
The Reductive Dehydroxylation Catalyzed by IspH, a Source of Inspiration for the Development of Novel Anti-Infectives

 , ,
, ,
Abstract
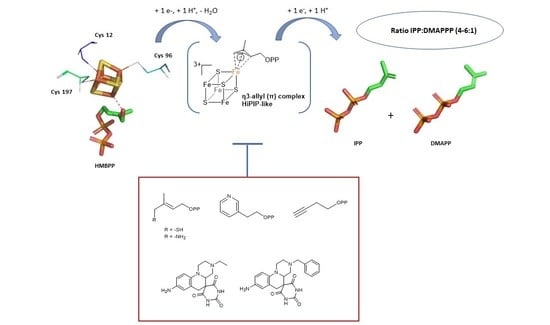
1. IspH, an Enzyme Involved in the Biosynthesis of Isoprenoids
2. Occurrence of IspH
3. The Discovery of the IspH Metalloenzyme
3.1. The Lytb Gene
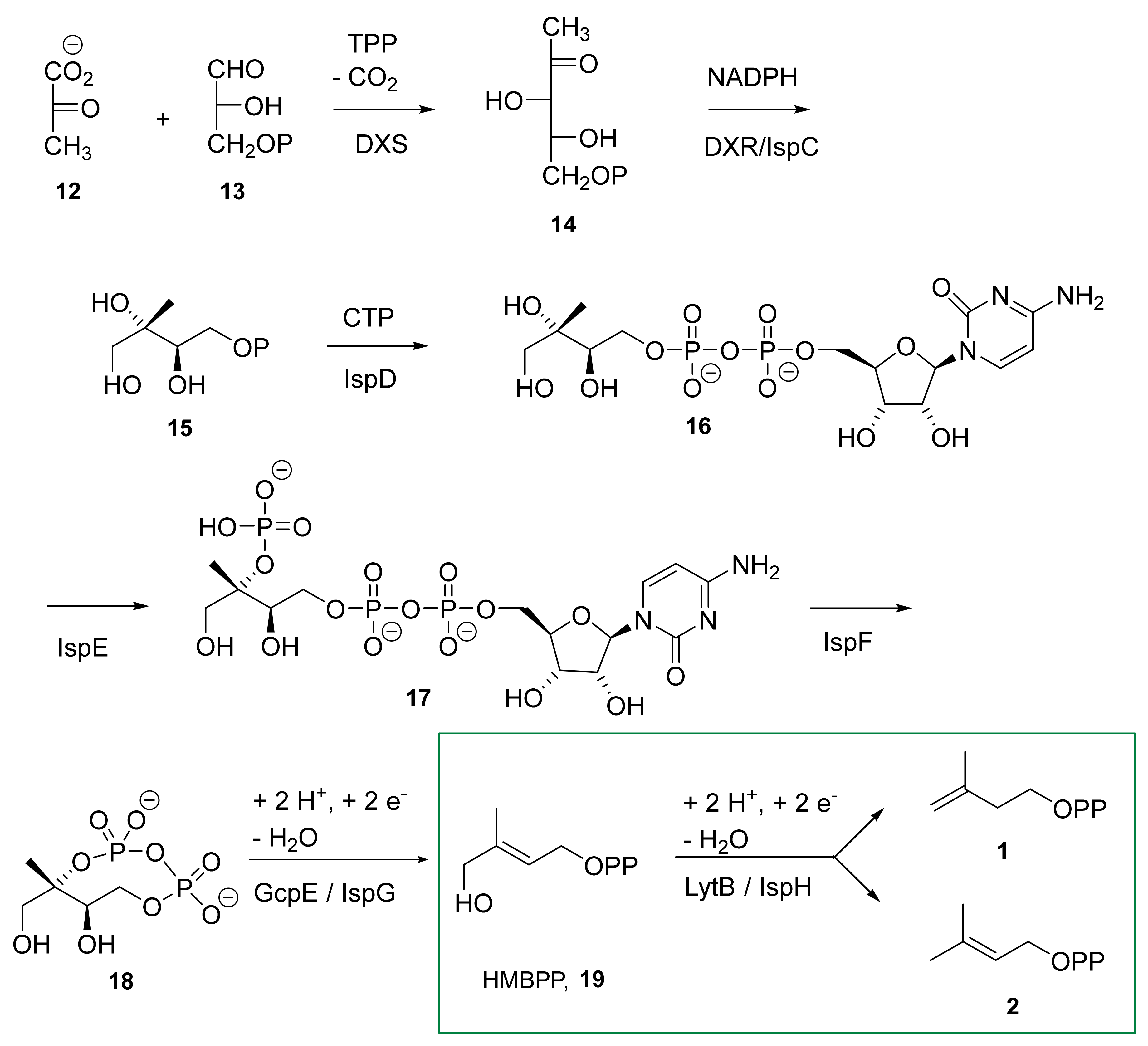
3.2. The IspH Catalyzed Reaction
3.3. IspH, a [4Fe-4S] Metalloenzyme
4. The Mechanism of the Reductive Dehydroxylation
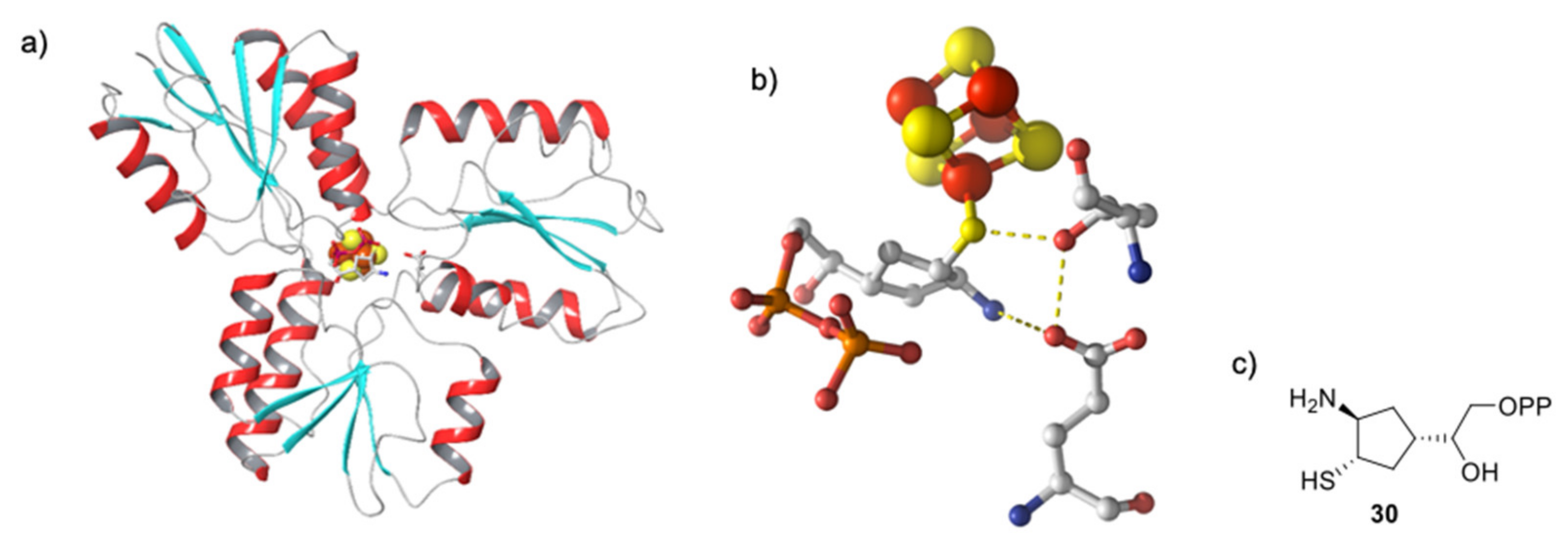
4.1. Protein Conformation in the Crystalline State
4.2. First Step: Binding of the Hydroxyl Group of HMBPP to the Iron-Sulfur Cluster
4.3. Second Step: Reduction of the [4Fe-4S]2+ Cluster and Rotation of the CH2OH Group
4.4. Third Step: Protonation of the Hydroxyl Group and Elimination of Water
4.5. Fourth Step: Formation of a HiPIP-like Cluster
4.6. Fifth Step: Protonation and Formation of the Two Products IPP and DMAPP
5. IspH Inhibitors
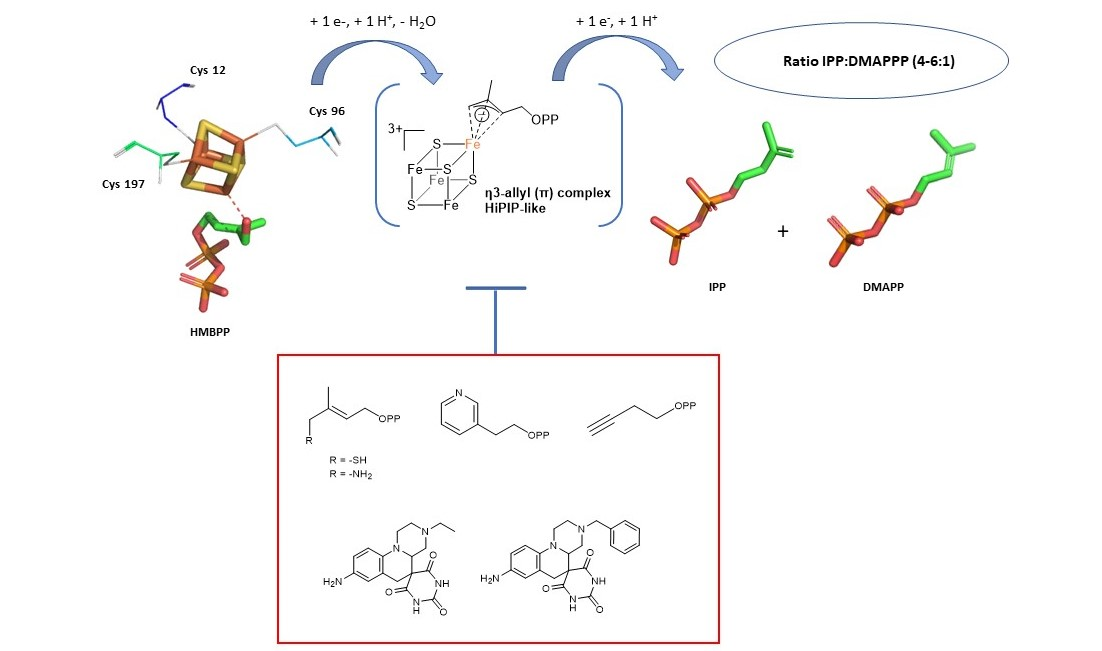
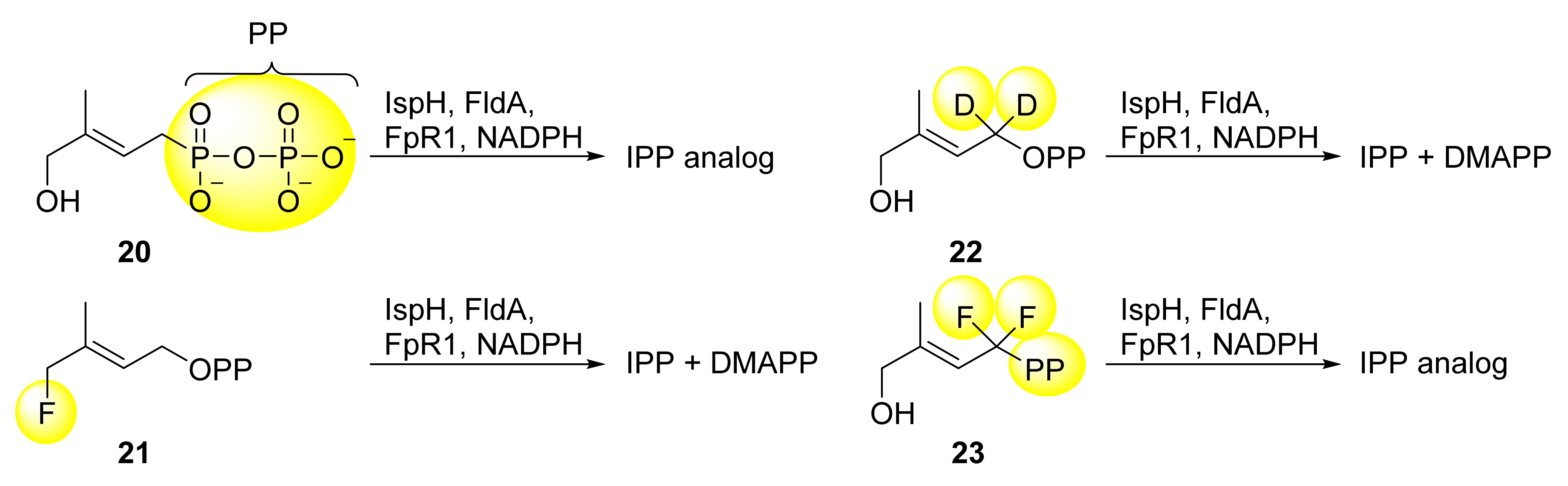
5.1. Analogs of the HMBPP Substrate as Inhibitors
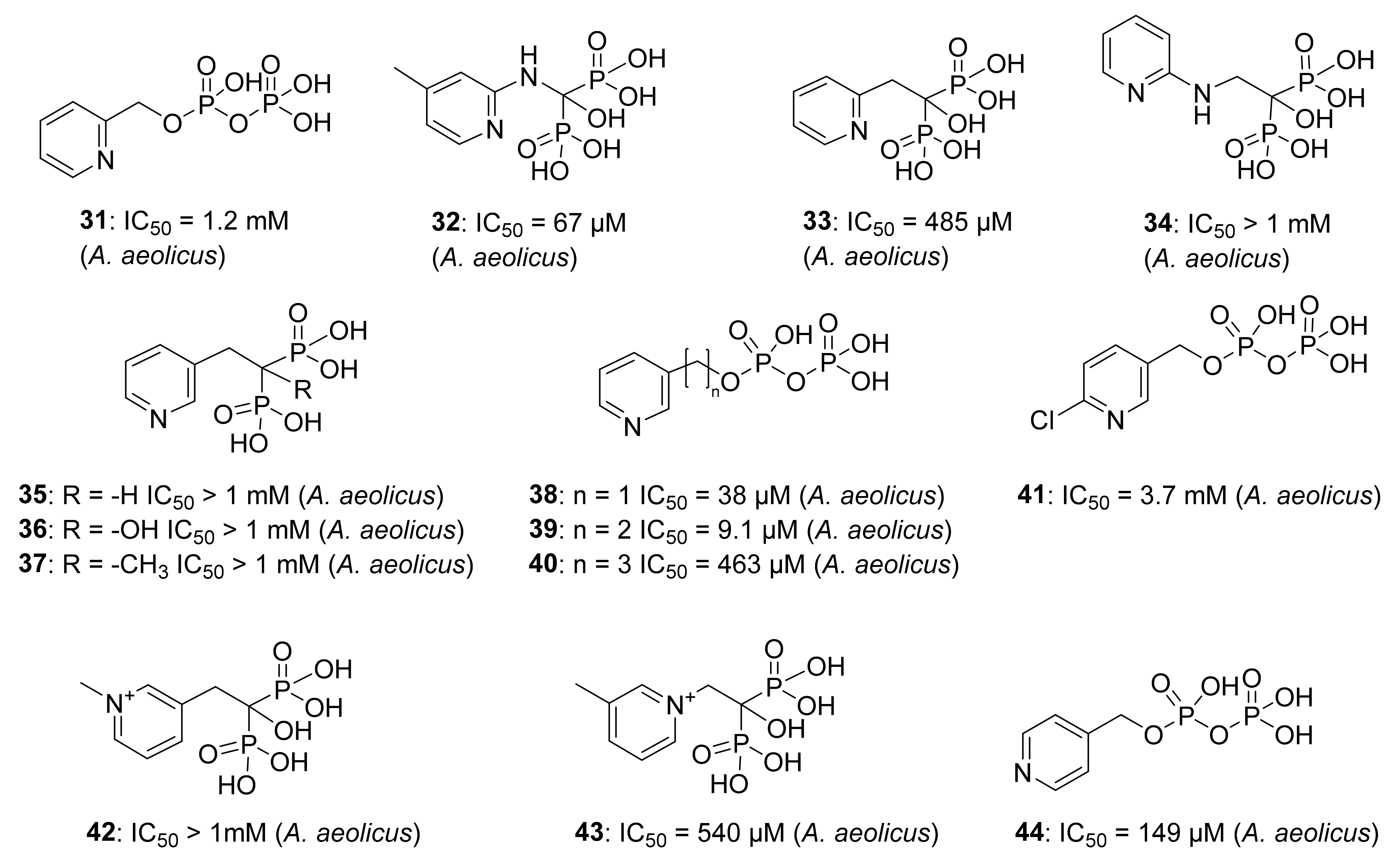
5.2. Pyridine Diphosphate as Inhibitors
5.3. Alkyne Derivatives as Inhibitors
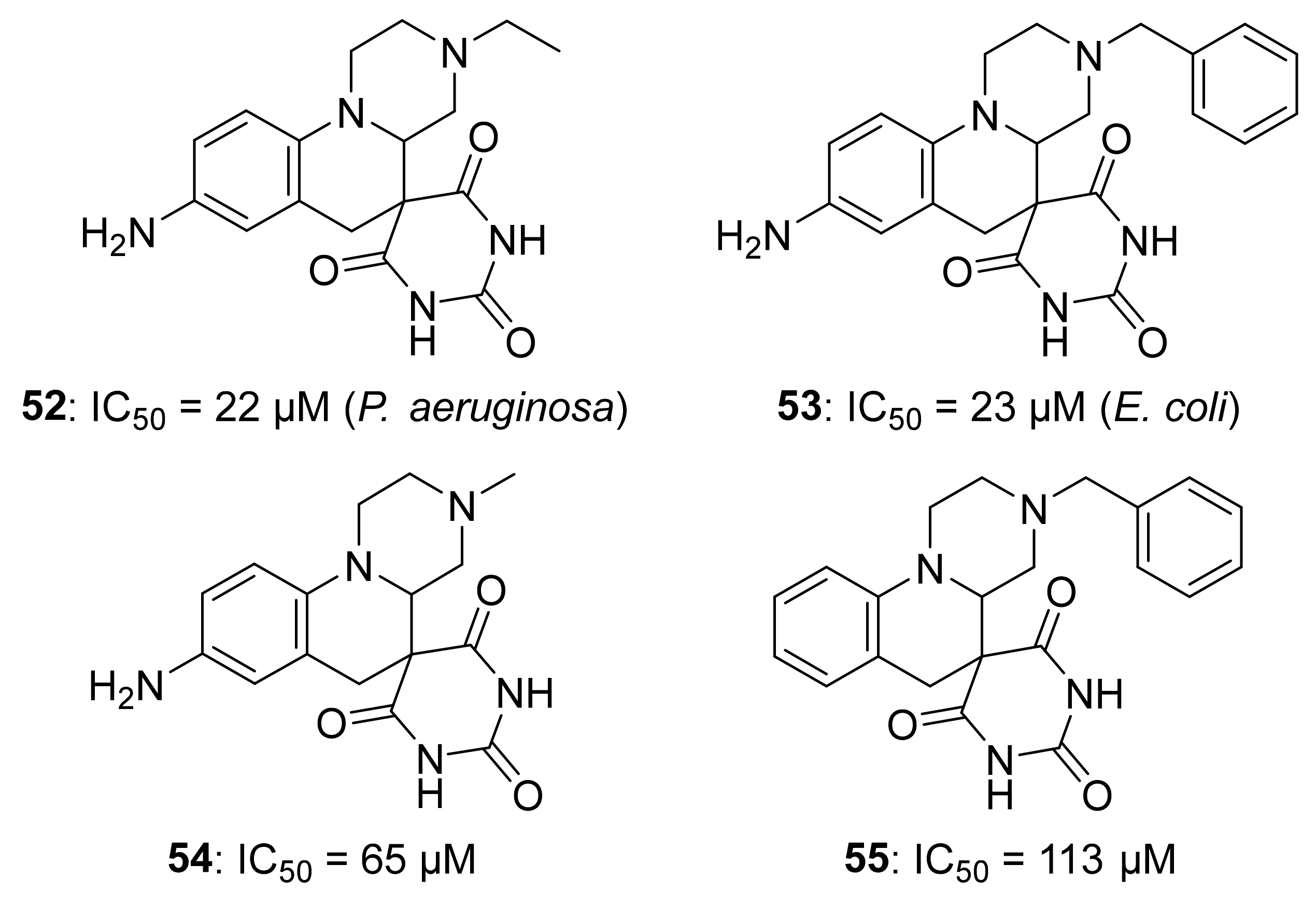
5.4. Non-Diphosphate Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cane, D.E.; Barton, D.H.R.; Nakanishi, K.; Meth-Cohn, O. (Eds.) Comprehensive Natural Products Chemistry: Isoprenoids, Including Carotenoids and Steroids; Elsevier: Oxford, UK, 1999; Volume 2. [Google Scholar]
- Sacchettini, J.C.; Poulter, C.D. Creating isoprenoid diversity. Science 1997, 277, 1788–1789. [Google Scholar] [CrossRef] [PubMed]
- Bloch, K. Sterol molecule: Structure, biosynthesis, and function. Steroids 1992, 57, 378–383. [Google Scholar] [CrossRef]
- Rohmer, M. The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Nat. Prod. Rep. 1999, 16, 565–574. [Google Scholar] [CrossRef]
- Eisenreich, W.; Schwarz, M.; Cartayrade, A.; Arigoni, D.; Zenk, M.H.; Bacher, A. The deoxyxylulose phosphate pathway of terpenoid biosynthesis in plants and microorganisms. Chem. Biol. 1998, 5, R221–R233. [Google Scholar] [CrossRef]
- Lichtenthaler, H.K. The 1-deoxy-D-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annu. Rev. Plant. Physiol. Plant. Mol. Biol. 1999, 50, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Rohmer, M.; Grosdemange-Billiard, C.; Seemann, M.; Tritsch, D. Isoprenoid biosynthesis as a novel target for antibacterial and antiparasitic drugs. Curr. Opin. Investig. Drugs 2004, 5, 154–162. [Google Scholar]
- Priorization of Pathogens to Guide Discovery, Research and Development of New Antibioitics for Drug-Resistant Bacterial Infections, Including Tuberculosis; Report No. WHO/EMP/IAU/2017.12; World Health Organization: Geneva, Switzerland, 2017.
- Miethke, M.; Pieroni, M.; Weber, T.; Brönstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef]
- Mann, F.M.; Xu, M.; Davenport, E.K.; Peters, R.J. Functional characterization and evolution of the isotuberculosinol operon in Mycobacterium tuberculosis and related mycobacteria. Front. Microbiol. 2012, 3, 368. [Google Scholar] [CrossRef]
