
Newly Emerging Strategies in Antiviral Drug Discovery: Dedicated to Prof. Dr. Erik De Clercq on Occasion of His 80th Anniversary

 , , and
, , and 
Abstract
:1. Introduction
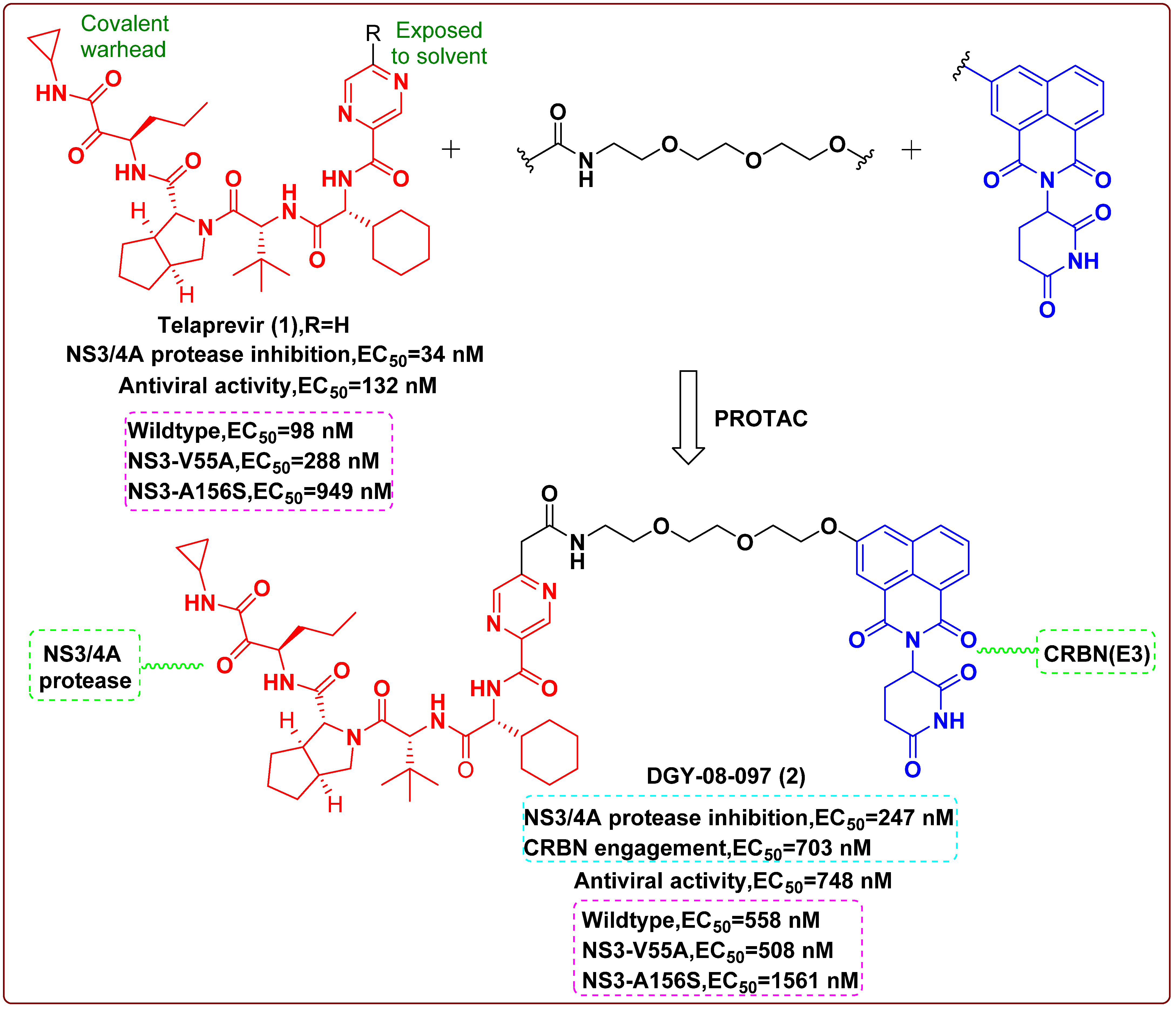
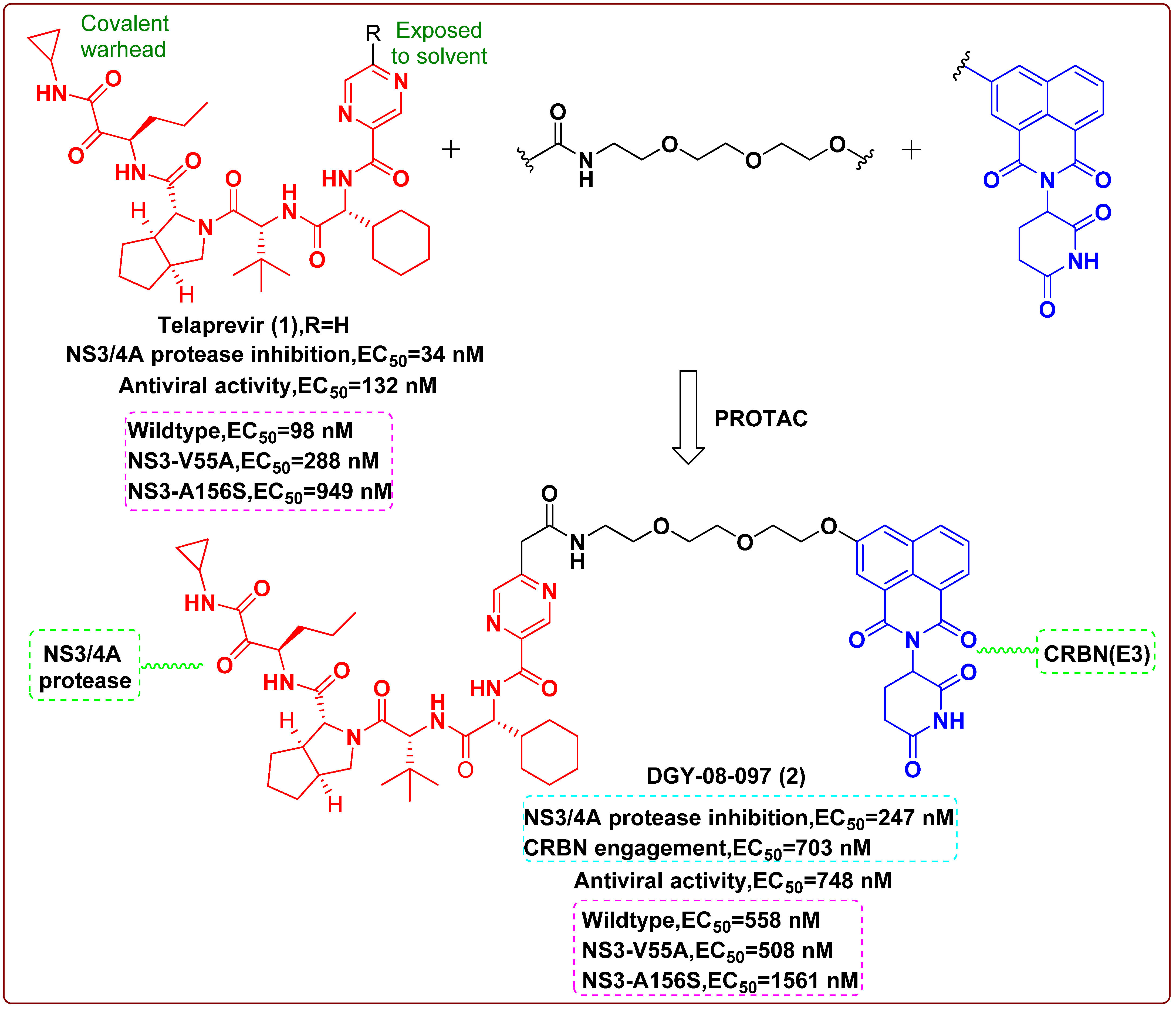
2. Proteolysis Targeting Chimera (PROTAC)
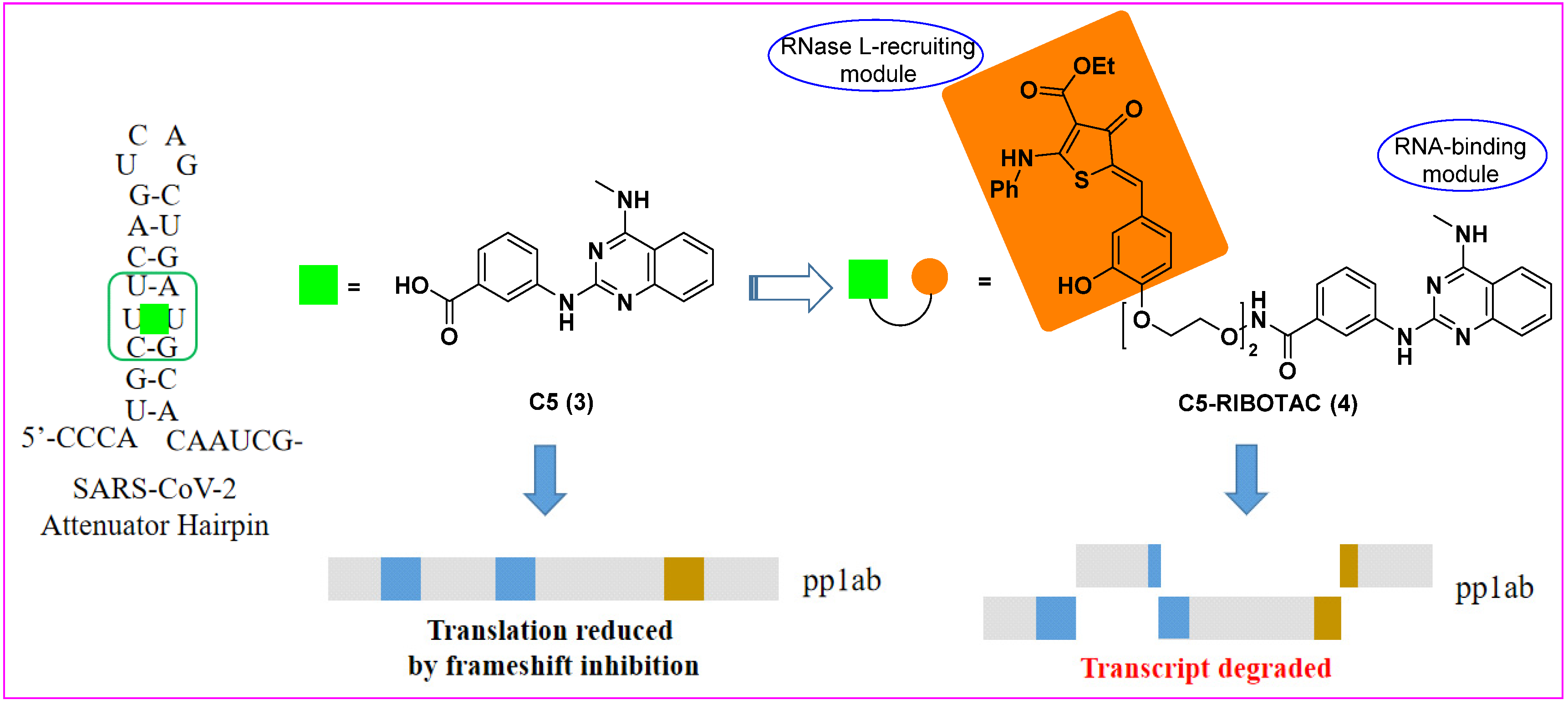
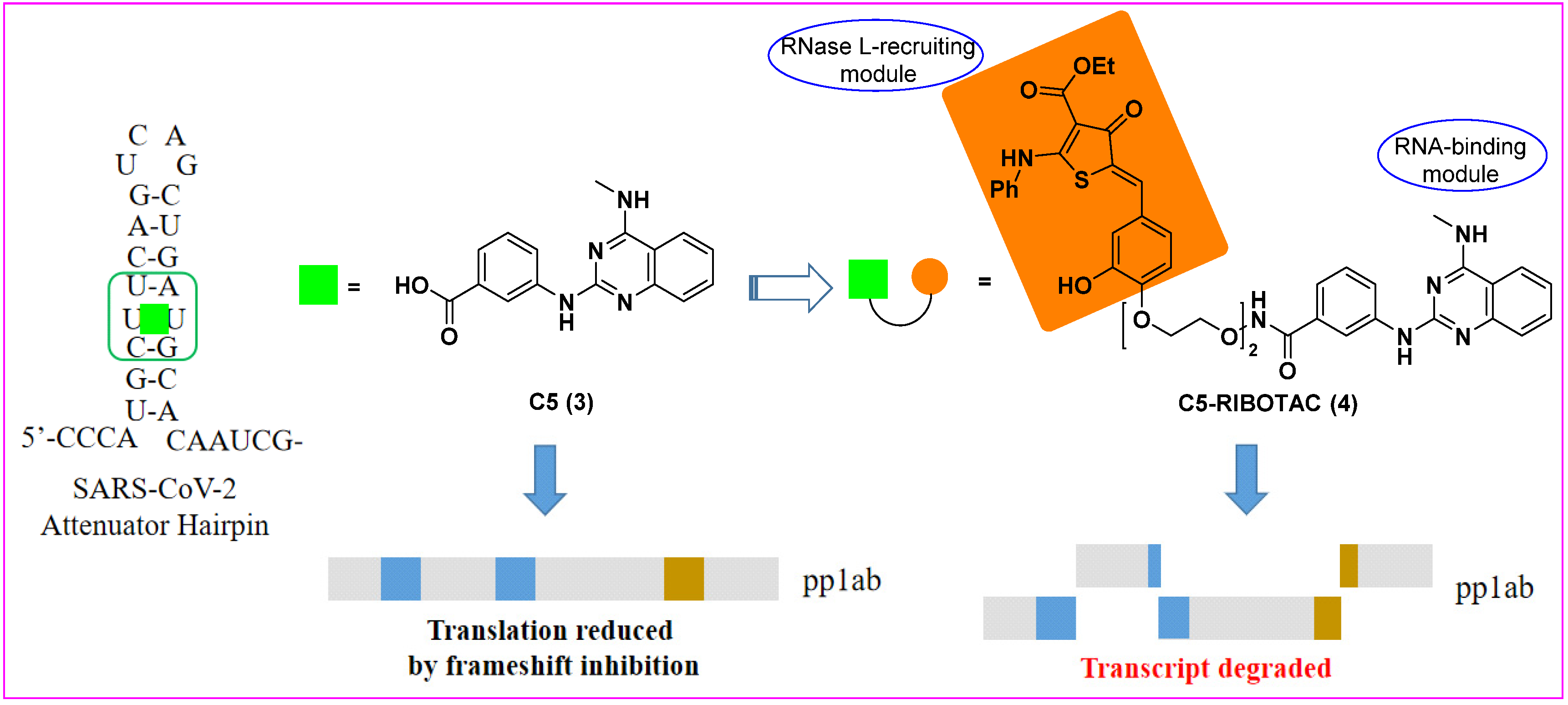
3. Ribonuclease Targeting Chimera (RIBOTAC)
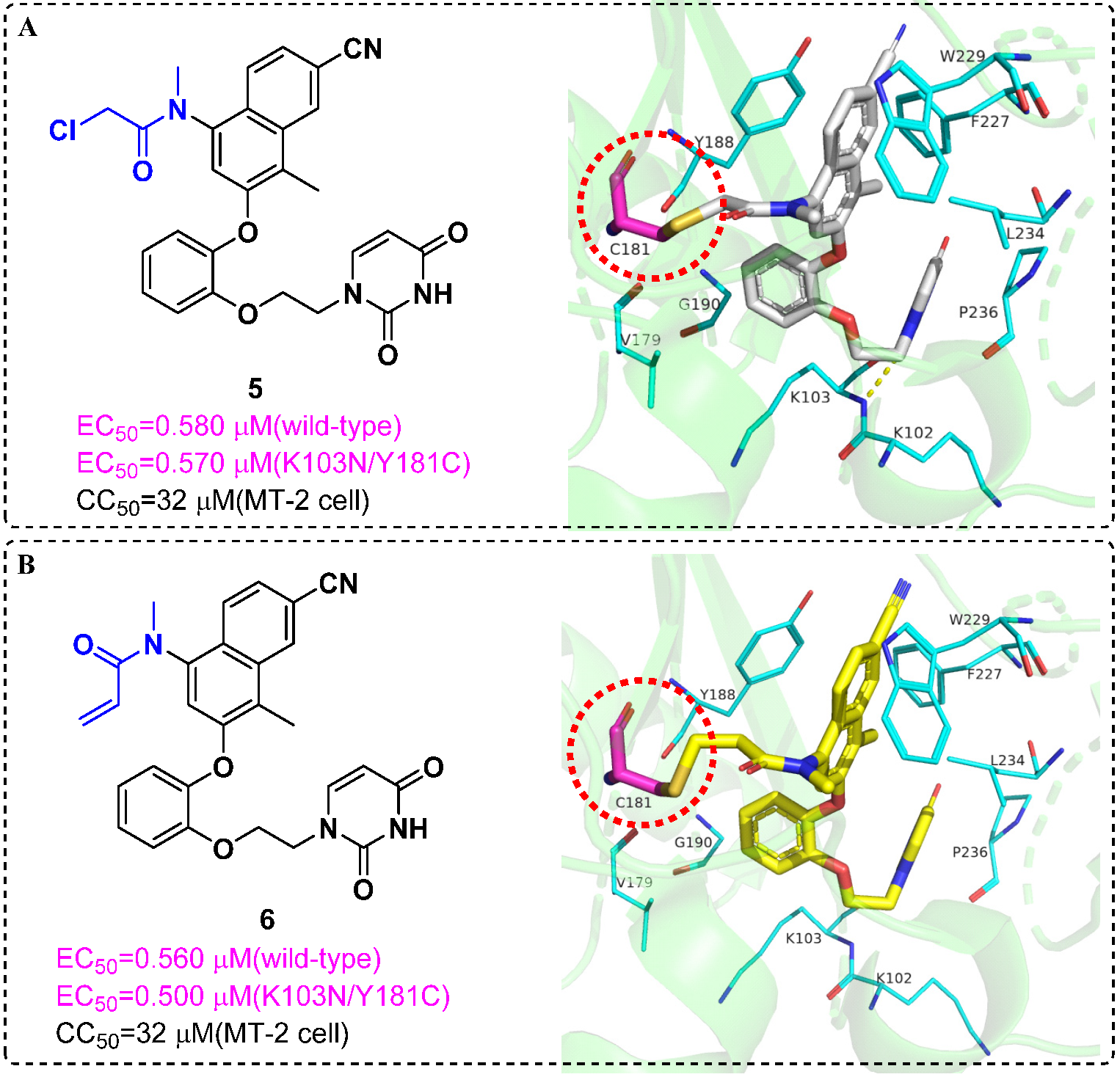
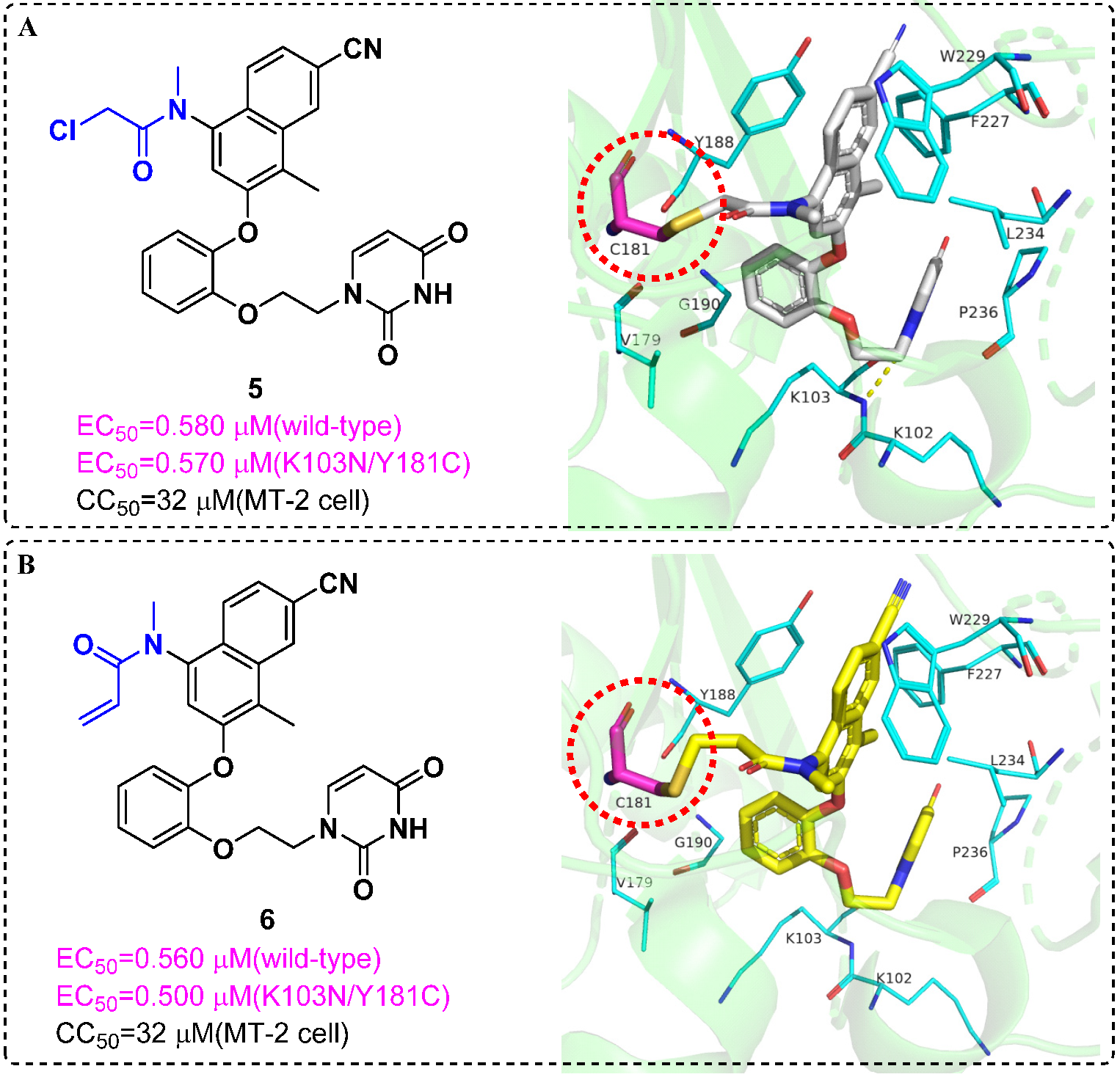
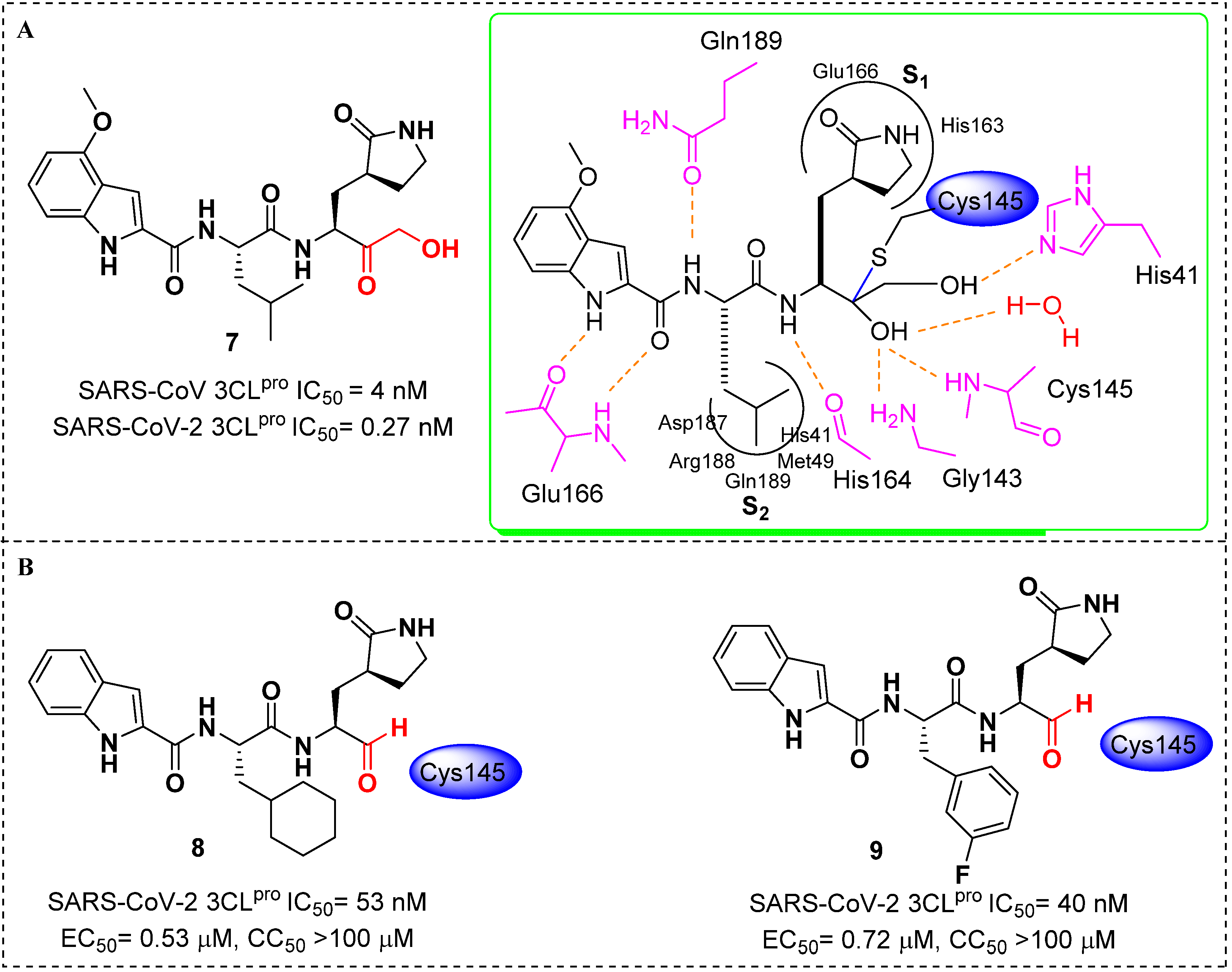
4. Targeted Covalent Inhibitors
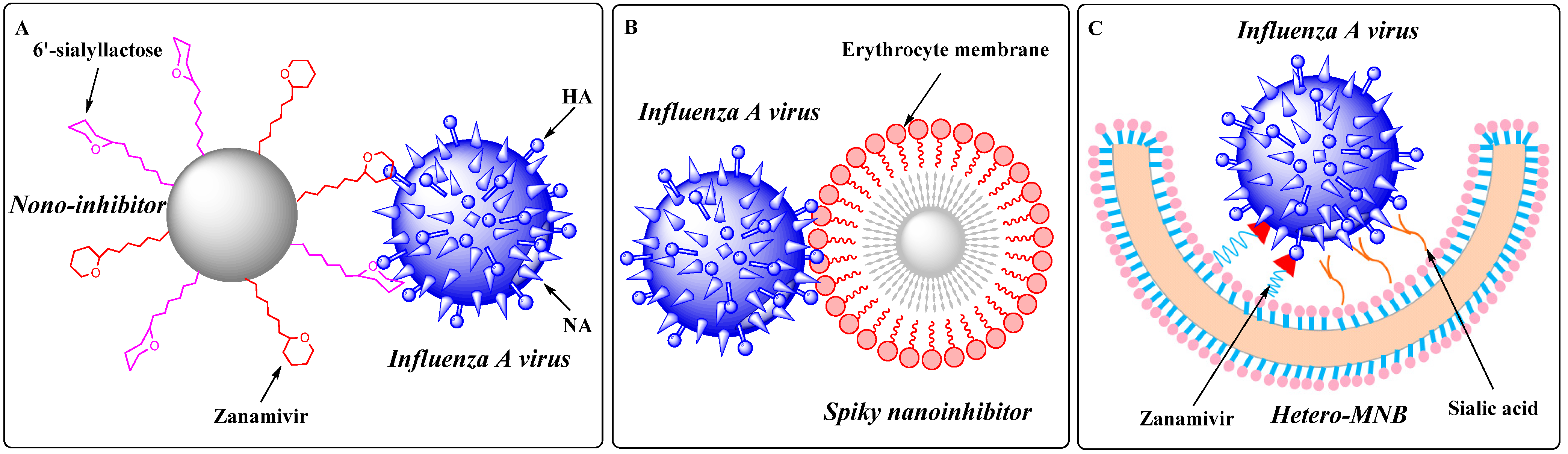
5. Topology-Matching Design
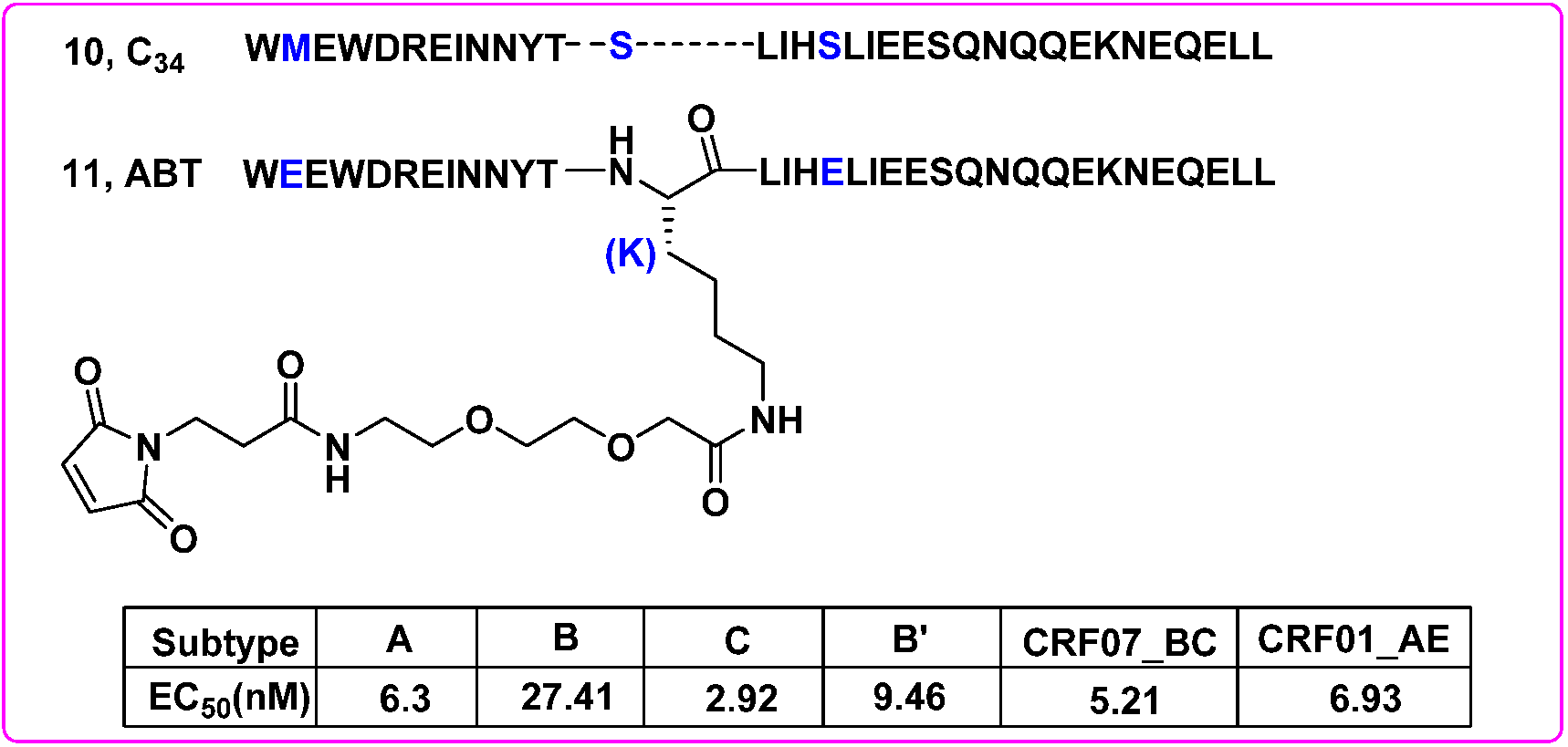
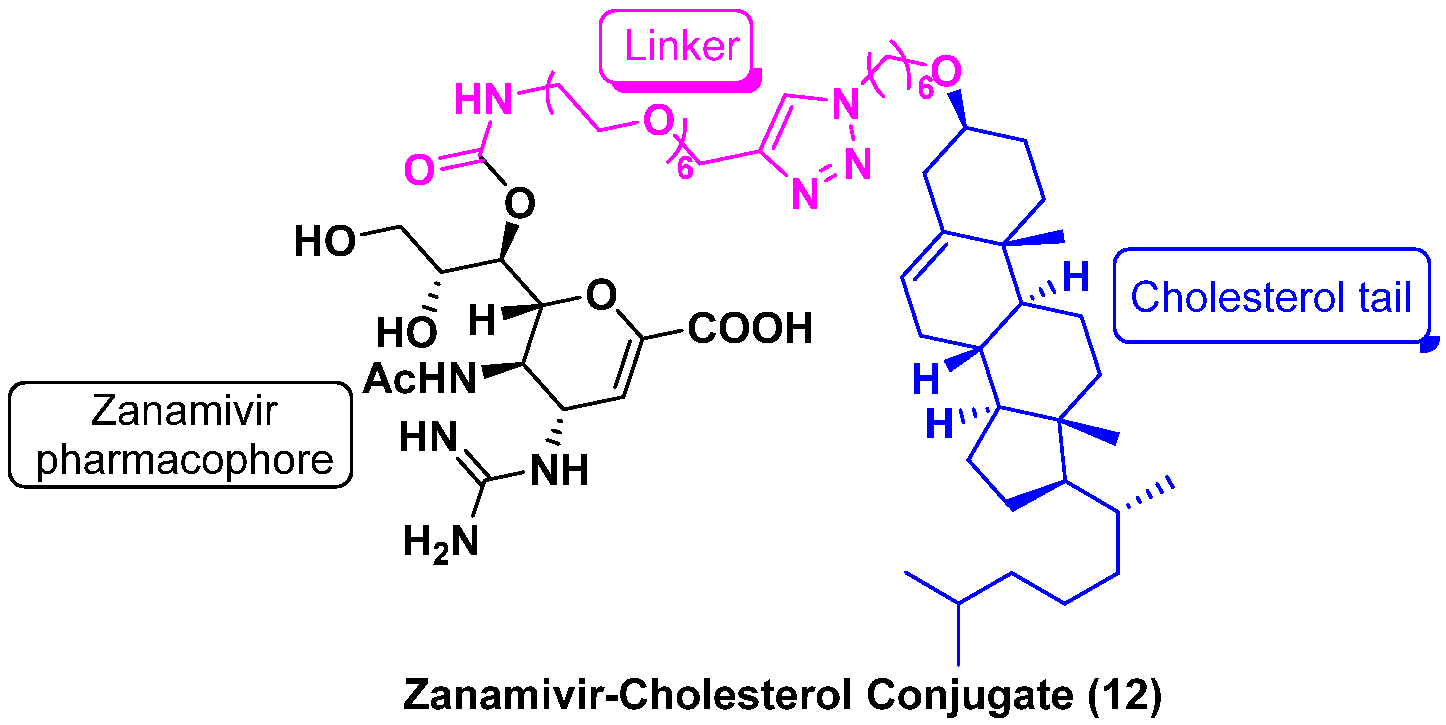
6. Antiviral Drug Delivery System
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Clercq, E. Antivirals: Past, present and future. Biochem. Pharmacol. 2013, 85, 727–744. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Fifty Years in Search of Selective Antiviral Drugs. J. Med. Chem. 2019, 62, 7322–7339. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2004, 2, 704–720. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Ehmann, R.; Smith, G.L. Smallpox in the Post-Eradication Era. Viruses 2020, 12, 138. [Google Scholar] [CrossRef] [Green Version]
- Trilla, A.; Trilla, G.; Daer, C. The 1918 “Spanish flu” in Spain. Clin. Infect. Dis. 2008, 47, 668–673. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Su, S.; Yang, H.; Jiang, S. Antivirals with common targets against highly pathogenic viruses. Cell. 2021, 184, 1604–1620. [Google Scholar] [CrossRef]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef]
- Morens, D.M.; Fauci, A.S. Emerging Pandemic Diseases: How We Got to COVID-19. Cell. 2020, 182, 1077–1092. [Google Scholar] [CrossRef]
- Gao, S.; Huang, T.; Song, L.; Xu, S.; Cheng, Y.; Cherukupalli, S.; Kang, D.; Zhao, T.; Sun, L.; Zhang, J.; et al. Medicinal chemistry strategies towards the development of effective SARS-CoV-2 inhibitors. Acta. Pharm. Sin. B. 2021. [Google Scholar] [CrossRef]
