
Investigating the Role of Obesity in Prostate Cancer and Identifying Biomarkers for Drug Discovery: Systems Biology and Deep Learning Approaches


Abstract
:1. Introduction
2. Results
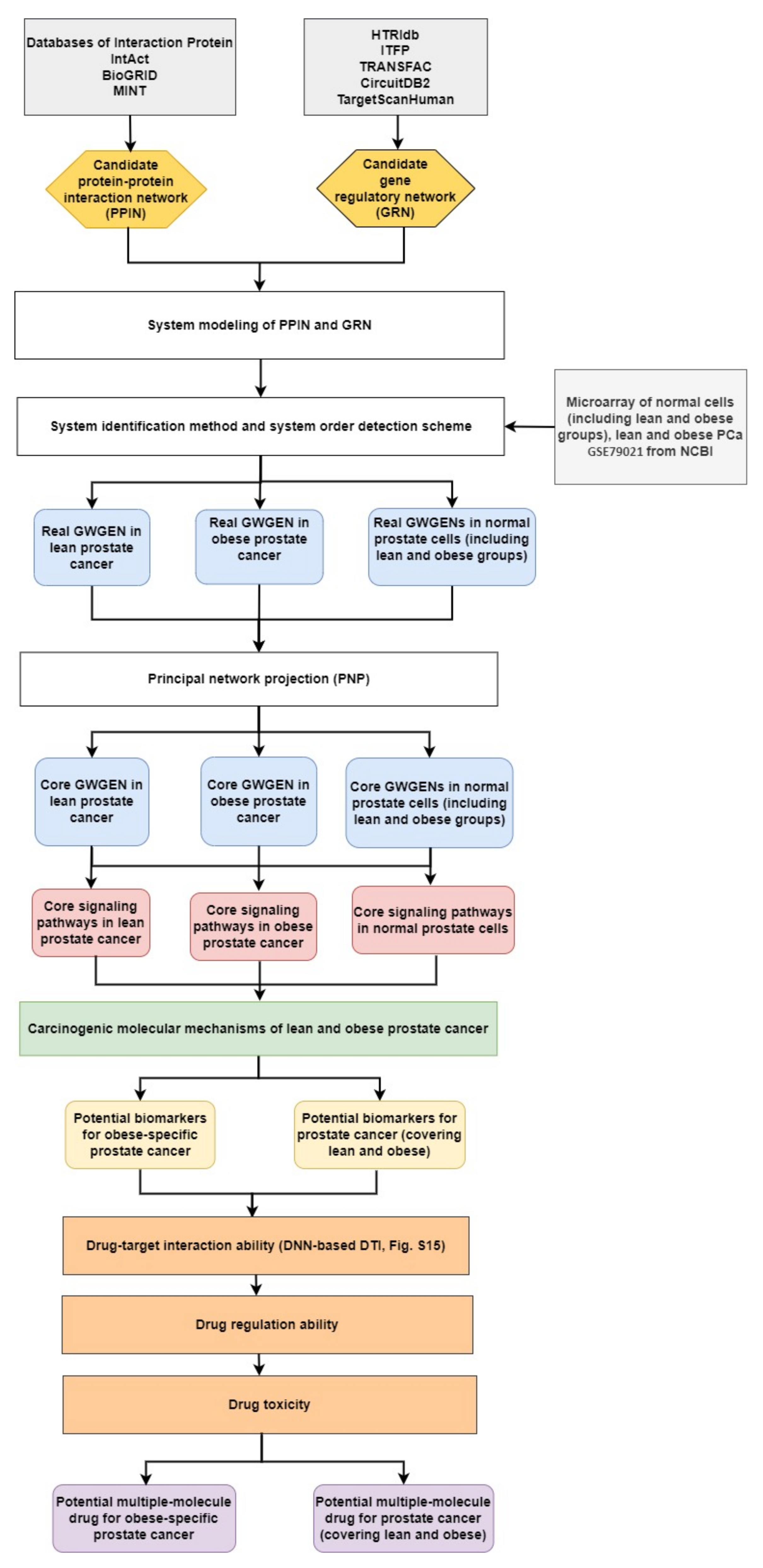
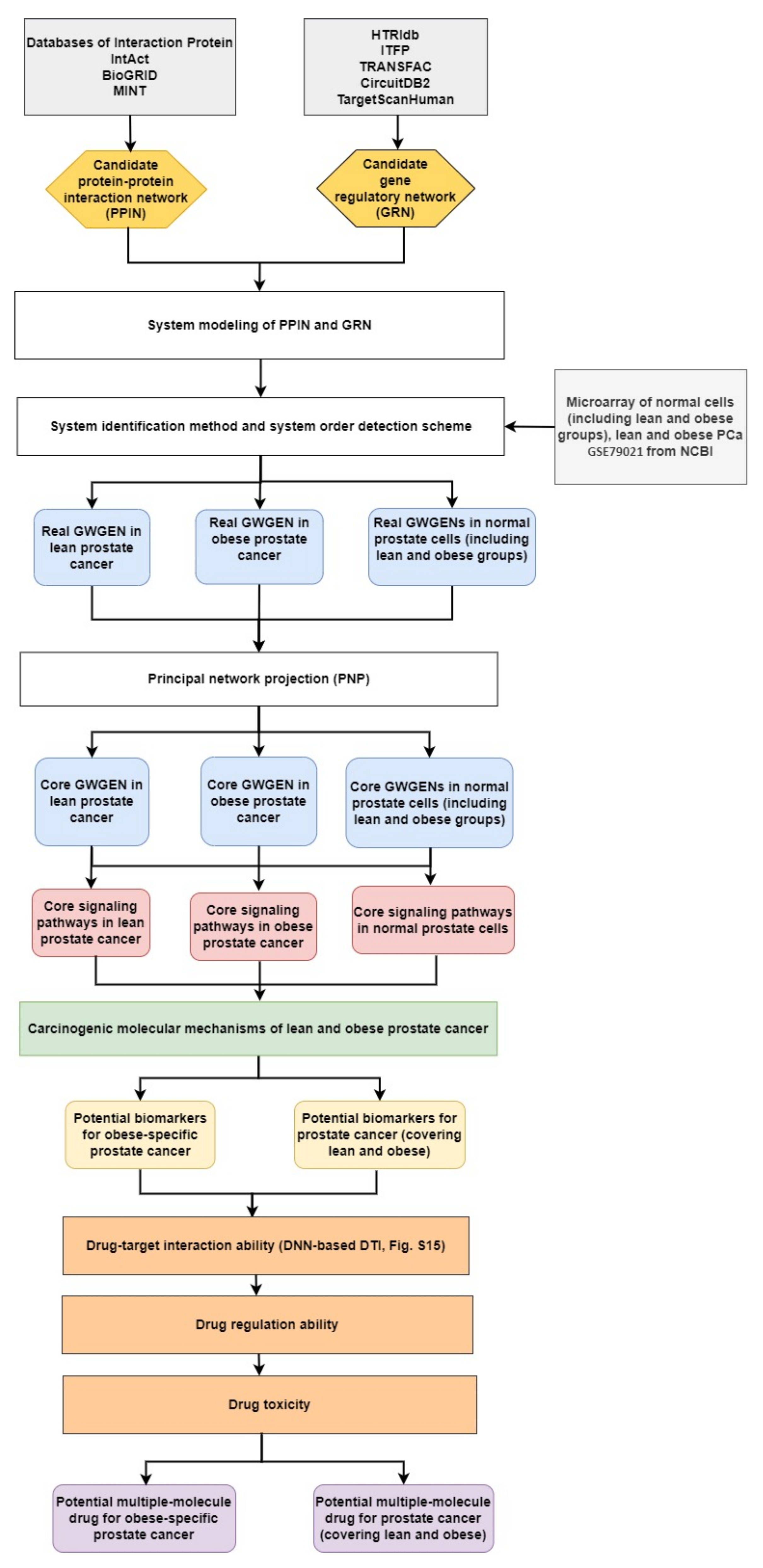
2.1. Overview of Systems Biology Approaches and Drug Design Specification
2.2. The Common Carcinogenic Molecular Mechanism between Lean and Obese PCa
2.3. The Specific Molecular Mechanism in Lean PCa
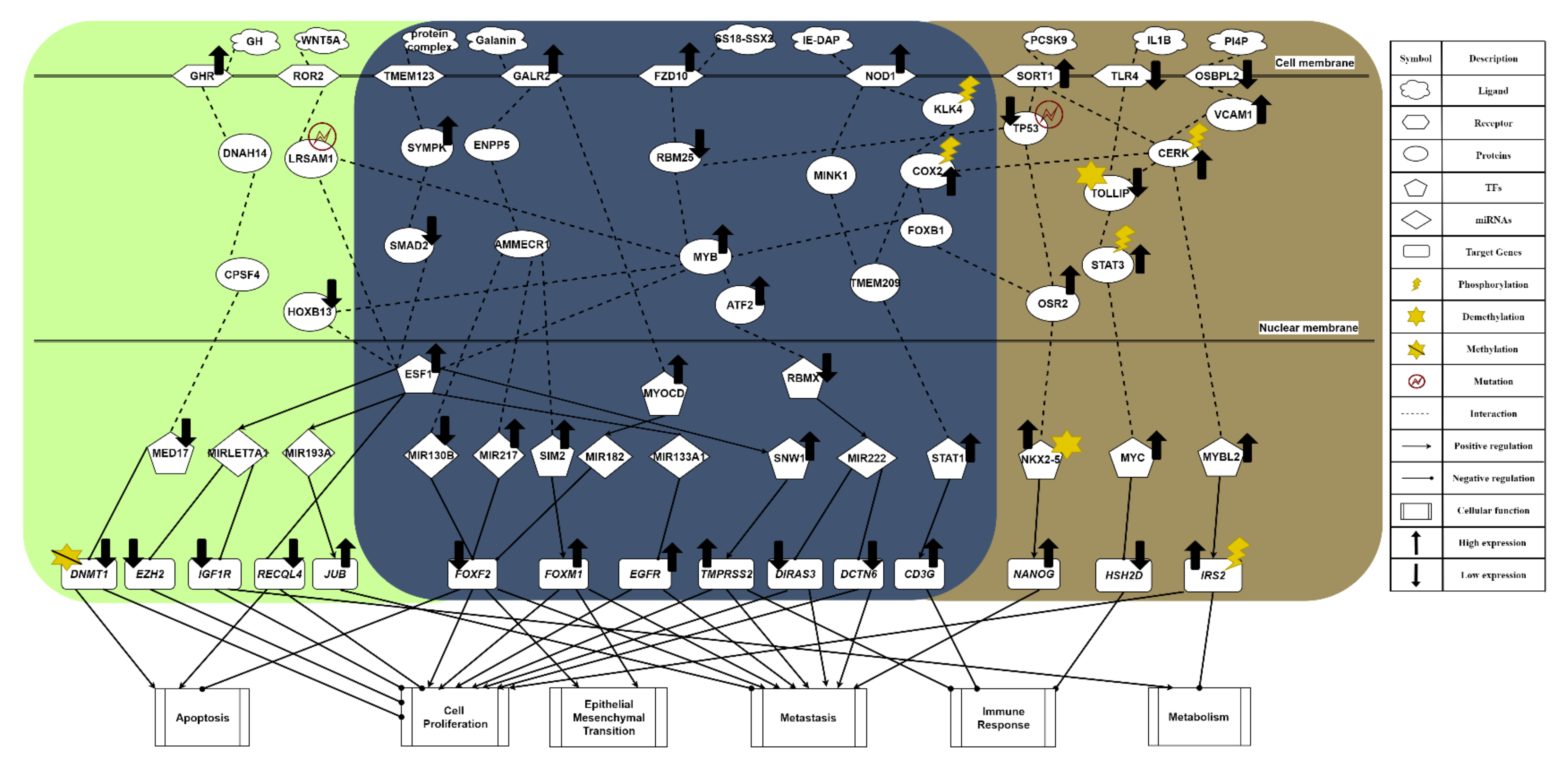
2.4. The Specific Carcinogenic Molecular Mechanism in Obese PCa
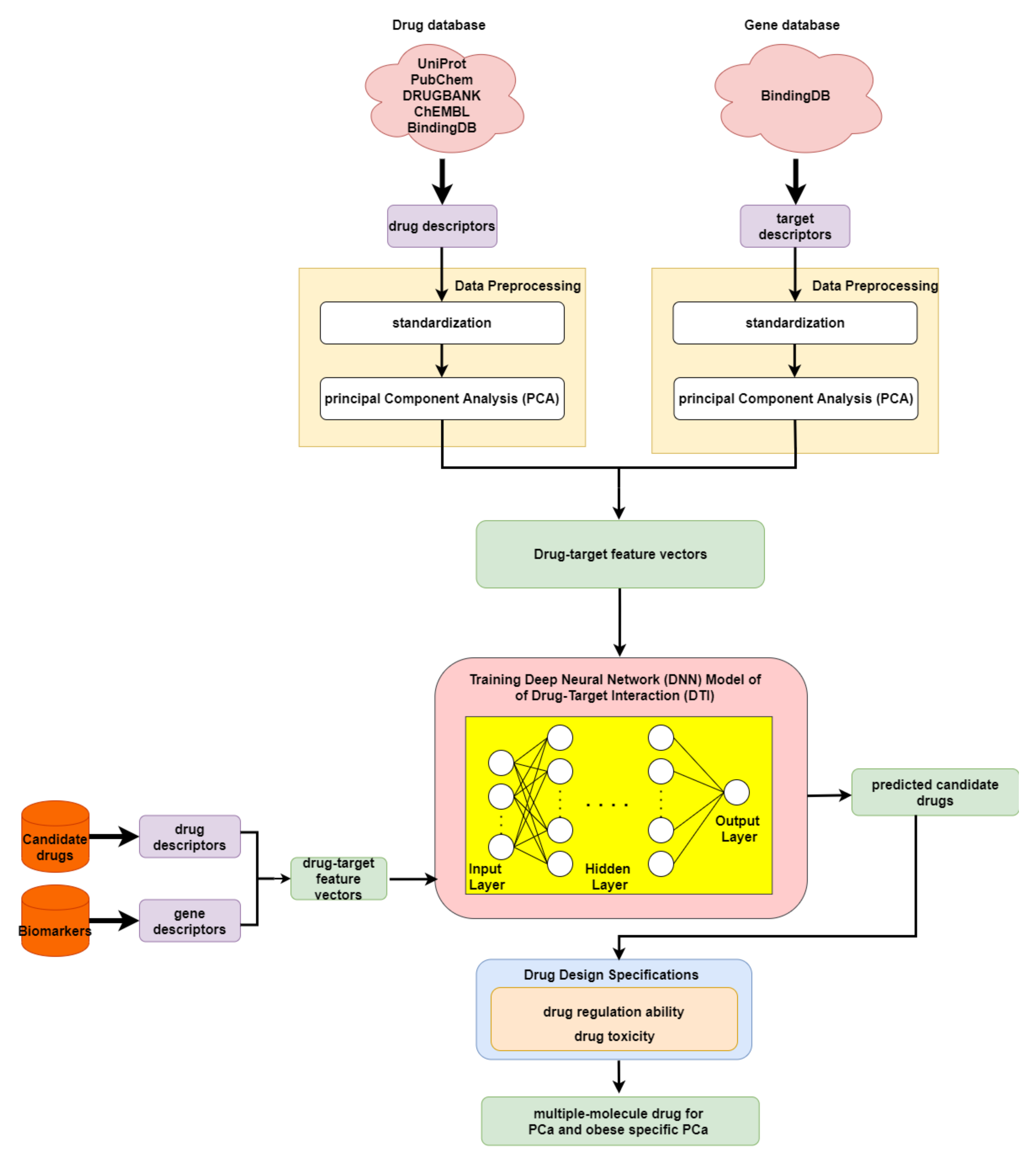
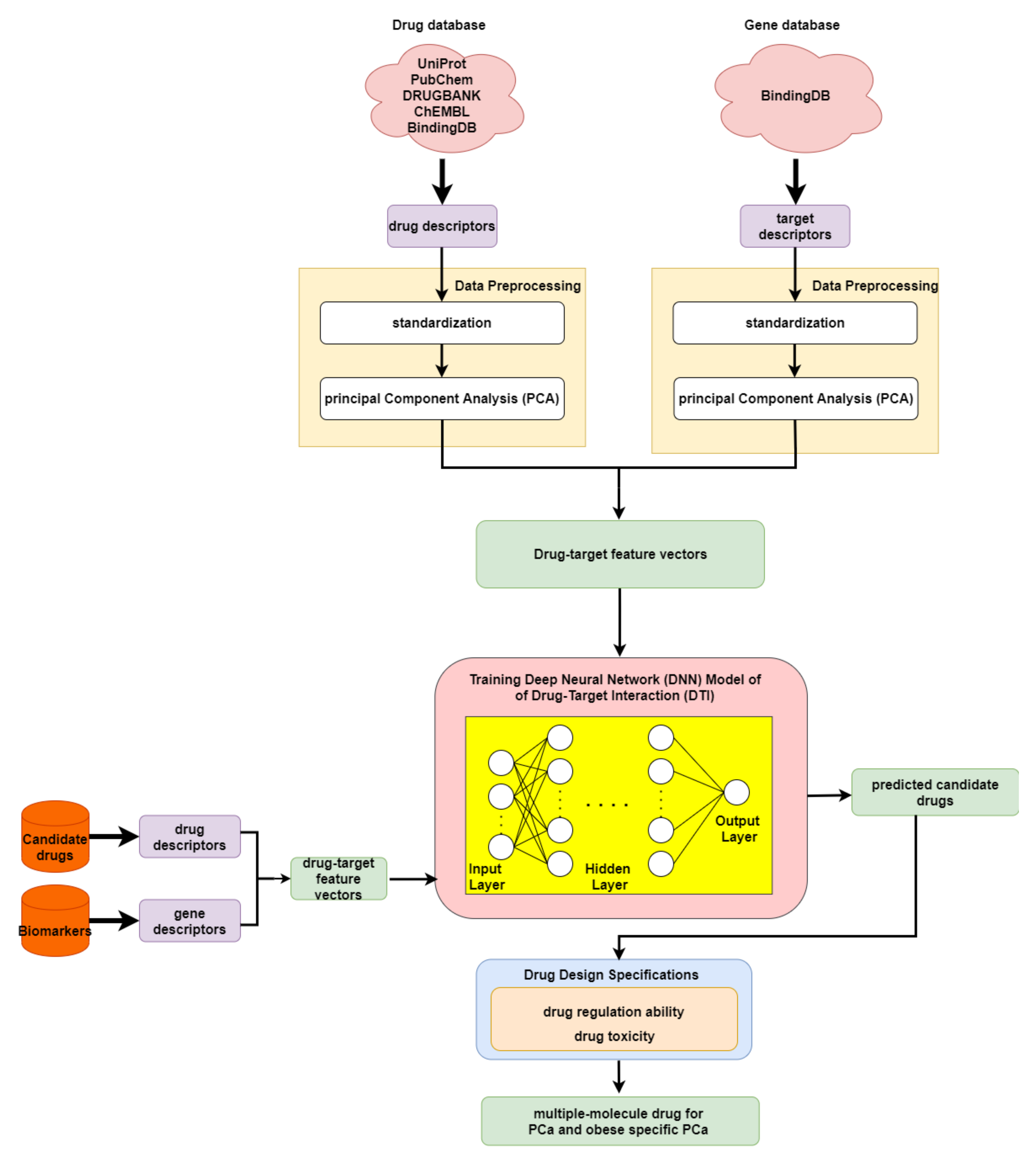
2.5. The Application of Deep Neural Network to Drug–Target Interaction Prediction and the Drug Design Specifications Considering Drug Regulation Ability and Drug Toxicity
3. Discussion