- Brown, A.C.; Kokoczka, R.; Parish, T. LytB1 and LytB2 of Mycobacterium tuberculosis are not genetically redundant. PLoS ONE 2015, 10, e0135638. [Google Scholar] [CrossRef]
- Kafai, N.M.; Odom John, A.R. Malaria in children. Infect. Dis. Clin. North Am. 2018, 32, 189–200. [Google Scholar] [CrossRef]
- Mombo-Ngoma, G.; Remppis, J.; Sievers, M.; Zoleko Manego, R.; Endamne, L.; Kabwende, L.; Veletzky, L.; Nguyen, T.T.; Groger, M.; Lötsch, F.; et al. Efficacy and safety of fosmidomycin–piperaquine as nonartemisinin-based combination therapy for uncomplicated falciparum malaria: A single-arm, age de-escalation proof-of-concept study in Gabon. Clin. Infect. Dis. 2018, 66, 1823–1830. [Google Scholar] [CrossRef]
- Hedl, M.; Sutherlin, A.; Wilding, E.I.; Mazzulla, M.; McDevitt, D.; Lane, P.; Burgner, J.W.; Lehnbeuter, K.R.; Stauffacher, C.V.; Gwynn, M.N.; et al. Enterococcus faecalis acetoacetyl-coenzyme a thiolase/3-hydroxy-3-methylglutaryl-coenzyme a reductase, a dual-function protein of isopentenyl diphosphate biosynthesis. J. Bacteriol. 2002, 184, 2116–2122. [Google Scholar] [CrossRef]
- Gustafson, C.E.; Kaul, S.; Ishiguro, E.E. Identification of the Escherichia coli lytB gene, which is involved in penicillin tolerance and control of the stringent response. J. Bacteriol. 1993, 175, 1203–1205. [Google Scholar] [CrossRef][Green Version]
- Potter, S.; Yang, X.; Boulanger, M.J.; Ishiguro, E.E. Occurrence of homologs of the Escherichia coli lytB gene in gram-negative bacterial species. J. Bacteriol. 1998, 180, 1959–1961. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.X.; Lafond, T.P.; Gantt, E. Evidence of a role for LytB in the nonmevalonate pathway of isoprenoid biosynthesis. J. Bacteriol. 2000, 182, 5841–5848. [Google Scholar] [CrossRef] [PubMed]
- Altincicek, B.; Kollas, A.-K.; Sanderbrand, S.; Wiesner, J.; Hintz, M.; Beck, E.; Jomaa, H. GcpE is involved in the 2-C-methyl-D-erythritol 4-phosphate pathway of isoprenoid biosynthesis in Escherichia coli. J. Bacteriol. 2001, 183, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- McAteer, S.; Coulson, A.; McLennan, N.; Masters, M. The lytB gene of Escherichia coli is essential and specifies a product needed for isoprenoid biosynthesis. J. Bacteriol. 2001, 183, 7403–7407. [Google Scholar] [CrossRef] [PubMed]
- Altincicek, B.; Kollas, A.-K.; Eberl, M.; Wiesner, J.; Sanderbrand, S.; Hintz, M.; Beck, E.; Jomaa, H. LytB, a novel gene of the 2-C-methyl-D-erythritol 4-phosphate pathway of isoprenoid biosynthesis in Escherichia coli. FEBS Lett. 2001, 499, 37–40. [Google Scholar] [CrossRef]
- Rohdich, F.; Hecht, S.; Gartner, K.; Adam, P.; Krieger, C.; Amslinger, S.; Arigoni, D.; Bacher, A.; Eisenreich, W. Studies on the nonmevalonate terpene biosynthetic pathway: Metabolic role of IspH (LytB) protein. Proc. Natl. Acad. Sci. USA 2002, 99, 1158–1163. [Google Scholar] [CrossRef]
- Hecht, S.; Eisenreich, W.; Adam, P.; Amslinger, S.; Kis, K.; Bacher, A.; Arigoni, D.; Rohdich, F. Studies on the nonmevalonate pathway to terpenes: The role of the GcpE (IspG) protein. Proc. Natl. Acad. Sci. USA 2001, 98, 14837–14842. [Google Scholar] [CrossRef]