- Zhan, P.; Pannecouque, C.; De Clercq, E.; Liu, X. Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J. Med. Chem. 2016, 59, 2849–2878. [Google Scholar] [CrossRef]
- Ma, Y.; Frutos-Beltran, E.; Kang, D.; Pannecouque, C.; De Clercq, E.; Menendez-Arias, L.; Liu, X.; Zhan, P. Medicinal chemistry strategies for discovering antivirals effective against drug-resistant viruses. Chem. Soc. Rev. 2021, 50, 4514–4540. [Google Scholar] [CrossRef]
- Wu, G.; Zhao, T.; Kang, D.; Zhang, J.; Song, Y.; Namasivayam, V.; Kongsted, J.; Pannecouque, C.; De Clercq, E.; Poongavanam, V.; et al. Overview of Recent Strategic Advances in Medicinal Chemistry. J. Med. Chem. 2019, 62, 9375–9414. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Paiva, S.L.; Crews, C.M. Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef]
- de Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 2019, 10, 3468. [Google Scholar] [CrossRef] [Green Version]
- Costales, M.G.; Aikawa, H.; Li, Y.; Childs-Disney, J.L.; Abegg, D.; Hoch, D.G.; Pradeep Velagapudi, S.; Nakai, Y.; Khan, T.; Wang, K.W.; et al. Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 2406–2411. [Google Scholar] [CrossRef] [Green Version]
- Costales, M.G.; Suresh, B.; Vishnu, K.; Disney, M.D. Targeted Degradation of a Hypoxia-Associated Non-coding RNA Enhances the Selectivity of a Small Molecule Interacting with RNA. Cell Chem. Biol. 2019, 26, 1180–1186.e1185. [Google Scholar] [CrossRef]
- Costales, M.G.; Matsumoto, Y.; Velagapudi, S.P.; Disney, M.D. Small Molecule Targeted Recruitment of a Nuclease to RNA. J. Am. Chem. Soc. 2018, 140, 6741–6744. [Google Scholar] [CrossRef]
- Haniff, H.S.; Tong, Y.; Liu, X.; Chen, J.L.; Suresh, B.M.; Andrews, R.J.; Peterson, J.M.; O’Leary, C.A.; Benhamou, R.I.; Moss, W.N.; et al. Targeting the SARS-CoV-2 RNA Genome with Small Molecule Binders and Ribonuclease Targeting Chimera (RIBOTAC) Degraders. ACS Cent. Sci. 2020, 6, 1713–1721. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef]
- Lonsdale, R.; Ward, R.A. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018, 47, 3816–3830. [Google Scholar] [CrossRef]