3.1. Systems Biology Approaches and Traditional Treatments for PCa
3.2. Multiple-Molecule Drugs for PCa and Obesity-Specific PCa
4. Materials and Methods
4.1. A General Review of Constructing Core Genome-Wide Genetic and Epigenetic Networks (GWGENs) of Normal Prostate Cells, and Lean and Obese PCa
- (1)
- Constructing the candidate GWGEN. Using big database mining, we constructed a candidate PPIN and a candidate GRN, including genes, miRNA, and lncRNA, as the first step. It is noted that the candidate GWGEN consists of a candidate PPIN and a candidate GRN.
- (2)
- Identifying real GWGENs. After performing system modeling for proteins, genes, miRNA, and lenRNA, we performed system identification by solving the constrained linear least squares estimation problem with the help of the microarray data for normal prostate cells (including lean and obese groups), and lean and obese PCa. We then used the system order detection scheme for computing the AIC, to prune the false-positive interactions in the candidate GWGEN.
- (3)
- Extracting the core GWGENs. To extract the core GWGENs, we applied the PNP approach. By doing so, we could compute a projection value for each node in the real GWGENs. The top 3000 elements with highest projection values remained.
- (4)
- Building and comparing the core pathways. The core signaling pathways for normal prostate cells (including lean and obese groups), and lean and obese PCa in the annotation of KEGG pathways could be found by referring to the projection values and the literature survey. We investigated the molecular mechanisms of carcinogenesis considering the microenvironmental factors of lean and obese PCa and their corresponding downstream core signaling pathways.
- (5)
- Identifying biomarkers (drug targets) for the design of multiple-molecule drugs. Based on the analysis of carcinogenic molecular mechanisms, we identified essential biomarkers for PCa (covering lean and obese) and obesity-specific PCa. Following the proposed drug design specifications, we considered drug–target interaction probability, drug regulation ability, and drug toxicity. One DNN-based DTI model was trained in advance for predicting candidate drugs targeting identified biomarkers. The aim of the drug regulation ability filter was to reverse the abnormal expression of biomarkers. The drug toxicity filter helped to find drugs with light toxicity. Consequently, we suggested two multiple-molecule drugs for PCa (covering lean and obese) and obesity-specific PCa.