- Adam, P.; Hecht, S.; Eisenreich, W.; Kaiser, J.; Gräwert, T.; Arigoni, D.; Bacher, A.; Rohdich, F. Biosynthesis of terpenes: Studies on 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase. Proc. Natl. Acad. Sci. USA 2002, 99, 12108–12113. [Google Scholar] [CrossRef]
- Rohdich, F.; Zepeck, F.; Adam, P.; Hecht, S.; Kaiser, J.; Laupitz, R.; Amslinger, S.; Eisenreich, W.; Bacher, A.; Arigoni, D. The deoxyxylulose phosphate pathway of isoprenoid biosynthesis: Studies on the mechanisms of the reactions catalyzed by IspG and IspH protein. Proc. Natl. Acad. Sci. USA 2002, 100, 1586–1591. [Google Scholar] [CrossRef]
- Altincicek, B.; Duin, E.C.; Reichenberg, A.; Hedderich, R.; Kollas, A.-K.; Hintz, M.; Wagner, S.; Wiesner, J.; Beck, E.; Jomaa, H. LytB protein catalyzes the terminal step of the 2-C-methyl-D-erythritol-4-phosphate pathway of isoprenoid biosynthesis. FEBS Lett. 2002, 532, 437–440. [Google Scholar] [CrossRef]
- Beinert, H. Iron-sulfur proteins: Ancient structures, still full of surprises. J. Biol. Inorg. Chem. 2000, 5, 2–15. [Google Scholar] [CrossRef]
- Seemann, M.; Tse Sum Bui, B.; Wolff, M.; Tritsch, D.; Campos, N.; Boronat, A.; Marquet, A.; Rohmer, M. Isoprenoid biosynthesis through the methylerythritol phosphate pathway: The (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase (GcpE) is a [4Fe–4S] protein. Angew. Chem. Int. Ed. 2002, 41, 4337–4339. [Google Scholar] [CrossRef]
- Wolff, M.; Seemann, M.; Tse Sum Bui, B.; Frapart, Y.; Tritsch, D.; Estrabot, A.G.; Rodríguez-Concepción, M.; Boronat, A.; Marquet, A.; Rohmer, M. Isoprenoid biosynthesis via the methylerythritol phosphate pathway: The (E)-4-hydroxy-3-methylbut-2-enyl diphosphate reductase (lytB/IspH) from Escherichia coli is a [4Fe-4S] protein. FEBS Lett. 2003, 541, 115–120. [Google Scholar] [CrossRef]
- Rupp, H.; Rao, K.K.; Hall, D.O.; Cammack, R. Electron spin relaxation of iron-sulphur proteins studied by microwave power saturation. Biochim. Biophys. Acta Protein Struct. 1978, 537, 255–269. [Google Scholar] [CrossRef]
- Ollagnier, S.; Mulliez, E.; Schmidt, P.P.; Eliasson, R.; Gaillard, J.; Deronzier, C.; Bergman, T.; Gräslund, A.; Reichard, P.; Fontecave, M. Activation of the anaerobic ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 1997, 272, 24216–24223. [Google Scholar] [CrossRef] [PubMed]
- Petrovich, R.M.; Ruzicka, F.J.; Reed, G.H.; Frey, P.A. Characterization of iron-sulfur clusters in lysine 2,3-aminomutase by electron paramagnetic resonance spectroscopy. Biochemistry 1992, 31, 10774–10781. [Google Scholar] [CrossRef]
- Gräwert, T.; Kaiser, J.; Zepeck, F.; Laupitz, R.; Hecht, S.; Amslinger, S.; Schramek, N.; Schleicher, E.; Weber, S.; Haslbeck, M.; et al. IspH protein of Escherichia coli: Studies on iron−sulfur cluster implementation and catalysis. J. Am. Chem. Soc. 2004, 126, 12847–12855. [Google Scholar] [CrossRef] [PubMed]
- Seemann, M.; Janthawornpong, K.; Schweizer, J.; Böttger, L.H.; Janoschka, A.; Ahrens-Botzong, A.; Tambou, E.N.; Rotthaus, O.; Trautwein, A.X.; Rohmer, M.; et al. Isoprenoid biosynthesis via the MEP pathway: In vivo Mössbauer spectroscopy identifies a [4Fe-4S]2+ center with unusual coordination sphere in the LytB protein. J. Am. Chem. Soc. 2009, 131, 13184–13185. [Google Scholar] [CrossRef]
