
Directed Evolution of a Nonheme Diiron N-oxygenase AzoC for Improving Its Catalytic Efficiency toward Nitrogen Heterocycle Substrates


Abstract
:1. Introduction
2. Results
2.1. The Substrate Scope Extension of AzoC
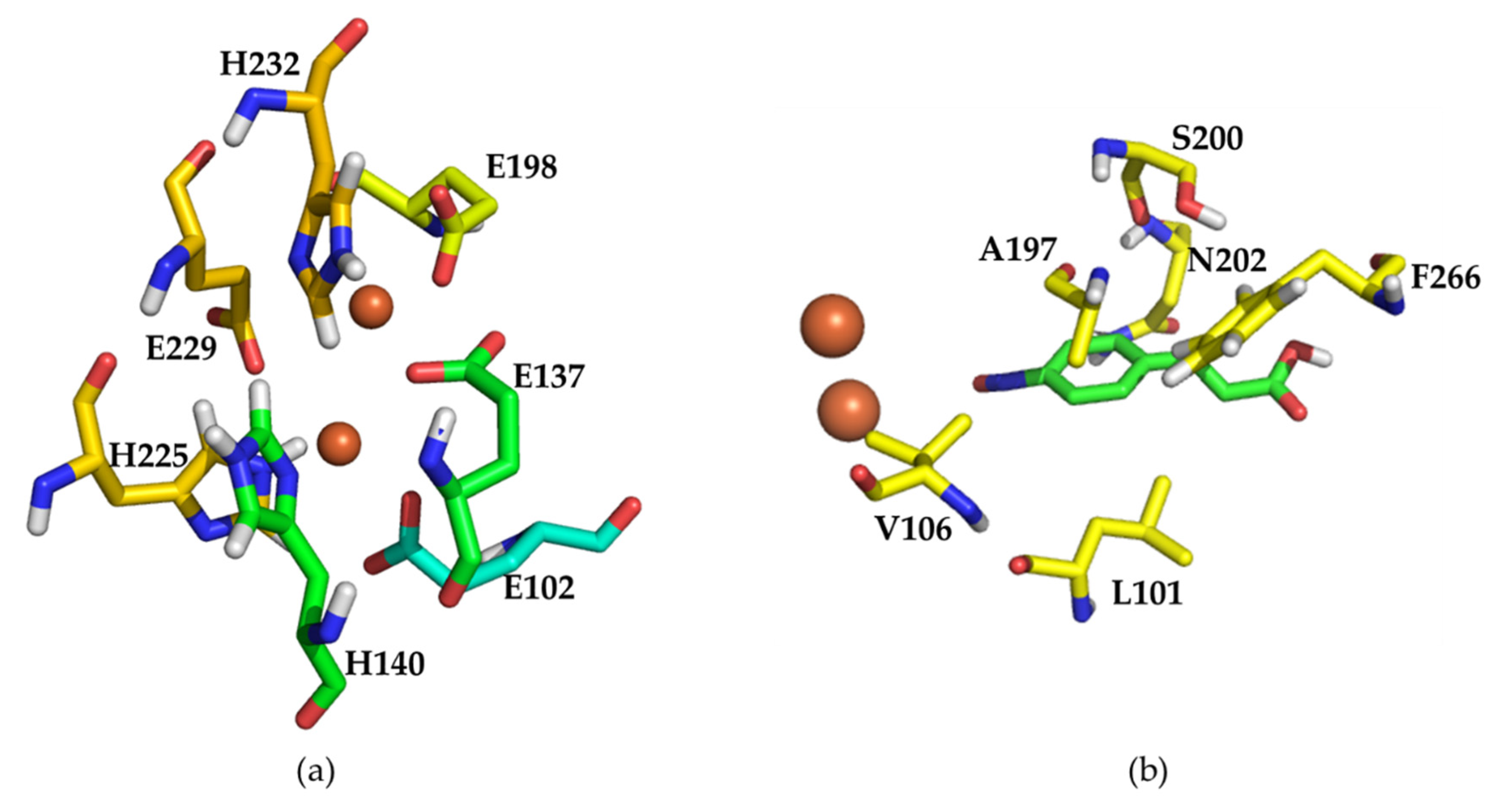
2.2. Mutational Analysis and Mutant Strain Construction
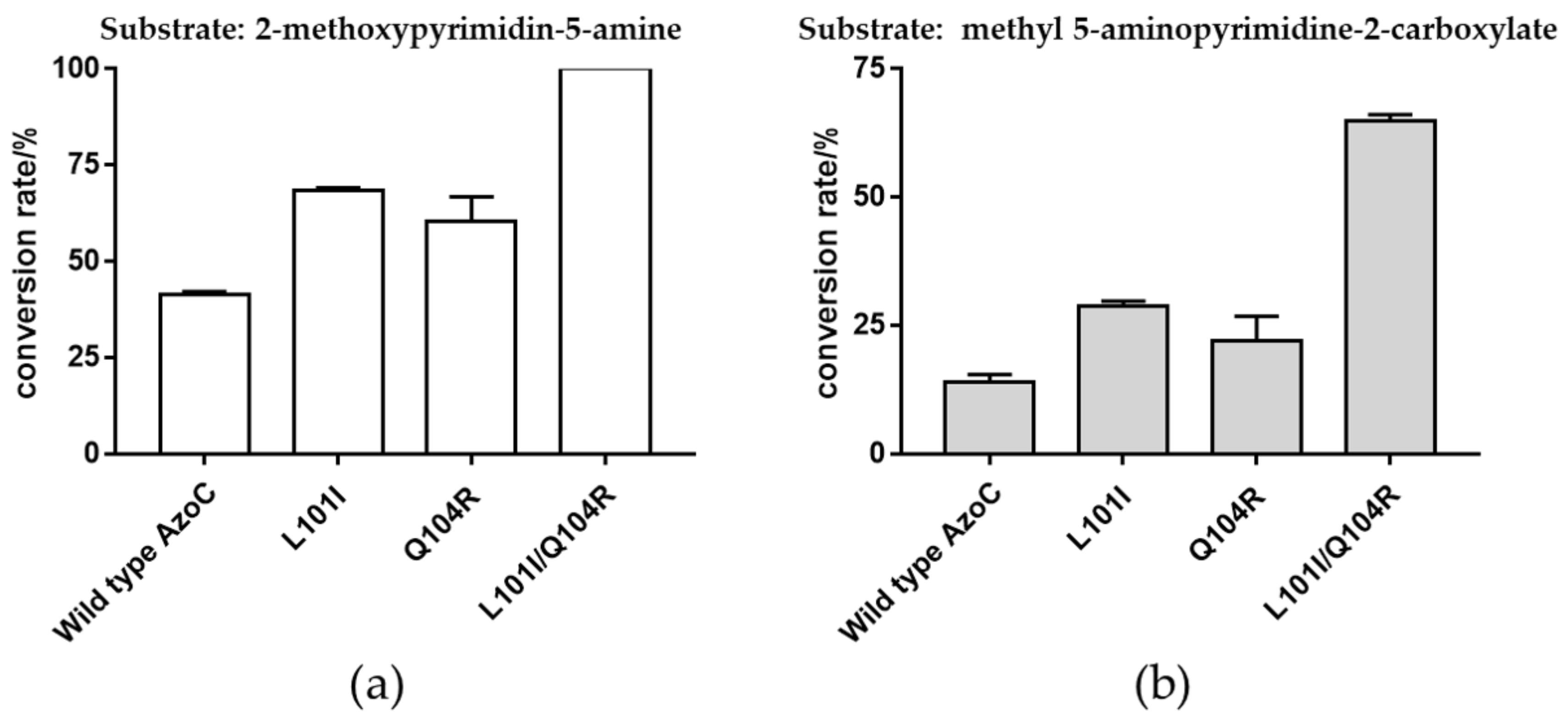
2.3. Beneficial Mutations with Improved Catalytic Efficiency toward 5-Aminopyrimidine-Type Compounds
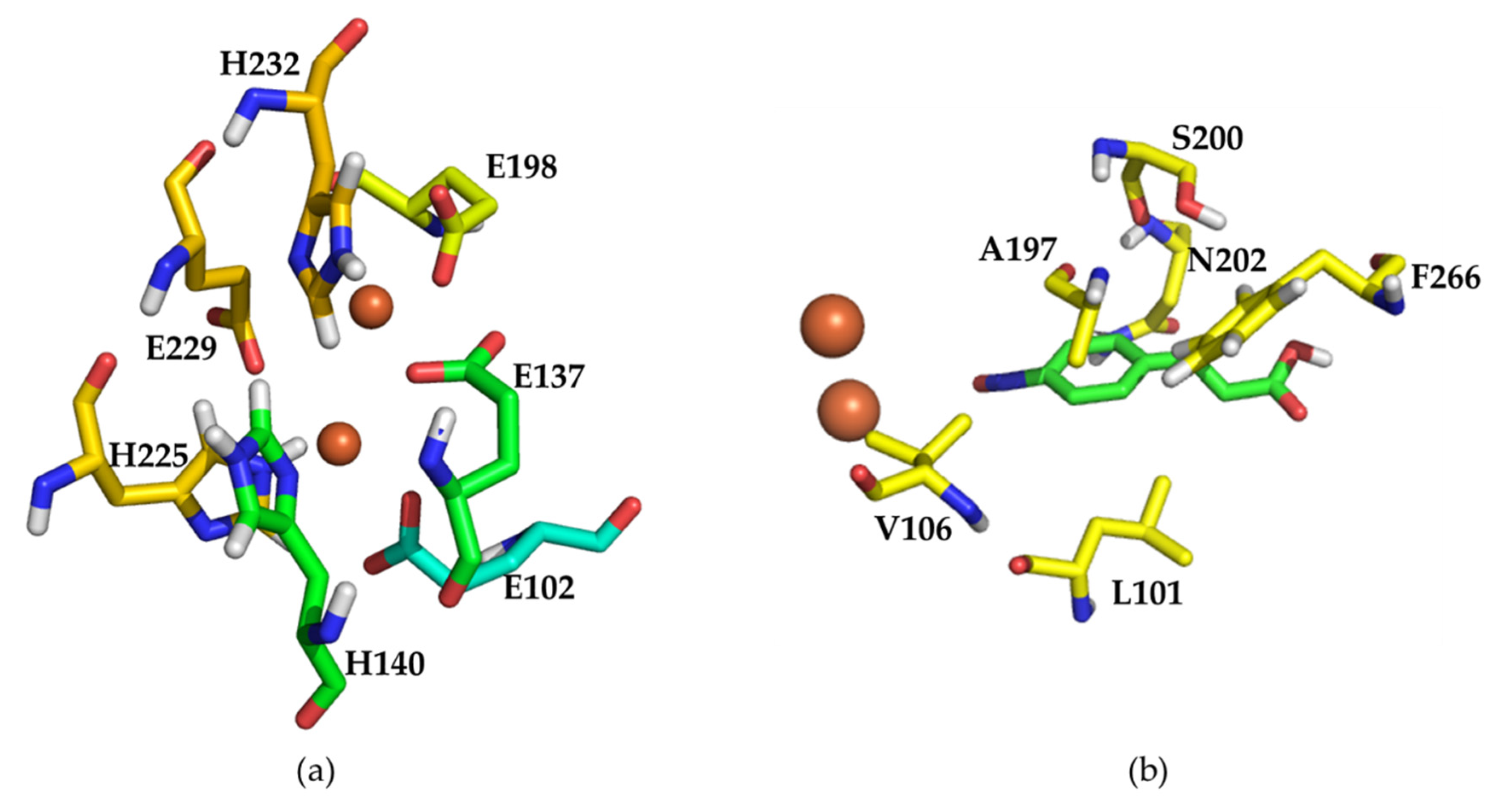
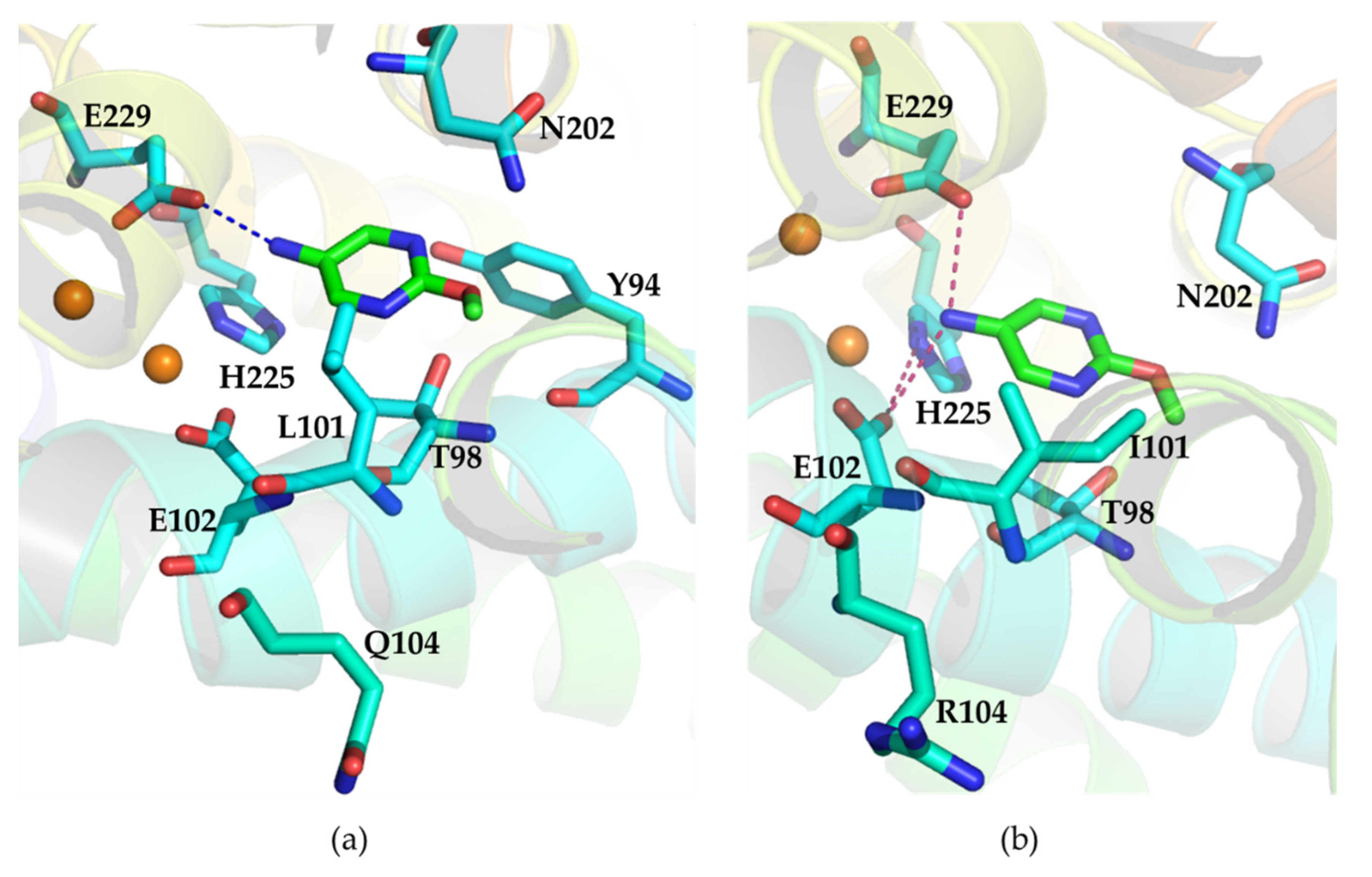
2.4. Structural Analysis of Wild Type AzoC and the Double Mutant L101I/Q104R
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. In Vitro Reaction System and LC-MS Analysis
4.3. Mutation Library Construction
4.4. The Catalytic Activity Assay of Crude Cell Lysates and the Purified Protein
4.5. Kinetic Data Analysis of Wild Type AzoC and the Mutant L101I, Q104R, L101I/Q104R
4.6. Molecular Modeling and Docking Analysis of Wild Type AzoC and the Mutant L101I/Q104R
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wibowo, M.; Ding, L. Chemistry and Biology of Natural Azoxy Compounds. J. Nat. Prod. 2020, 83, 3482–3491. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.M.; Sperry, J. Natural Products Containing a Nitrogen–Nitrogen Bond. J. Nat. Prod. 2013, 76, 794–812. [Google Scholar] [CrossRef] [PubMed]
