi, H.; Khan, A.U. Insight into the binding mechanism of imipenem to human serum albumin by spectroscopic and computational approaches. Mol. Pharm. 2014, 11, 1785–1797. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Tabrez, S.; Islam, B.U.; Rehman, M.T.; Alsenaidy, A.M.; AlAjmi, M.F.; Khan, R.A.; Alsenaidy, M.A.; Khan, M.S. Characterization of colchicine binding with normal and glycated albumin: In vitro and molecular docking analysis. J. Biomol. Struct. Dyn. 2018, 36, 3453–3462. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.T.; Ahmed, S.; Khan, A.U. Interaction of meropenem with ‘N’ and ‘B’ isoforms of human serum albumin: A spectroscopic and molecular docking study. J. Biomol. Struct. Dyn. 2016, 34, 1849–1864. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).