- Xiao, Y.; Chu, L.; Sanakis, Y.; Liu, P. Revisiting the IspH catalytic system in the deoxyxylulose phosphate pathway: Achieving high activity. J. Am. Chem. Soc. 2009, 131, 9931–9933. [Google Scholar] [CrossRef] [PubMed]
- Schünemann, V.; Winkler, H. Structure and dynamics of biomolecules studied by Mössbauer spectroscopy. Rep. Prog. Phys. 2000, 63, 263–353. [Google Scholar] [CrossRef]
- Rekittke, I.; Wiesner, J.; Röhrich, R.; Demmer, U.; Warkentin, E.; Xu, W.; Troschke, K.; Hintz, M.; No, J.H.; Duin, E.C.; et al. Structure of (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate reductase, the terminal enzyme of the non-mevalonate pathway. J. Am. Chem. Soc. 2008, 130, 17206–17207. [Google Scholar] [CrossRef] [PubMed]
- Gräwert, T.; Span, I.; Eisenreich, W.; Rohdich, F.; Eppinger, J.; Bacher, A.; Groll, M. Probing the reaction mechanism of IspH protein by X-ray structure analysis. Proc. Natl. Acad. Sci. USA 2010, 107, 1077–1081. [Google Scholar] [CrossRef]
- Faus, I.; Reinhard, A.; Rackwitz, S.; Wolny, J.A.; Schlage, K.; Wille, H.-C.; Chumakov, A.; Krasutsky, S.; Chaignon, P.; Poulter, C.D.; et al. Isoprenoid biosynthesis in pathogenic bacteria: Nuclear resonance vibrational spectroscopy provides insight into the unusual [4Fe-4S] cluster of the E. Coli LytB/IspH protein. Angew. Chem. Int. Ed. 2015, 54, 12584–12587. [Google Scholar] [CrossRef] [PubMed]
- Scheidt, W.; Durbin, S.; Sage, J. Nuclear resonance vibrational spectroscopy? NRVS. J. Inorg. Biochem. 2005, 99, 60–71. [Google Scholar] [CrossRef]
- Xu, W.; Lees, N.S.; Hall, D.; Welideniya, D.; Hoffman, B.M.; Duin, E.C. A closer look at the spectroscopic properties of possible reaction intermediates in wild-type and mutant (E)-4-hydroxy-3-methylbut-2-enyl diphosphate reductase. Biochemistry 2012, 51, 4835–4849. [Google Scholar] [CrossRef]
- Röhrich, R.C.; Englert, N.; Troschke, K.; Reichenberg, A.; Hintz, M.; Seeber, F.; Balconi, E.; Aliverti, A.; Zanetti, G.; Köhler, U.; et al. Reconstitution of an apicoplast-localised electron transfer pathway involved in the isoprenoid biosynthesis of Plasmodium falciparum. FEBS Lett. 2005, 579, 6433–6438. [Google Scholar] [CrossRef] [PubMed]
- Gräwert, T.; Rohdich, F.; Span, I.; Bacher, A.; Eisenreich, W.; Eppinger, J.; Groll, M. Structure of active IspH enzyme from Escherichia coli provides mechanistic insights into substrate reduction. Angew. Chem. Int. Ed. 2009, 48, 5756–5759. [Google Scholar] [CrossRef]
- Rekittke, I.; Olkhova, E.; Wiesner, J.; Demmer, U.; Warkentin, E.; Jomaa, H.; Ermler, U. Structure of the (E)-4-hydroxy-3-methyl-but-2-enyl-diphosphate reductase from Plasmodium falciparum. FEBS Lett. 2013, 587, 3968–3972. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.; Groll, M. The methylerythritol phosphate pathway to isoprenoids. Chem. Rev. 2017, 117, 5675–5703. [Google Scholar] [CrossRef]
- Span, I.; Gräwert, T.; Bacher, A.; Eisenreich, W.; Groll, M. Crystal structures of mutant IspH proteins reveal a rotation of the substrate’s hydroxymethyl group during catalysis. J. Mol. Biol. 2012, 416, 1–9. [Google Scholar] [CrossRef]