- Laatsch, H. AntiBase: The Natural Compound Identifier; Wiley-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Dai, Y.; Li, C.; Shen, Y.; Lim, T.; Xu, J.; Li, Y.; Niemantsverdriet, H.; Besenbacher, F.; Lock, N.; Su, R. Light-tuned selective photosynthesis of azo- and azoxy-aromatics using graphitic C3N. Nat. Commun. 2018, 9, 60. [Google Scholar] [CrossRef]
- Chilaya, G.; Chanishvili, A.; Petriashvili, G.; Barberi, R.; Bartolino, R.; Cipparrone, G.; Mazzulla, A.; Shibaev, P.V. Reversible Tuning of Lasing in Cholesteric Liquid Crystals Controlled by Light-Emitting Diodes. Adv. Mater. 2007, 19, 565–568. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Fujii, A.; Mori, H. Selective synthesis of azoxybenzenes from nitrobenzenes by visible light irradiation under continuous flow conditions. React. Chem. Eng. 2019, 4, 2055–2059. [Google Scholar] [CrossRef]
- Wang, Y.; Li, S.; Li, Y.; Zhang, R.; Wang, D.; Pang, S. A comparative study of the structure, energetic performance and stability of nitro-NNO-azoxy substituted explosives. J. Mater. Chem. A 2014, 2, 20806–20813. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, W.; Xiao, H. Comparative Theoretical Studies of Energetic Substituted Carbon- and Nitrogen-Bridged Difurazans. J. Phys. Chem. A 2009, 114, 603–612. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, Y.; Xiao, Q.; Zhu, H. Selective reduction of nitroaromatics to azoxy compounds on supported Ag–Cu alloy nanoparticles through visible light irradiation. Green Chem. 2015, 18, 817–825. [Google Scholar] [CrossRef]
- Kariya, Y.; Kubota, T.; Fromont, J.; Kobayashi, J. Pyrinadine A, a novel pyridine alkaloid with an azoxy moiety from sponge Cribrochalina sp. Tetrahedron Lett. 2006, 47, 997–998. [Google Scholar] [CrossRef]
- Kariya, Y.; Kubota, T.; Fromont, J.; Kobayashi, J. Pyrinadines B–G, new bis-pyridine alkaloids with an azoxy moiety from sponge Cribrochalina sp. Bioorganic Med. Chem. 2006, 14, 8415–8419. [Google Scholar] [CrossRef]
- Anwar, M.; Lee, V. A short biomimetic synthesis of marine sponge alkaloid Pyrinadine A. Tetrahedron 2009, 65, 5834–5837. [Google Scholar] [CrossRef]