- Chan, A.H.; Lee, W.G.; Spasov, K.A.; Cisneros, J.A.; Kudalkar, S.N.; Petrova, Z.O.; Buckingham, A.B.; Anderson, K.S.; Jorgensen, W.L. Covalent inhibitors for eradication of drug-resistant HIV-1 reverse transcriptase: From design to protein crystallography. Proc. Natl. Acad. Sci. USA 2017, 114, 9725–9730. [Google Scholar] [CrossRef] [Green Version]
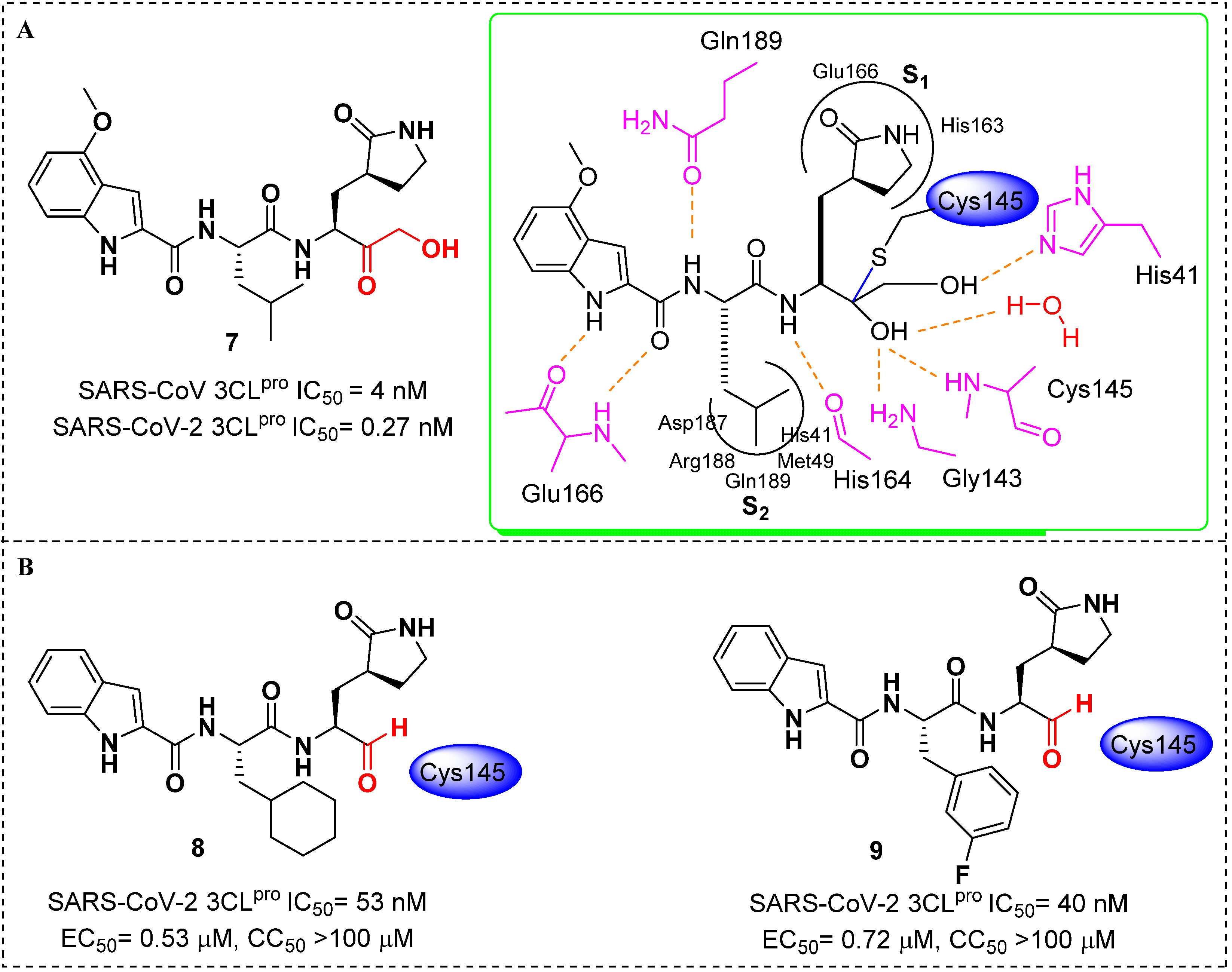
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Calder, L.J.; Rosenthal, P.B. Cryomicroscopy provides structural snapshots of influenza virus membrane fusion. Nat. Struct. Mol. Biol. 2016, 23, 853–858. [Google Scholar] [CrossRef]
- Amaro, R.E.; Ieong, P.U.; Huber, G.; Dommer, A.; Steven, A.C.; Bush, R.M.; Durrant, J.D.; Votapka, L.W. A Computational Assay that Explores the Hemagglutinin/Neuraminidase Functional Balance Reveals the Neuraminidase Secondary Site as a Novel Anti-Influenza Target. ACS Cent. Sci. 2018, 4, 1570–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
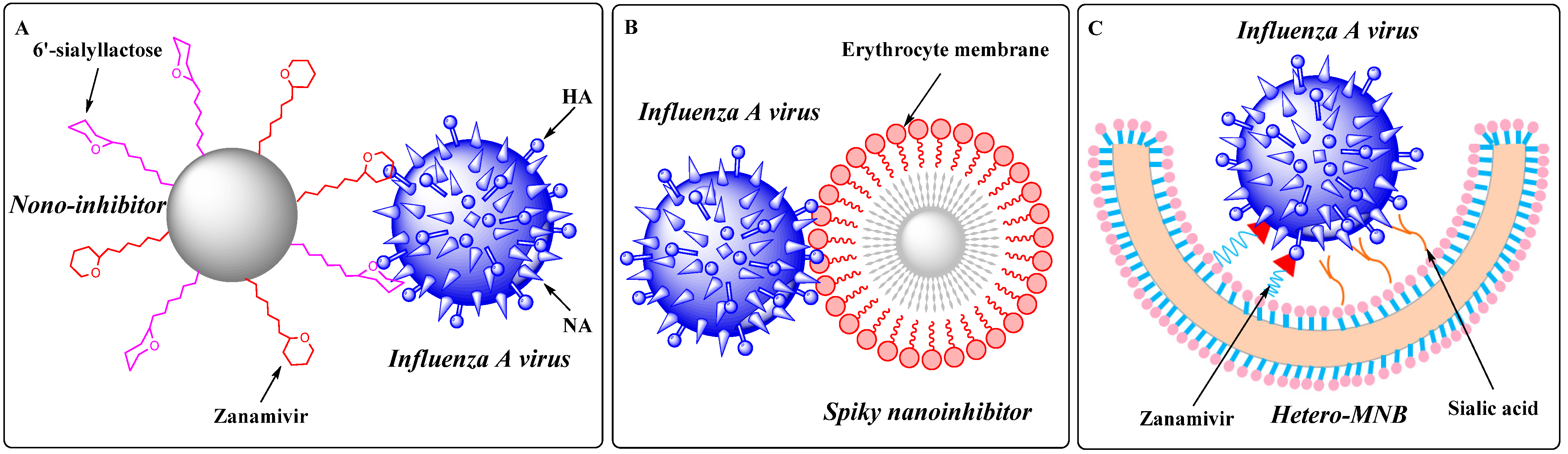
- Nie, C.; Parshad, B.; Bhatia, S.; Cheng, C.; Stadtmuller, M.; Oehrl, A.; Kerkhoff, Y.; Wolff, T.; Haag, R. Topology-Matching Design of an Influenza-Neutralizing Spiky Nanoparticle-Based Inhibitor with a Dual Mode of Action. Angew. Chem. Int. Ed. Engl. 2020, 59, 15532–15536. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Stadtmuller, M.; Yang, H.; Xia, Y.; Wolff, T.; Cheng, C.; Haag, R. Spiky Nanostructures with Geometry-matching Topography for Virus Inhibition. Nano. Lett. 2020, 20, 5367–5375. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Stadtmüller, M.; Parshad, B.; Wallert, M.; Ahmadi, V.; Kerkhoff, Y.; Bhatia, S.; Block, S.; Cheng, C.; Wolff, T.; et al. Heteromultivalent topology-matched nanostructures as potent and broad-spectrum influenza A virus inhibitors. Sci. Adv. 2021, 7, eabd3803. [Google Scholar] [CrossRef]
- Matos, M.J. Learning from nature: The role of albumin in drug delivery. Future Med. Chem. 2018, 10, 983–985. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, X. Simple bioconjugate chemistry serves great clinical advances: Albumin as a versatile platform for diagnosis and precision therapy. Chem. Soc. Rev. 2016, 45, 1432–1456. [Google Scholar] [CrossRef] [Green Version]