4.2. Data Preprocessing for Constructing the Candidate GWGEN
4.3. System Modeling for Normal Prostate Cells and PCa
4.4. Utilizing System Identification and System Order Detection Methods to Identify Real GWGENs from the Candidate GWGEN
4.5. Extracting Core GWGENs from the Real GWGENs Using the Principal Network Projection Method
4.6. Deep-Neural-Network-Based Drug–Target Interaction Prediction Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Pramono, L.A.; Setiati, S.; Soewondo, P.; Subekti, I.; Adisasmita, A.; Kodim, N.; Sutrisna, B.J.A. Prevalence and predictors of undiagnosed diabetes mellitus in Indonesia. Age 2010, 46, 100. [Google Scholar]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, S.P.; Graubard, B.I.; Andreotti, G.; Younes, N.; Cleary, S.D.; Cook, M.B. Prediagnostic Body Mass Index Trajectories in Relation to Prostate Cancer Incidence and Mortality in the PLCO Cancer Screening Trial. J. Natl. Cancer Inst. 2017, 109, djw225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantarutti, A.; Bonn, S.E.; Adami, H.O.; Gronberg, H.; Bellocco, R.; Balter, K. Body mass index and mortality in men with prostate cancer. Prostate 2015, 75, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Freedland, S.J.; Deka, A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Thun, M.J.; Calle, E.E.J.C.E.; Biomarkers, P. Body mass index, weight change, and risk of prostate cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiol. Prev. Biomark. 2007, 16, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Giovannucci, E.; Liu, Y.; Platz, E.A.; Stampfer, M.J.; Willett, W.C. Risk factors for prostate cancer incidence and progression in the health professionals follow-up study. Int. J. Cancer 2007, 121, 1571–1578. [Google Scholar] [CrossRef] [Green Version]
- Stocks, T.; Hergens, M.P.; Englund, A.; Ye, W.; Stattin, P. Blood pressure, body size and prostate cancer risk in the Swedish Construction Workers cohort. Int. J. Cancer 2010, 127, 1660–1668. [Google Scholar] [CrossRef]
- Udager, A.M.; Tomlins, S.A. Molecular biomarkers in the clinical management of prostate cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030601. [Google Scholar] [CrossRef]
- Divella, R.; De Luca, R.; Abbate, I.; Naglieri, E.; Daniele, A. Obesity and cancer: The role of adipose tissue and adipo-cytokines-induced chronic inflammation. J. Cancer 2016, 7, 2346. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, E.J.; LeRoith, D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol. Metab. 2010, 21, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Lane, B.R.; Stephenson, A.J.; Magi-Galluzzi, C.; Lakin, M.M.; Klein, E.A. Low testosterone and risk of biochemical recurrence and poorly differentiated prostate cancer at radical prostatectomy. Urology 2008, 72, 1240–1245. [Google Scholar] [CrossRef] [PubMed]
- Kaaks, R.; Stattin, P. Obesity, endogenous hormone metabolism, and prostate cancer risk: A conundrum of “highs” and “lows”. Cancer Prev. Res. 2010, 3, 259–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, H.; Gu, J.; Meng, Q.H.; Kim, J.; Strom, S.; Davis, J.W.; He, Y.; Wagar, E.A.; Thompson, T.C.; Logothetis, C.J. Low serum testosterone is associated with tumor aggressiveness and poor prognosis in prostate cancer. Oncol. Lett. 2017, 13, 1949–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tafuri, A.; Sebben, M.; Shakir, A.; Pirozzi, M.; Processali, T.; Rizzetto, R.; Amigoni, N.; Brunelli, M.; Migliorini, F.; Siracusano, S.; et al. Endogenous testosterone mirrors prostate cancer aggressiveness: Correlation between basal testosterone serum levels and prostate cancer European Urology Association clinical risk classes in a large cohort of Caucasian patients. Int. Urol. Nephrol. 2020, 52, 1261–1269. [Google Scholar] [CrossRef]
- Cabral, P.H.O.; Iwamoto, M.W.; Fanni, V.S.S.; Barros, L.d.R.; Cardoso, S.N.; Mello, L.F.; Glina, S. Study of testosterone as a predictor of tumor aggressiveness in patients with prostate cancer. Int. Braz. J. Urol. 2013, 39, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klap, J.; Schmid, M.; Loughlin, K.R. The relationship between total testosterone levels and prostate cancer: A review of the continuing controversy. J. Urol. 2015, 193, 403–414. [Google Scholar] [CrossRef]
- Laurent, V.; Guérard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 1–15. [Google Scholar] [CrossRef]
- Zhang, T.; Tseng, C.; Zhang, Y.; Sirin, O.; Corn, P.G.; Li-Ning-Tapia, E.M.; Troncoso, P.; Davis, J.; Pettaway, C.; Ward, J. CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumour microenvironment. Nat. Commun. 2016, 7, 1–16. [Google Scholar] [CrossRef]
- Parikesit, D.; Mochtar, C.A.; Umbas, R.; Hamid, A.R.A.H. The impact of obesity towards prostate diseases. Prostate Int. 2016, 4, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.J.; Heo, S.K.; Song, E.J.; Kim, D.J.; Han, S.Y.; Han, J.H.; Kim, B.Y.; Park, J.H. Activation of Nod1 and Nod2 induces innate immune responses of prostate epithelial cells. Prostate 2012, 72, 1351–1358. [Google Scholar] [CrossRef] [Green Version]