- Yin, P.; Zhang, Q.; Shreeve, J.M. Dancing with Energetic Nitrogen Atoms: Versatile N-Functionalization Strategies for N-Heterocyclic Frameworks in High Energy Density Materials. Accounts Chem. Res. 2015, 49, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.P.; Ma, Y.; Hoyt, J.C.; Parry, R.J. Molecular characterization and analysis of the biosynthetic gene cluster for the azoxy antibiotic valanimycin. Mol. Microbiol. 2002, 46, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.-Y.; Li, H.; Zhou, Z.-X.; Mao, X.-M.; Tang, Y.; Chen, X.; Jiang, X.-H.; Liu, Y.; Jiang, H.; Li, Y.-Q. Identification and Biosynthetic Characterization of Natural Aromatic Azoxy Products from Streptomyces chattanoogensis L. Org. Lett. 2015, 17, 6114–6117. [Google Scholar] [CrossRef]
- Guo, Y.-Y.; Li, Z.-H.; Xia, T.-Y.; Du, Y.-L.; Mao, X.-M.; Li, Y.-Q. Molecular mechanism of azoxy bond formation for azoxymycins biosynthesis. Nat. Commun. 2019, 10, 4420. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Denard, C.A.; Ren, H.; Zhao, H. Improving and repurposing biocatalysts via directed evolution. Curr. Opin. Chem. Biol. 2015, 25, 55–64. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, P.; Cao, M.; Yu, T.; Lane, S.T.; Zhao, H. Directed Evolution: Methodologies and Applications. Chem. Rev. 2021, 121, 12384–12444. [Google Scholar] [CrossRef]