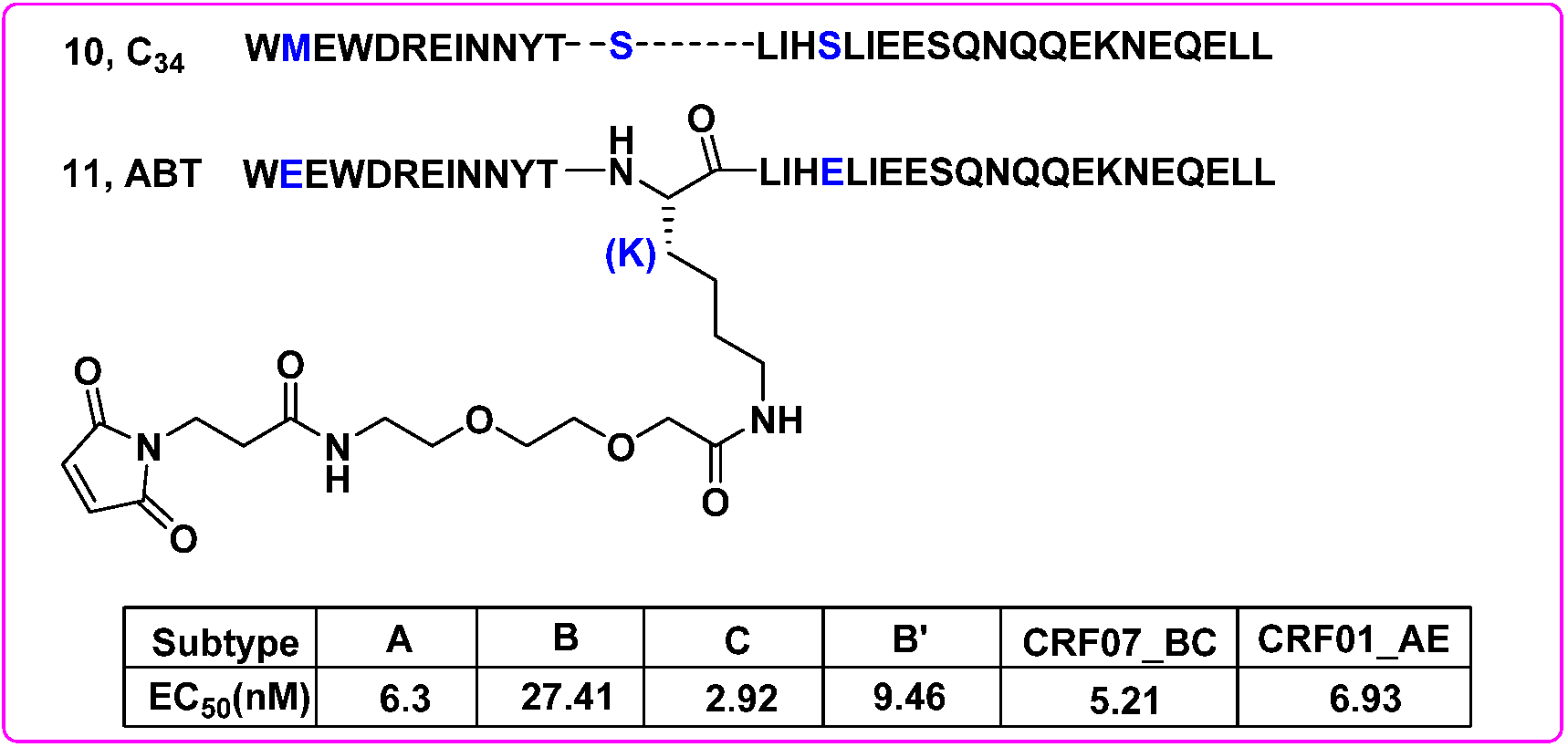
- Chong, H.; Yao, X.; Zhang, C.; Cai, L.; Cui, S.; Wang, Y.; He, Y. Biophysical property and broad anti-HIV activity of albuvirtide, a 3-maleimimidopropionic acid-modified peptide fusion inhibitor. PLoS ONE 2012, 7, e32599. [Google Scholar] [CrossRef] [Green Version]
- Su, B.; Yao, C.; Zhao, Q.X.; Cai, W.P.; Wang, M.; Lu, H.Z.; Chen, Y.Y.; Liu, L.; Wang, H.; He, Y.; et al. Efficacy and safety of the long-acting fusion inhibitor albuvirtide in antiretroviral-experienced adults with human immunodeficiency virus-1: Interim analysis of the randomized, controlled, phase 3, non-inferiority TALENT study. Chin. Med. J. 2020, 133, 2919–2927. [Google Scholar] [CrossRef]
- Tai, W.; Zhao, P.; Gao, X. Cytosolic delivery of proteins by cholesterol tagging. Sci. Adv. 2020, 6, eabb0310. [Google Scholar] [CrossRef]
- von Itzstein, M. The war against influenza: Discovery and development of sialidase inhibitors. Nat. Rev. Drug. Discov. 2007, 6, 967–974. [Google Scholar] [CrossRef]
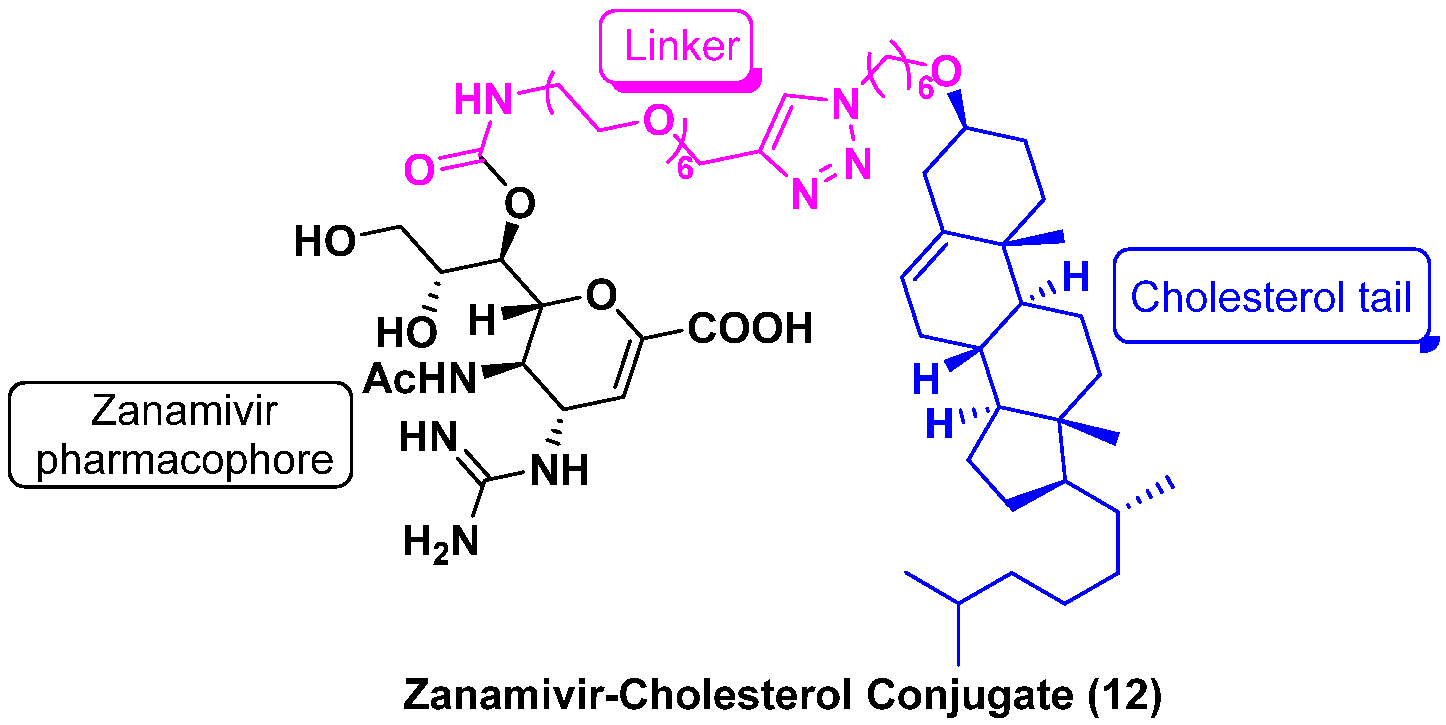
- Lv, X.; Wang, P.; Li, C.; Cheng, S.; Bi, Y.; Li, X. Zanamivir-Cholesterol Conjugate: A Long-Acting Neuraminidase Inhibitor with Potent Efficacy against Drug-Resistant Influenza Viruses. J. Med. Chem. 2021, 64, 17403–17412. [Google Scholar] [CrossRef]
- Chamakuri, S.; Lu, S.; Ucisik, M.N.; Bohren, K.M.; Chen, Y.C.; Du, H.C.; Faver, J.C.; Jimmidi, R.; Li, F.; Li, J.Y.; et al. DNA-encoded chemistry technology yields expedient access to SARS-CoV-2 M(pro) inhibitors. Proc. Natl. Acad. Sci. USA 2021, 118, e2111172118. [Google Scholar] [CrossRef]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature. 2020, 578, 229–236. [Google Scholar] [CrossRef]
- Sun, M.; Liu, S.; Wei, X.; Wan, S.; Huang, M.; Song, T.; Lu, Y.; Weng, X.; Lin, Z.; Chen, H.; et al. Aptamer Blocking Strategy Inhibits SARS-CoV-2 Virus Infection. Angew. Chem. Int. Ed. Engl. 2021, 60, 10266–10272. [Google Scholar] [CrossRef] [PubMed]