- Mukai, S.; Yorita, K.; Yamasaki, K.; Nagai, T.; Kamibeppu, T.; Sugie, S.; Kida, K.; Onizuka, C.; Tsukino, H.; Kamimura, T. Expression of human kallikrein 1-related peptidase 4 (KLK4) and MET phosphorylation in prostate cancer tissue: Immunohistochemical analysis. Hum. Cell 2015, 28, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Alexanian, A.; Miller, B.; Chesnik, M.; Mirza, S.; Sorokin, A. Post-translational regulation of COX2 activity by FYN in prostate cancer cells. Oncotarget 2014, 5, 4232. [Google Scholar] [CrossRef] [PubMed]
- Siu, M.K.Y.; Wong, E.S.Y.; Kong, D.S.H.; Chan, H.Y.; Jiang, L.; Wong, O.G.W.; Lam, E.W.F.; Chan, K.K.L.; Ngan, H.Y.S.; Le, X.F.; et al. Stem cell transcription factor NANOG controls cell migration and invasion via dysregulation of E-cadherin and FoxJ1 and contributes to adverse clinical outcome in ovarian cancers. Oncogene 2013, 32, 3500–3509. [Google Scholar] [CrossRef] [Green Version]
- Vitkin, N.; Nersesian, S.; Siemens, D.R.; Koti, M.J.F.i.i. The tumor immune contexture of prostate cancer. Front. Immunol. 2019, 10, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Wu, H.; Warden, C.; Steele, L.; Liu, X.; Iterson, M.V.; Wu, X.; Nelson, R.; Liu, Z.; Yuan, Y.-C. Gene expression differences in prostate cancers between young and old men. PLoS Genet. 2016, 12, e1006477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murillo-Garzón, V.; Gorroño-Etxebarria, I.; Åkerfelt, M.; Puustinen, M.C.; Sistonen, L.; Nees, M.; Carton, J.; Waxman, J.; Kypta, R.M. Frizzled-8 integrates Wnt-11 and transforming growth factor-β signaling in prostate cancer. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Qu, C.-B.; Zhang, Y.; Zhang, W.-F.; Wang, D.-D.; Gao, C.-C.; Ma, L.; Chen, J.-S.; Liu, K.-L.; Zheng, B.J.O. Dysregulation of p53-RBM25-mediated circAMOTL1L biogenesis contributes to prostate cancer progression through the circAMOTL1L-miR-193a-5p-Pcdha pathway. Oncogene 2019, 38, 2516–2532. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.K.; Bhardwaj, A.; Singh, S.; Arora, S.; McClellan, S.; Grizzle, W.E.; Reed, E.; Singh, A.P. Myb overexpression overrides androgen depletion–induced cell cycle arrest and apoptosis in prostate cancer cells, and confers aggressive malignant traits: Potential role in castration resistance. Carcinogenesis 2012, 33, 1149–1157. [Google Scholar] [CrossRef] [Green Version]
- Höflmayer, D.; Willich, C.; Hube-Magg, C.; Simon, R.; Lang, D.; Neubauer, E.; Jacobsen, F.; Hinsch, A.; Luebke, A.M.; Tsourlakis, M.C. SNW1 is a prognostic biomarker in prostate cancer. Diagn. Pathol. 2019, 14, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, Y.; Zhang, J.; Hu, Q.; Zhi, F.; Zhang, S.; Mao, D.; Zhang, Y.; Liang, H. Significance of the TMPRSS2: ERG gene fusion in prostate cancer. Mol. Med. Rep. 2017, 16, 5450–5458. [Google Scholar] [CrossRef] [Green Version]
- Elliott, D.J.; Dalgliesh, C.; Hysenaj, G.; Ehrmann, I. RBMX family proteins connect the fields of nuclear RNA processing, disease and sex chromosome biology. Int. J. Biochem. Cell Biol. 2019, 108, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Amankwah, E.K.; Anegbe, E.; Park, H.; Pow-Sang, J.; Hakam, A.; Park, J.Y. miR-21, miR-221 and miR-222 expression and prostate cancer recurrence among obese and non-obese cases. Asian J. Androl. 2013, 15, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.; Kwabi-Addo, B.; Ittmann, M.; Jelinek, J.; Shen, L.; Yu, Y.; Issa, J.-P.J. Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling. PLoS ONE 2008, 3, e2079. [Google Scholar] [CrossRef]
- Kanwal, R.; Plaga, A.R.; Liu, X.; Shukla, G.C.; Gupta, S. MicroRNAs in prostate cancer: Functional role as biomarkers. Cancer Lett. 2017, 407, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Asara, J.M.; Sanda, M.G.; Arredouani, M.S. The role of the transcription factor SIM2 in prostate cancer. PLoS ONE 2011, 6, e28837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elian, F.A.; Yan, E.; Walter, M.A. FOXC1, the new player in the cancer sandbox. Oncotarget 2018, 9, 8165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérot, G.; Derré, J.; Coindre, J.-M.; Tirode, F.; Lucchesi, C.; Mariani, O.; Gibault, L.; Guillou, L.; Terrier, P.; Aurias, A. Strong smooth muscle differentiation is dependent on myocardin gene amplification in most human retroperitoneal leiomyosarcomas. Cancer Res. 2009, 69, 2269–2278. [Google Scholar] [CrossRef] [Green Version]
- Hirata, H.; Ueno, K.; Shahryari, V.; Deng, G.; Tanaka, Y.; Tabatabai, Z.L.; Hinoda, Y.; Dahiya, R. MicroRNA-182-5p promotes cell invasion and proliferation by down regulating FOXF2, RECK and MTSS1 genes in human prostate cancer. PLoS ONE 2013, 8, e55502. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Nyalwidhe, J.O.; Guo, S.; Drake, R.R.; Semmes, O.J.; Proteomics, C. Targeted identification of metastasis-associated cell-surface sialoglycoproteins in prostate cancer. Mol. Cell. Proteom. 2011, 10, M110.007294. [Google Scholar] [CrossRef] [Green Version]