- Span, I.; Wang, K.; Wang, W.; Zhang, Y.; Bacher, A.; Eisenreich, W.; Li, K.; Schulz, C.; Oldfield, E.; Groll, M. Discovery of acetylene hydratase activity of the iron–sulphur protein IspH. Nat. Commun. 2012, 3, 1042. [Google Scholar] [CrossRef] [PubMed]
- Borel, F.; Barbier, E.; Krasutsky, S.; Janthawornpong, K.; Chaignon, P.; Poulter, C.D.; Ferrer, J.-L.; Seemann, M. Further insight into crystal structures of Escherichia coli IspH/lytB in complex with two potent inhibitors of the MEP pathway: A starting point for rational design of new antimicrobials. ChemBioChem 2017, 18, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- Span, I.; Wang, K.; Wang, W.; Jauch, J.; Eisenreich, W.; Bacher, A.; Oldfield, E.; Groll, M. Structures of fluoro, amino, and thiol inhibitors bound to the [Fe4S4] protein IspH. Angew. Chem. Int. Ed. 2013, 52, 2118–2121. [Google Scholar] [CrossRef]
- Wang, W.; Wang, K.; Span, I.; Jauch, J.; Bacher, A.; Groll, M.; Oldfield, E. Are free radicals involved in IspH catalysis? An EPR and crystallographic investigation. J. Am. Chem. Soc. 2012, 134, 11225–11234. [Google Scholar] [CrossRef]
- Span, I.; Wang, K.; Eisenreich, W.; Bacher, A.; Zhang, Y.; Oldfield, E.; Groll, M. Insights into the binding of pyridines to the iron–sulfur enzyme IspH. J. Am. Chem. Soc. 2014, 136, 7926–7932. [Google Scholar] [CrossRef]
- Brown, A.C.; Suess, D.L.M. Controlling substrate binding to Fe4S4 clusters through remote steric effects. Inorg. Chem. 2019, 58, 5273–5280. [Google Scholar] [CrossRef]
- Blachly, P.G.; Sandala, G.M.; Giammona, D.A.; Bashford, D.; McCammon, J.A.; Noodleman, L. Broken-symmetry DFT computations for the reaction pathway of IspH, an iron–sulfur enzyme in pathogenic bacteria. Inorg. Chem. 2015, 54, 6439–6461. [Google Scholar] [CrossRef]
- Blachly, P.G.; Sandala, G.M.; Giammona, D.A.; Liu, T.; Bashford, D.; McCammon, J.A.; Noodleman, L. Use of broken-symmetry density functional theory to characterize the IspH oxidized state: Implications for IspH mechanism and inhibition. J. Chem. Theory Comput. 2014, 10, 3871–3884. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, K.; Liu, Y.-L.; No, J.-H.; Li, J.; Nilges, M.J.; Oldfield, E. Bioorganometallic mechanism of action, and inhibition, of IspH. Proc. Natl. Acad. Sci. USA 2010, 107, 4522–4527. [Google Scholar] [CrossRef] [PubMed]
- Fuss, J.O.; Tsai, C.-L.; Ishida, J.P.; Tainer, J.A. Emerging critical roles of Fe–S clusters in DNA replication and repair. Bioch. Biophys. Acta Mol. Cell Res. 2015, 1853, 1253–1271. [Google Scholar] [CrossRef]
- Ter Beek, J.; Parkash, V.; Bylund, G.O.; Osterman, P.; Sauer-Eriksson, A.E.; Johansson, E. Structural evidence for an essential Fe–S cluster in the catalytic core domain of DNA polymerase ϵ. Nucleic Acids Res. 2019, 47, 5712–5722. [Google Scholar] [CrossRef]