- Davids, T.; Schmidt, M.; Böttcher, D.; Bornscheuer, U.T. Strategies for the discovery and engineering of enzymes for biocatalysis. Curr. Opin. Chem. Biol. 2013, 17, 215–220. [Google Scholar] [CrossRef]
- Lane, M.D.; Seelig, B. Advances in the directed evolution of proteins. Curr. Opin. Chem. Biol. 2014, 22, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Mate, D.; Alcalde, M. Laccase engineering: From rational design to directed evolution. Biotechnol. Adv. 2015, 33, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chica, R.A.; Doucet, N.; Pelletier, J.N. Semi-rational approaches to engineering enzyme activity: Combining the benefits of directed evolution and rational design. Curr. Opin. Biotechnol. 2005, 16, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S. Beyond directed evolution—semi-rational protein engineering and design. Curr. Opin. Biotechnol. 2010, 21, 734–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voskarides, K. Directed Evolution. The Legacy of a Nobel Prize. J. Mol. Evol. 2020, 89, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Jasniewski, A.J.; Que, J.L. Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev. 2018, 118, 2554–2592. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Osés, G.; Osuna, S.; Gao, X.; Sawaya, M.; Gilson, L.; Collier, S.J.; Huisman, G.W.; Yeates, T.O.; Tang, Y.; Houk, K.N. The role of distant mutations and allosteric regulation on LovD active site dynamics. Nat. Chem. Biol. 2014, 10, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Acevedo-Rocha, C.G.; Li, A.; D’Amore, L.; Hoebenreich, S.; Sanchis, J.; Lubrano, P.; Ferla, M.; Garcia-Borràs, M.; Osuna, S.; Reetz, M.T. Pervasive cooperative mutational effects on multiple catalytic enzyme traits emerge via long-range conformational dynamics. Nat. Commun. 2021, 12, 1261. [Google Scholar] [CrossRef]
- Romero-Rivera, A.; Garcia-Borràs, M.; Osuna, S. Role of Conformational Dynamics in the Evolution of Retro-Aldolase Activity. ACS Catal. 2017, 7, 8524–8532. [Google Scholar] [CrossRef]
- Ma, N.; Chen, Z.; Chen, J.; Chen, J.; Wang, C.; Zhou, H.; Yao, L.; Shoji, O.; Watanabe, Y.; Cong, Z. Dual-Functional Small Molecules for Generating an Efficient Cytochrome P450BM3 Peroxygenase. Angew. Chem. Int. 2018, 57, 7628–7633. [Google Scholar] [CrossRef]