- Takekoshi, T.; Tada, Y.; Watanabe, T.; Sugaya, M.; Hoashi, T.; Komine, M.; Kawashima, T.; Shimizu, T.; Hau, C.S.; Asahina, A. Identification of a novel marker for dendritic cell maturation, mouse transmembrane protein 123. J. Biol. Chem. 2010, 285, 31876–31884. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wahdan-Alaswad, R.; Danielpour, D. Critical role of smad2 in tumor suppression and transforming growth factor-β–induced apoptosis of prostate epithelial cells. Cancer Res. 2009, 69, 2185–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss-Messer, E.; Merom, O.; Adi, A.; Karry, R.; Bidosee, M.; Ber, R.; Kaploun, A.; Stein, A.; Barkey, R.J. Growth hormone (GH) receptors in prostate cancer: Gene expression in human tissues and cell lines and characterization, GH signaling and androgen receptor regulation in LNCaP cells. Mol. Cell. Endocrinol. 2004, 220, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Vijayvargia, R.; May, M.S.; Fondell, J.D. A coregulatory role for the mediator complex in prostate cancer cell proliferation and gene expression. Cancer Res. 2007, 67, 4034–4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yegnasubramanian, S. Prostate cancer epigenetics and its clinical implications. Asian J. Androl. 2016, 18, 549. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.T.; Kwon, S.J.; Kim, J.; Kwon, Y.S.; Lee, N.; Hong, J.H.; Jamieson, C.; Kim, W.-J.; Kim, I.Y. WNT5A induces castration-resistant prostate cancer via CCL2 and tumour-infiltrating macrophages. Br. J. Cancer 2018, 118, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Brechka, H.; Bhanvadia, R.R.; VanOpstall, C.; Vander Griend, D.J. HOXB13 mutations and binding partners in prostate development and cancer: Function, clinical significance, and future directions. Genes Dis. 2017, 4, 75–87. [Google Scholar] [CrossRef]
- Wang, L.-N.; Chen, W.-W.; Zhang, J.; Li, C.-Y.; Liu, C.-Y.; Xue, J.; Zhang, P.-J.; Jiang, A.-L. The miRNA let-7a1 inhibits the expression of insulin-like growth factor 1 receptor (IGF1R) in prostate cancer PC-3 cells. Asian J. Androl. 2013, 15, 753. [Google Scholar] [CrossRef] [Green Version]
- Pietrangelo, A.; Ridgway, N.D. Golgi localization of oxysterol binding protein-related protein 4L (ORP4L) is regulated by ligand binding. J. Cell Sci. 2018, 131, jcs215335. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.-C.; Chen, P.-C.; Lin, Y.-F.; Su, C.-M.; Liu, J.-F.; Lin, T.-H.; Chuang, S.-M.; Tang, C.-H. Osteoblast-secreted WISP-1 promotes adherence of prostate cancer cells to bone via the VCAM-1/integrin α4β1 system. Cancer Lett. 2018, 426, 47–56. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Musa, J.; Aynaud, M.-M.; Mirabeau, O.; Delattre, O.; Grünewald, T.G. MYBL2 (B-Myb): A central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017, 8, e2895. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, E.J.; Li, L. Toll-interacting protein in resolving and non-resolving inflammation. Front. Immunol. 2017, 8, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Clarke, P.A.; Davis, R.; Mumuni, S.; Kwabi-Addo, B. Sex steroid-induced DNA methylation changes and inflammation response in prostate cancer. Cytokine 2016, 86, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Don-Doncow, N.; Marginean, F.; Coleman, I.; Nelson, P.S.; Ehrnström, R.; Krzyzanowska, A.; Morrissey, C.; Hellsten, R.; Bjartell, A. Expression of STAT3 in prostate cancer metastases. Eur. Urol. 2017, 71, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and prostate cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Aryankalayil, M.J.; Makinde, A.Y.; Gameiro, S.R.; Hodge, J.W.; Rivera-Solis, P.P.; Palayoor, S.T.; Ahmed, M.M.; Coleman, C.N. Defining molecular signature of pro-immunogenic radiotherapy targets in human prostate cancer cells. Radiat. Res. 2014, 182, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Johnson, I.R.; Parkinson-Lawrence, E.J.; Keegan, H.; Spillane, C.D.; Barry-O’Crowley, J.; Watson, W.R.; Selemidis, S.; Butler, L.M.; O’Leary, J.J.; Brooks, D.A. Endosomal gene expression: A new indicator for prostate cancer patient prognosis? Oncotarget 2015, 6, 37919. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Wang, J.; Sadar, M.D. Crosstalk between the androgen receptor and β-catenin in castrate-resistant prostate cancer. Cancer Res. 2008, 68, 9918–9927. [Google Scholar] [CrossRef] [Green Version]
- Jeter, C.R.; Liu, B.; Lu, Y.; Chao, H.-P.; Zhang, D.; Liu, X.; Chen, X.; Li, Q.; Rycaj, K.; Calhoun-Davis, T. NANOG reprograms prostate cancer cells to castration resistance via dynamically repressing and engaging the AR/FOXA1 signaling axis. Cell Discov. 2016, 2, 1–19. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- Stathias, V.; Turner, J.; Koleti, A.; Vidovic, D.; Cooper, D.; Fazel-Najafabadi, M.; Pilarczyk, M.; Terryn, R.; Chung, C.; Umeano, A.; et al. LINCS Data Portal 2.0: Next generation access point for perturbation-response signatures. Nucleic Acids Res. 2019, 48, D431–D439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, K.; Akakura, K. Pros and cons of active surveillance for low-risk prostate cancer. Gan Kagaku Ryoho. Cancer Chemother. 2011, 38, 2538–2541. [Google Scholar]