- Citron, C.A.; Brock, N.L.; Rabe, P.; Dickschat, J.S. The stereochemical course and mechanism of the IspH reaction. Angew. Chem. Int. Ed. 2012, 51, 4053–4057. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-I.; Igarashi, R.Y.; Laryukhin, M.; Doan, P.E.; Dos Santos, P.C.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. An organometallic intermediate during alkyne reduction by nitrogenase. J. Am. Chem. Soc. 2004, 126, 9563–9569. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-I.; Sørlie, M.; Christiansen, J.; Yang, T.-C.; Shao, J.; Dean, D.R.; Hales, B.J.; Hoffman, B.M. Electron inventory, kinetic assignment (En), structure, and bonding of nitrogenase turnover intermediates with C2H2 and CO. J. Am. Chem. Soc. 2005, 127, 15880–15890. [Google Scholar] [CrossRef] [PubMed]
- Chaignon, P.; Petit, B.E.; Vincent, B.; Allouche, L.; Seemann, M. Methylerythritol phosphate pathway: Enzymatic evidence for a rotation in the Lytb/IspH catalyzed reaction. Chem. Eur. J. 2020, 26, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Hard and soft acids and bases. J. Am. Chem. Soc. 1963, 85, 7. [Google Scholar] [CrossRef]
- Rao, P.V.; Holm, R.H. Synthetic analogues of the active sites of iron−sulfur proteins. Chem. Rev. 2004, 104, 527–559. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhao, Z.K.; Liu, P. Mechanistic studies of IspH in the deoxyxylulose phosphate pathway: Heterolytic C-O bond cleavage at C4 position. J. Am. Chem. Soc. 2008, 130, 2164–2165. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, P. IspH protein of the deoxyxylulose phosphate pathway: Mechanistic studies with C1 -deuterium-labeled substrate and fluorinated analogue. Angew. Chem. Int. Ed. 2008, 47, 9722–9725. [Google Scholar] [CrossRef]
- Abdel-Azeim, S.; Jedidi, A.; Eppinger, J.; Cavallo, L. Mechanistic insights into the reductive dehydroxylation pathway for the biosynthesis of isoprenoids promoted by the IspH enzyme. Chem. Sci. 2015, 6, 5643–5651. [Google Scholar] [CrossRef]
- Walters, E.M.; Garcia-Serres, R.; Jameson, G.N.L.; Glauser, D.A.; Bourquin, F.; Manieri, W.; Schürmann, P.; Johnson, M.K.; Huynh, B.H. Spectroscopic characterization of site-specific [Fe4S4] cluster chemistry in ferredoxin:thioredoxin reductase: Implications for the catalytic mechanism. J. Am. Chem. Soc. 2005, 127, 9612–9624. [Google Scholar] [CrossRef]
- Belinskii, M. Spin coupling model for tetrameric iron clusters in ferredoxins. I. Theory, exchange levels, g-factors. Chem. Phys. 1993, 172, 189–211. [Google Scholar] [CrossRef]
- Li, J.; Wang, K.; Smirnova, T.I.; Khade, R.L.; Zhang, Y.; Oldfield, E. Isoprenoid biosynthesis: Ferraoxetane or allyl anion mechanism for IspH catalysis? Angew. Chem. Int. Ed. 2013, 52, 6522–6525. [Google Scholar] [CrossRef] [PubMed]
- Charon, L.; Pale-Grosdemange, C.; Rohmer, M. On the reduction steps in the mevalonate independent 2-C-methyi-D-erythritol 4-phosphate (MEP) pathway for isoprenoid biosynthesis in the bacterium Zymomonas Mobilis. Tetrahedron Lett. 1999, 40, 7231–7234. [Google Scholar] [CrossRef]
- Charon, L.; Hoeffler, J.-F.; Pale-Grosdemange, C.; Lois, L.-M.; Campos, N.; Boronat, A.; Rohmer, M. Deuterium-labelled isotopomers of 2-C-methyl-D-erythritol as tools for the elucidation of the 2-C-methyl-D-erythritol 4-phosphate pathway for isoprenoid biosynthesis. Biochem. J. 2000, 346, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Hoeffler, J.-F.; Hemmerlin, A.; Grosdemange-Billiard, C.; Bach, T.J.; Rohmer, M. Isoprenoid biosynthesis in higher plants and in Escherichia coli: On the branching in the methylerythritol phosphate pathway and the independent biosynthesis of isopentenyl diphosphate and dimethylallyl diphosphate. Biochem. J. 2002, 366, 573–583. [Google Scholar] [CrossRef]