- Watkins, D.; Jenkins, J.; Grayson, K.J.; Wood, N.; Steventon, J.W.; Le Vay, K.K.; Goodwin, M.I.; Mullen, A.S.; Bailey, H.J.; Crump, M.P.; et al. Construction and in vivo assembly of a catalytically proficient and hyperthermostable de novo enzyme. Nat. Commun. 2017, 8, 358. [Google Scholar] [CrossRef] [Green Version]
- Reig, A.; Pires, M.M.; Snyder, R.A.; Wu, Y.; Jo, H.; Kulp, D.W.; Butch, S.E.; Calhoun, J.R.; Szyperski, T.; Solomon, E.I.; et al. Alteration of the oxygen-dependent reactivity of de novo Due Ferri proteins. Nat. Chem. 2012, 4, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Nastri, F.; Chino, M.; Maglio, O.; Bhagi-Damodaran, A.; Lu, Y.; Lombardi, A. Design and engineering of artificial oxygen-activating metalloenzymes. Chem. Soc. Rev. 2016, 45, 5020–5054. [Google Scholar] [CrossRef] [PubMed]
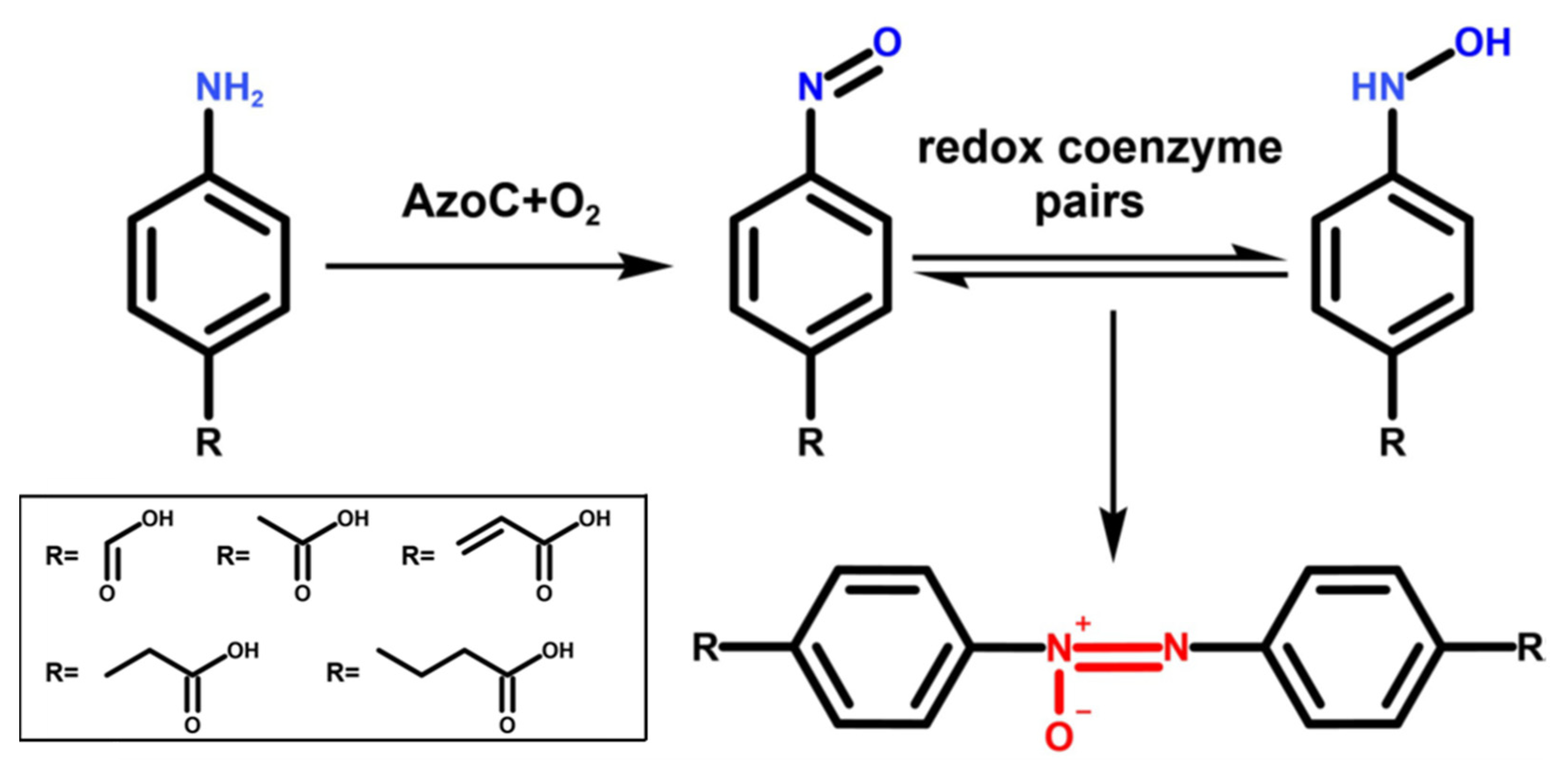




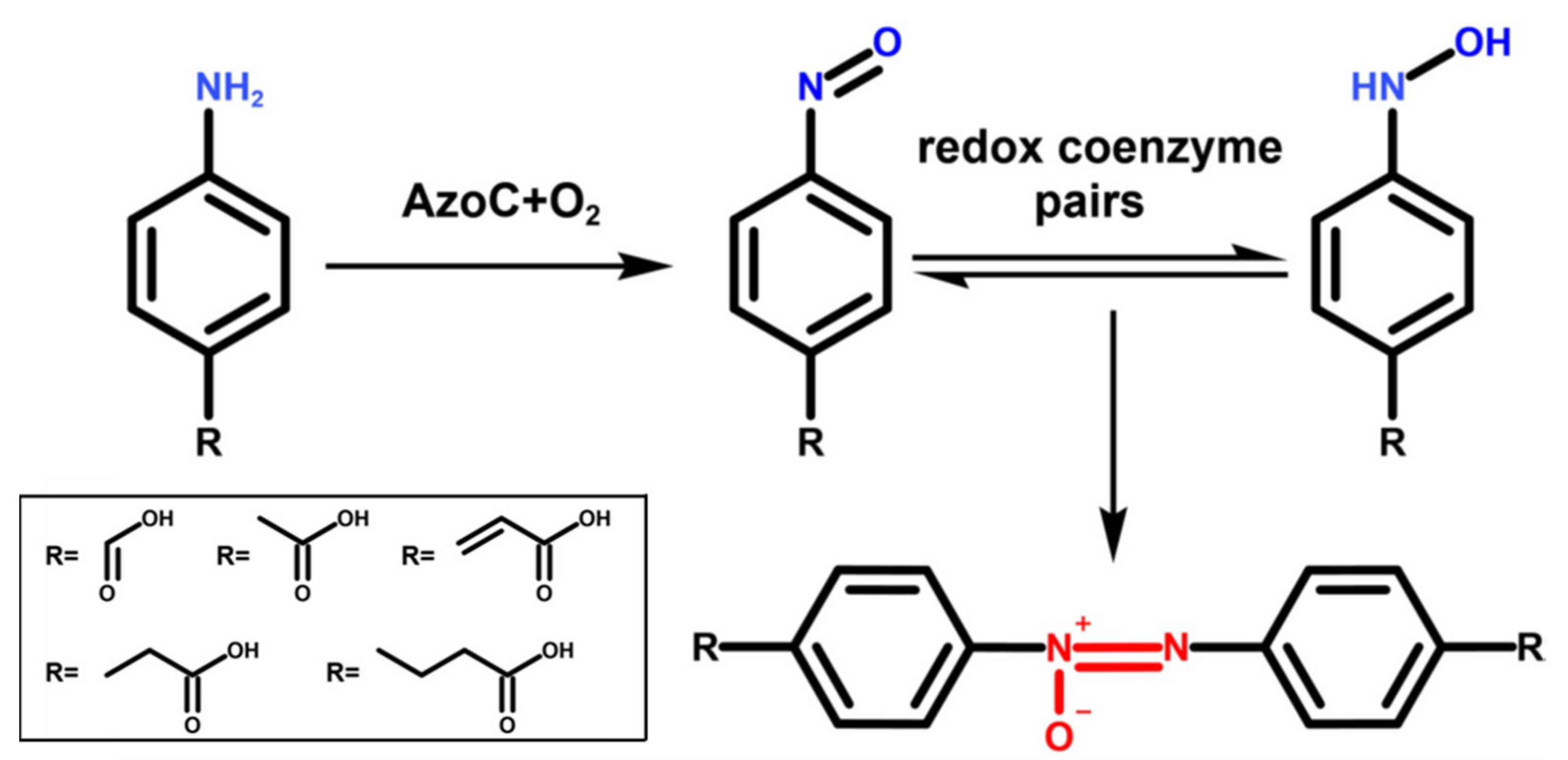{kind=link}
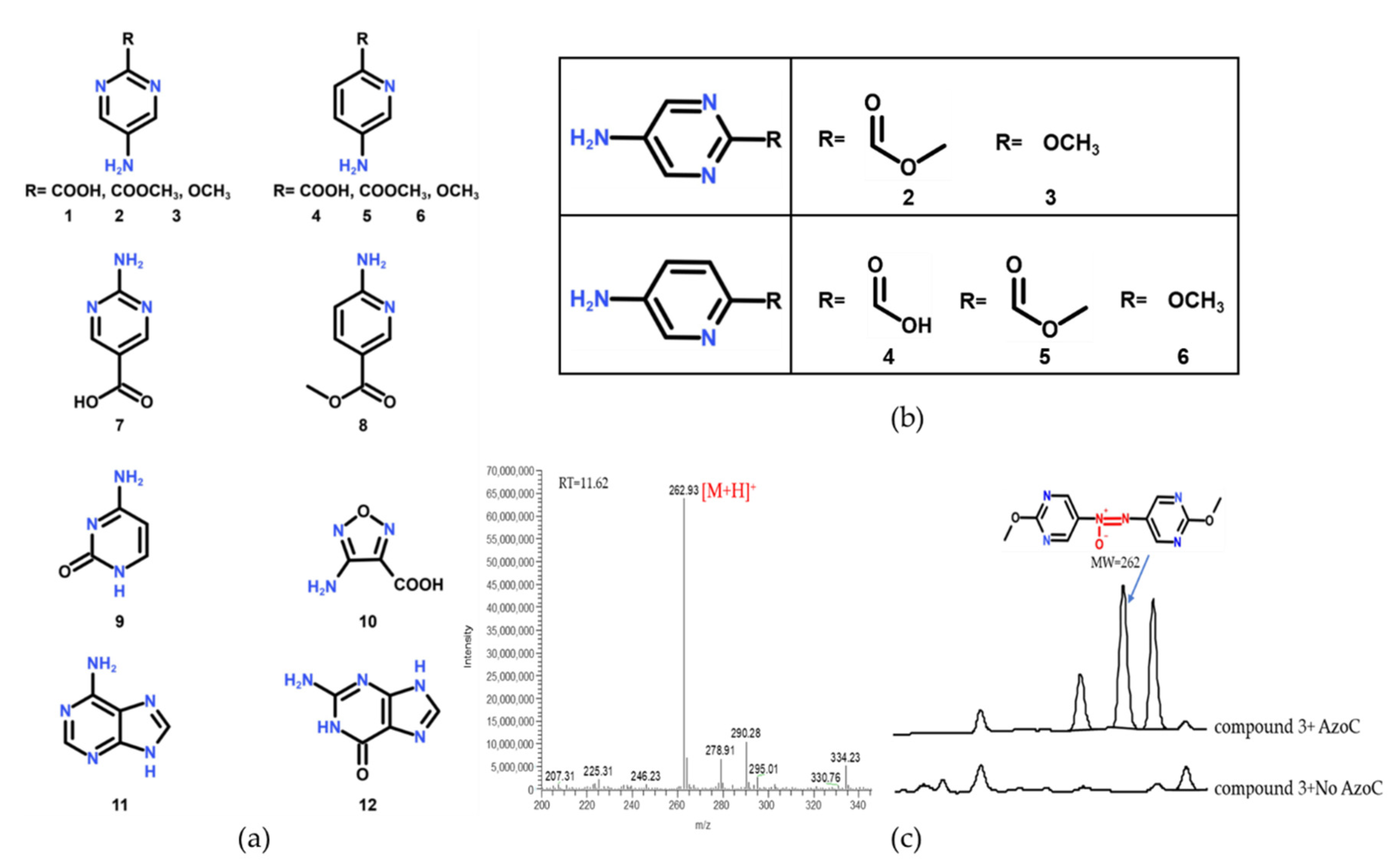{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Substrate: 2-methoxypyrimidin-5-amine | |||
|---|---|---|---|---|
| Km (mM) | kcat (s−1) | kcat/Km (M−1s−1) | Improvement | |
| Wild type | 0.65 ± 0.12 | 0.018 ± 0.0014 | 27.7 | 1 |
| L101I | 0.51 ± 0.059 | 0.031 ± 0.0013 | 60.8 | 2.19-fold |