- Paulson, D.F. Impact of radical prostatectomy in the management of clinically localized disease. J. Urol. 1994, 152, 1826–1830. [Google Scholar] [CrossRef]
- Prasad, K.; Cole, W.; Kumar, B. Pros and cons of antioxidant use during radiation therapy. Cancer Treat. Rev. 2002, 28, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B.; Novellino, E. The therapeutic potential of apigenin. Int. J. Mol. Sci. 2019, 20, 1305. [Google Scholar] [CrossRef] [Green Version]
- Kaapu, K.J.; Murtola, T.J.; Talala, K.; Taari, K.; Tammela, T.L.; Auvinen, A. Digoxin and prostate cancer survival in the Finnish Randomized Study of Screening for Prostate Cancer. Br. J. Cancer 2016, 115, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- Sheikhzade, M.; Akbari, A.; Karkhaneh, R.; Ghassemi, F.; Sadeghi, A.; Kavakani, S.; Mobini, G.R.; Mohebbi, M.; Heidari, M. Digoxin Inhibits Retinoblastoma through Suppressing a Non-canonical TGFβ Signaling Pathway. Br. J. Med. Med Res. 2016, 15, 1–10. [Google Scholar] [CrossRef]
- Wright, C.; Iyer, A.K.V.; Kaushik, V.; Azad, N. Anti-tumorigenic potential of a novel orlistat-AICAR combination in prostate cancer cells. J. Cell. Biochem. 2017, 118, 3834–3845. [Google Scholar] [CrossRef]
- Yeh, S.-J.; Lin, C.-Y.; Li, C.-W.; Chen, B.-S. Systems Biology Approaches to Investigate Genetic and Epigenetic Molecular Progression Mechanisms for Identifying Gene Expression Signatures in Papillary Thyroid Cancer. Int. J. Mol. Sci. 2019, 20, 2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatr-Aryamontri, A.; Breitkreutz, B.-J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015, 43, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef]
- Bovolenta, L.A.; Acencio, M.L.; Lemke, N. HTRIdb: An open-access database for experimentally verified human transcriptional regulation interactions. Nat. Précéd. 2012, 13, 1–10. [Google Scholar]
- Zheng, G.; Tu, K.; Yang, Q.; Xiong, Y.; Wei, C.; Xie, L.; Zhu, Y.; Li, Y. ITFP: An integrated platform of mammalian transcription factors. Bioinformatics 2008, 24, 2416–2417. [Google Scholar] [CrossRef]
- Wingender, E. The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief. Bioinform. 2008, 9, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Friard, O.; Re, A.; Taverna, D.; De Bortoli, M.; Corá, D. CircuitsDB: A database of mixed microRNA/transcription factor feed-forward regulatory circuits in human and mouse. BMC Bioinform. 2010, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2. 0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.-S.; Wu, C.-C. Systems biology as an integrated platform for bioinformatics, systems synthetic biology, and systems metabolic engineering. Cells 2013, 2, 635–688. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Ishiguro, M.; Kitagawa, G. Akaike Information Criterion Statistics; D. Reidel: Dordrecht, The Netherlands, 1986; Volume 81, p. 26853. [Google Scholar]
- Dong, J.; Yao, Z.-J.; Zhang, L.; Luo, F.; Lin, Q.; Lu, A.-P.; Chen, A.F.; Cao, D.-S. PyBioMed: A python library for various molecular representations of chemicals, proteins and DNAs and their interactions. J. Cheminform. 2018, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringnér, M. What is principal component analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Hecht-Nielsen, R. Theory of the backpropagation neural network. In Neural Networks for Perception; Elsevier: Amsterdam, The Netherlands, 1992; pp. 65–93. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Disease | Drug Targets |
|---|---|
| PCa (covering lean and obese) | STAT1, FOXF2, SIM2, SMAD2, MYB, EGFR |
| Obesity-specific PCa | STAT1, FOXF2, SIM2, SMAD2, CERK, STAT3, TP53 |
| Targets | STAT1 | FOXF2 | SIM2 | SMAD2 | MYB | EGFR | |
|---|---|---|---|---|---|---|---|
| Drugs | |||||||
| Apigenin | ▪ | ▪ | ▪ | ▪ | |||
| Digoxin | ▪ | ▪ | |||||
| Targets | STAT1 | FOXF2 | SIM2 | SMAD2 | CERK | STAT3 | TP53 | |
|---|---|---|---|---|---|---|---|---|
| Drugs | ||||||||
| Apigenin | ▪ | ▪ | ▪ | ▪ | ||||
| Digoxin | ▪ | ▪ | ▪ | ▪ | ||||
| Orlistat | ▪ | ▪ | ▪ | ▪ | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, S.-J.; Chung, Y.-C.; Chen, B.-S. Investigating the Role of Obesity in Prostate Cancer and Identifying Biomarkers for Drug Discovery: Systems Biology and Deep Learning Approaches. Molecules 2022, 27, 900. https://doi.org/10.3390/molecules27030900
Yeh S-J, Chung Y-C, Chen B-S. Investigating the Role of Obesity in Prostate Cancer and Identifying Biomarkers for Drug Discovery: Systems Biology and Deep Learning Approaches. Molecules. 2022; 27(3):900. https://doi.org/10.3390/molecules27030900
Chicago/Turabian StyleYeh, Shan-Ju, Yun-Chen Chung, and Bor-Sen Chen. 2022. "Investigating the Role of Obesity in Prostate Cancer and Identifying Biomarkers for Drug Discovery: Systems Biology and Deep Learning Approaches" Molecules 27, no. 3: 900. https://doi.org/10.3390/molecules27030900
APA StyleYeh, S.-J., Chung, Y.-C., & Chen, B.-S. (2022). Investigating the Role of Obesity in Prostate Cancer and Identifying Biomarkers for Drug Discovery: Systems Biology and Deep Learning Approaches. Molecules, 27(3), 900. https://doi.org/10.3390/molecules27030900