- Giner, J.-L.; Jaun, B.; Arigoni, D. Biosynthesis of isoprenoids in Escherichia coli: The fate of the 3-H and 4-H atoms of 1-deoxy-d-xylulose. Chem. Commun. 1998, 1857–1858. [Google Scholar] [CrossRef]
- Rieder, C.; Jaun, B.; Arigoni, D. On the early steps of cineol biosynthesis in Eucalyptus globulus. Helv. Chim. Acta 2000, 83, 2504–2513. [Google Scholar] [CrossRef]
- Arigoni, D.; Eisenreich, W.; Latzel, C.; Sagner, S.; Radykewicz, T.; Zenk, M.H.; Bacher, A. Dimethylallyl pyrophosphate is not the committed precursor of isopentenyl pyrophosphate during terpenoid biosynthesis from 1-deoxyxylulose in higher plants. Proc. Natl. Acad. Sci. USA 1999, 96, 1309–1314. [Google Scholar] [CrossRef]
- Laupitz, R.; Gräwert, T.; Rieder, C.; Zepeck, F.; Bacher, A.; Arigoni, D.; Rohdich, F.; Eisenreich, W. Stereochemical studies on the making and unmaking of isopentenyl diphosphate in different biological systems. Chem. Biodivers. 2004, 1, 1367–1376. [Google Scholar] [CrossRef]
- Chang, W.; Xiao, Y.; Liu, H.; Liu, P. Mechanistic studies of an IspH-catalyzed reaction: Implications for substrate binding and protonation in the biosynthesis of isoprenoids. Angew. Chem. Int. Ed. 2011, 50, 12304–12307. [Google Scholar] [CrossRef] [PubMed]
- Masini, T.; Kroezen, B.S.; Hirsch, A.K.H. Druggability of the enzymes of the non-mevalonate-pathway. Drug Discov. Today 2013, 18, 1256–1262. [Google Scholar] [CrossRef]
- Janthawornpong, K.; Krasutsky, S.; Chaignon, P.; Rohmer, M.; Poulter, C.D.; Seemann, M. Inhibition of IspH, a [4Fe–4S]2+ enzyme involved in the biosynthesis of isoprenoids via the methylerythritol phosphate pathway. J. Am. Chem. Soc. 2013, 135, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Ahrens-Botzong, A.; Janthawornpong, K.; Wolny, J.A.; Tambou, E.N.; Rohmer, M.; Krasutsky, S.; Poulter, C.D.; Schünemann, V.; Seemann, M. Biosynthesis of isoprene units: Mössbauer spectroscopy of substrate and inhibitor binding to the [4Fe-4S] cluster of the LytB/IspH enzyme. Angew. Chem. Int. Ed. 2011, 50, 11976–11979. [Google Scholar] [CrossRef]
- Van Hoof, S.; Lacey, C.J.; Röhrich, R.C.; Wiesner, J.; Jomaa, H.; Van Calenbergh, S. Synthesis of analogues of (E)-1-hydroxy-2-methylbut-2-enyl 4-diphosphate, an isoprenoid precursor and human Γδ T cell activator. J. Org. Chem. 2008, 73, 1365–1370. [Google Scholar] [CrossRef]
- Wang, K.; Wang, W.; No, J.-H.; Zhang, Y.; Zhang, Y.; Oldfield, E. Inhibition of the Fe4S4 -cluster-containing protein IspH (lytB): Electron paramagnetic resonance, metallacycles, and mechanisms. J. Am. Chem. Soc. 2010, 132, 6719–6727. [Google Scholar] [CrossRef]
- Elliott, T.S.; Slowey, A.; Ye, Y.; Conway, S.J. The use of phosphate bioisosteres in medicinal chemistry and chemical biology. Med. Chem. Commun. 2012, 3, 735. [Google Scholar] [CrossRef]