| Q104R | 0.42 ± 0.082 | 0.016 ± 0.0012 | 38.1 | 1.38-fold |
| L101I/Q104R | 0.55 ± 0.060 | 0.046 ± 0.0019 | 83.6 | 3.02-fold |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Liu, X.-F.; Chen, X.-A.; Li, Y.-Q. Directed Evolution of a Nonheme Diiron N-oxygenase AzoC for Improving Its Catalytic Efficiency toward Nitrogen Heterocycle Substrates. Molecules 2022, 27, 868. https://doi.org/10.3390/molecules27030868
Xu Y, Liu X-F, Chen X-A, Li Y-Q. Directed Evolution of a Nonheme Diiron N-oxygenase AzoC for Improving Its Catalytic Efficiency toward Nitrogen Heterocycle Substrates. Molecules. 2022; 27(3):868. https://doi.org/10.3390/molecules27030868
Chicago/Turabian StyleXu, Ye, Xiao-Fang Liu, Xin-Ai Chen, and Yong-Quan Li. 2022. "Directed Evolution of a Nonheme Diiron N-oxygenase AzoC for Improving Its Catalytic Efficiency toward Nitrogen Heterocycle Substrates" Molecules 27, no. 3: 868. https://doi.org/10.3390/molecules27030868
APA StyleXu, Y., Liu, X.-F., Chen, X.-A., & Li, Y.-Q. (2022). Directed Evolution of a Nonheme Diiron N-oxygenase AzoC for Improving Its Catalytic Efficiency toward Nitrogen Heterocycle Substrates. Molecules, 27(3), 868. https://doi.org/10.3390/molecules27030868