- Tauber, C.; Wamser, R.; Arkona, C.; Tugend, M.; Abdul Aziz, U.B.; Pach, S.; Schulz, R.; Jochmans, D.; Wolber, G.; Neyts, J.; et al. Chemical Evolution of Antivirals Against Enterovirus D68 through Protein-Templated Knoevenagel Reactions. Angew. Chem. Int. Ed. Engl. 2021, 60, 13294–13301. [Google Scholar] [CrossRef] [PubMed]
- Ekiert, H.M.; Szopa, A. Biological Activities of Natural Products. Molecules 2020, 25, 5769. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Sun, L.; Huang, B.; Liu, X.; Zhan, P. Medicinal chemistry strategies of targeting HIV-1 capsid protein for antiviral treatment. Future Med. Chem. 2020, 12, 1281–1284. [Google Scholar] [CrossRef]
- Kasprzyk, R.; Spiewla, T.J.; Smietanski, M.; Golojuch, S.; Vangeel, L.; De Jonghe, S.; Jochmans, D.; Neyts, J.; Kowalska, J.; Jemielity, J. Identification and evaluation of potential SARS-CoV-2 antiviral agents targeting mRNA cap guanine N7-Methyltransferase. Antiviral Res. 2021, 193, 105142. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef]
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021, 371, 926–931. [Google Scholar] [CrossRef]
- De Clercq, E. Emerging antiviral drugs. Expert Opin. Emerg. Drugs. 2008, 13, 393–416. [Google Scholar] [CrossRef]
- De Clercq, E. In search of a selective therapy of viral infections. Antiviral Res. 2010, 85, 19–24. [Google Scholar] [CrossRef]
- De Clercq, E. Highlights in the discovery of antiviral drugs: A personal retrospective. J. Med. Chem. 2010, 53, 1438–1450. [Google Scholar] [CrossRef]
- De Clercq, E. A 40-year journey in search of selective antiviral chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 1–24. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E. Highlights in antiviral drug research: Antivirals at the horizon. Med. Res. Rev. 2013, 33, 1215–1248. [Google Scholar] [CrossRef]
- De Clercq, E. Dancing with chemical formulae of antivirals: A personal account. Biochem. Pharmacol. 2013, 86, 711–725. [Google Scholar] [CrossRef]
- De Clercq, E. Curious discoveries in antiviral drug development: The role of serendipity. Med. Res. Rev. 2015, 35, 698–719. [Google Scholar] [CrossRef]
- De Clercq, E. Curious (Old and New) Antiviral Nucleoside Analogues with Intriguing Therapeutic Potential. Curr. Med. Chem. 2015, 22, 3866–3880. [Google Scholar] [CrossRef]
- De Clercq, E. Current treatment of hepatitis B virus infections. Rev. Med. Virol. 2015, 25, 354–365. [Google Scholar] [CrossRef]
- De Clercq, E. An Odyssey in antiviral drug development-50 years at the Rega Institute: 1964–2014. Acta. Pharm. Sin. B. 2015, 5, 520–543. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Chen, W.; Zhao, T.; Li, Z.; Jiang, X.; Ginex, T.; Vílchez, D.; Luque, F.J.; Kang, D.; Gao, P.; et al. Exploiting the Tolerant Region I of the Non-Nucleoside Reverse Transcriptase Inhibitor (NNRTI) Binding Pocket: Discovery of Potent Diarylpyrimidine-Typed HIV-1 NNRTIs against Wild-Type and E138K Mutant Virus with Significantly Improved Water Solubility and Favorable Safety Profiles. J. Med. Chem. 2019, 62, 2083–2098. [Google Scholar]
- Huang, B.; Ginex, T.; Luque, F.J.; Jiang, X.; Gao, P.; Zhang, J.; Kang, D.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; et al. Structure-Based Design and Discovery of Pyridyl-Bearing Fused Bicyclic HIV-1 Inhibitors: Synthesis, Biological Characterization, and Molecular Modeling Studies. J. Med. Chem. 2021, 64, 13604–13621. [Google Scholar] [CrossRef]
- Kang, D.; Fang, Z.; Huang, B.; Lu, X.; Zhang, H.; Xu, H.; Huo, Z.; Zhou, Z.; Yu, Z.; Meng, Q.; et al. Structure-Based Optimization of Thiophene[3,2-d]pyrimidine Derivatives as Potent HIV-1 Non-nucleoside Reverse Transcriptase Inhibitors with Improved Potency against Resistance-Associated Variants. J. Med. Chem. 2017, 60, 4424–4443. [Google Scholar] [CrossRef]
- Kang, D.; Fang, Z.; Li, Z.; Huang, B.; Zhang, H.; Lu, X.; Xu, H.; Zhou, Z.; Ding, X.; Daelemans, D.; et al. Design, Synthesis, and Evaluation of Thiophene[3,2-d]pyrimidine Derivatives as HIV-1 Non-nucleoside Reverse Transcriptase Inhibitors with Significantly Improved Drug Resistance Profiles. J. Med. Chem. 2016, 59, 7991–8007. [Google Scholar] [CrossRef]
- Kang, D.; Feng, D.; Ginex, T.; Zou, J.; Wei, F.; Zhao, T.; Huang, B.; Sun, Y.; Desta, S.; De Clercq, E.; et al. Exploring the hydrophobic channel of NNIBP leads to the discovery of novel piperidine-substituted thiophene[3,2-d]pyrimidine derivatives as potent HIV-1 NNRTIs. Acta Pharm. Sin. B. 2020, 10, 878–894. [Google Scholar] [CrossRef]
- Kang, D.; Feng, D.; Sun, Y.; Fang, Z.; Wei, F.; De Clercq, E.; Pannecouque, C.; Liu, X.; Zhan, P. Structure-Based Bioisosterism Yields HIV-1 NNRTIs with Improved Drug-Resistance Profiles and Favorable Pharmacokinetic Properties. J. Med. Chem. 2020, 63, 4837–4848. [Google Scholar] [CrossRef]
- Kang, D.; Ruiz, F.X.; Feng, D.; Pilch, A.; Zhao, T.; Wei, F.; Wang, Z.; Sun, Y.; Fang, Z.; De Clercq, E.; et al. Discovery and Characterization of Fluorine-Substituted Diarylpyrimidine Derivatives as Novel HIV-1 NNRTIs with Highly Improved Resistance Profiles and Low Activity for the hERG Ion Channel. J. Med. Chem. 2020, 63, 1298–1312. [Google Scholar] [CrossRef]
- Kang, D.; Ruiz, F.X.; Sun, Y.; Feng, D.; Jing, L.; Wang, Z.; Zhang, T.; Gao, S.; Sun, L.; De Clercq, E.; et al. 2,4,5-Trisubstituted Pyrimidines as Potent HIV-1 NNRTIs: Rational Design, Synthesis, Activity Evaluation, and Crystallographic Studies. J. Med. Chem. 2021, 64, 4239–4256. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Zhang, H.; Wang, Z.; Zhao, T.; Ginex, T.; Luque, F.J.; Yang, Y.; Wu, G.; Feng, D.; Wei, F.; et al. Identification of Dihydrofuro[3,4- d]pyrimidine Derivatives as Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors with Promising Antiviral Activities and Desirable Physicochemical Properties. J. Med. Chem. 2019, 62, 1484–1501. [Google Scholar] [CrossRef]
- Sun, L.; Dick, A.; Meuser, M.E.; Huang, T.; Zalloum, W.A.; Chen, C.H.; Cherukupalli, S.; Xu, S.; Ding, X.; Gao, P.; et al. Design, Synthesis, and Mechanism Study of Benzenesulfonamide-Containing Phenylalanine Derivatives as Novel HIV-1 Capsid Inhibitors with Improved Antiviral Activities. J. Med. Chem. 2020, 63, 4790–4810. [Google Scholar] [CrossRef]
- Wang, Z.; Zalloum, W.A.; Wang, W.; Jiang, X.; De Clercq, E.; Pannecouque, C.; Kang, D.; Zhan, P.; Liu, X. Discovery of Novel Dihydrothiopyrano[4,3-d]pyrimidine Derivatives as Potent HIV-1 NNRTIs with Significantly Reduced hERG Inhibitory Activity and Improved Resistance Profiles. J. Med. Chem. 2021, 64, 13658–13675. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZNV | ZNV–Cholesterol Conjugate (12) | |||
|---|---|---|---|---|
| IC50 (nM) | EC50 (nM) | IC50 (nM) | EC50 (nM) | |
| H1N1 | 0.9 ± 0.4 | 62.7 ± 4.1 | 28.0 ± 4.0 | 36.8 ± 2.1 |
| H3N2 | 0.7 ± 0.2 | 87.1 ± 11.9 | 16.1 ± 8.1 | 32.1 ± 6.7 |
| H5N1 | 0.3 ± 0.2 | 123.4 ± 7.3 | 24.3 ± 10.1 | 35.4 ± 10.0 |
| H1N1H275Y | 1.0 ± 0.2 | 26.6 ± 6.3 | 22.0 ± 6.4 | 22.0 ± 6.2 |
| H3N2E119V | 10.0 ± 0.7 | 5240.0 ± 996.3 | 162.4 ± 8.8 | 421.7 ± 110.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Ding, D.; Zhang, X.; Sun, L.; Kang, D.; Huang, B.; Liu, X.; Zhan, P. Newly Emerging Strategies in Antiviral Drug Discovery: Dedicated to Prof. Dr. Erik De Clercq on Occasion of His 80th Anniversary. Molecules 2022, 27, 850. https://doi.org/10.3390/molecules27030850
Xu S, Ding D, Zhang X, Sun L, Kang D, Huang B, Liu X, Zhan P. Newly Emerging Strategies in Antiviral Drug Discovery: Dedicated to Prof. Dr. Erik De Clercq on Occasion of His 80th Anniversary. Molecules. 2022; 27(3):850. https://doi.org/10.3390/molecules27030850
Chicago/Turabian StyleXu, Shujing, Dang Ding, Xujie Zhang, Lin Sun, Dongwei Kang, Boshi Huang, Xinyong Liu, and Peng Zhan. 2022. "Newly Emerging Strategies in Antiviral Drug Discovery: Dedicated to Prof. Dr. Erik De Clercq on Occasion of His 80th Anniversary" Molecules 27, no. 3: 850. https://doi.org/10.3390/molecules27030850
APA StyleXu, S., Ding, D., Zhang, X., Sun, L., Kang, D., Huang, B., Liu, X., & Zhan, P. (2022). Newly Emerging Strategies in Antiviral Drug Discovery: Dedicated to Prof. Dr. Erik De Clercq on Occasion of His 80th Anniversary. Molecules, 27(3), 850. https://doi.org/10.3390/molecules27030850