- Wang, W.; Li, J.; Wang, K.; Smirnova, T.I.; Oldfield, E. Pyridine inhibitor binding to the 4Fe-4S protein, A. Aeolicus IspH (LytB): A HYSCORE Investigation. J. Am. Chem. Soc. 2011, 133, 6525–6528. [Google Scholar] [CrossRef]
- O’Dowd, B.; Williams, S.; Wang, H.; No, J.H.; Rao, G.; Wang, W.; McCammon, J.A.; Cramer, S.P.; Oldfield, E. Spectroscopic and computational investigations of ligand binding to IspH: Discovery of non-diphosphate inhibitors. ChemBioChem 2017, 18, 914–920. [Google Scholar] [CrossRef]
- Miller, A.A.; Bundy, G.L.; Mott, J.E.; Skepner, J.E.; Boyle, T.P.; Harris, D.W.; Hromockyj, A.E.; Marotti, K.R.; Zurenko, G.E.; Munzner, J.B.; et al. Discovery and characterization of QPT-1, the progenitor of a new class of bacterial topoisomerase inhibitors. Antimicrob. Agents Chemother. 2008, 52, 2806–2812. [Google Scholar] [CrossRef] [PubMed]
- Brandstetter, H.; Grams, F.; Glitz, D.; Lang, A.; Huber, R.; Bode, W.; Krell, H.-W.; Engh, R.A. The 1.8-Å crystal structure of a matrix metalloproteinase 8-barbiturate inhibitor complex reveals a previously unobserved mechanism for collagenase substrate recognition. J. Biol. Chem. 2001, 276, 17405–17412. [Google Scholar] [CrossRef] [PubMed]
- Dunten, P.; Kammlott, U.; Crowther, R.; Levin, W.; Foley, L.H.; Wang, P.; Palermo, R. X-ray structure of a novel matrix metalloproteinase inhibitor complexed to stromelysin. Protein Sci. 2001, 10, 923–926. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Py, B.; Gerez, C.; Huguenot, A.; Vidaud, C.; Fontecave, M.; Ollagnier de Choudens, S.; Barras, F. The ErpA/NfuA complex builds an oxidation-resistant Fe-S cluster delivery pathway. J. Biol. Chem. 2018, 293, 7689–7702. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Priority | Bacteria | IspH |
|---|---|---|
| critical | Acinetobacter baumannii, carbapenem-resistant | + |
| Pseudomonas aeruginosa, carbapenem-resistant | + | |
| Enterobacteriaceae, carbapenem-resistant, ESBL-producing | + | |
| high | Enterococcus faecium, vancomycin-resistant | − a |
| Staphylococcus aureus, methicillin-resistant, vancomycin-intermediate and resistant | − | |
| Helicobacter pylori, clarithromycin-resistant | + | |
| Campylobacter spp., fluoroquinolone-resistant | + | |
| Salmonellae, fluoroquinolone-resistant | + | |
| Neisseria gonorrhoeae, cephalosporin-resistant, fluoroquinolone-resistant | + | |
| medium | Streptococcus pneumoniae, penicillin-non-susceptible | − |
| Haemophilus influenzae, ampicillin-resistant | + | |
| Shigella spp., fluoroquinolone-resistant | + |
| ΔEQ [mm s−1] | δ [mm s−1] | Oxidation State and Coordination | |
|---|---|---|---|
| Component 1 (50%) | 1.21 (1.33) | 0.42 (0.42) | Tetrahedrally coordinated Fe2.5+, mixed valence iron pairs, 3 S2− and 1 Cys ligand |
| Component 2 (25%) | 0.89 (0.92) | 0.37 (0.38) | Tetrahedrally coordinated high-spin Fe3+, 3 S2− and 1 Cys ligand |
| Component 3 (25%) | 1.97 (1.00) | 0.89 (0.64) | Hexa-coordinated high-spin Fe2+, 3 S2− and 3 H2O (tetrahedrally coordinated, 3 S2− and OHHMBPP) |
| PDB ID | Release Date | Source Organism | Crystallization Method (Vapor difusion) | Crystal Growth Procedure | pH | Space Group | Res. (Å) | Cluster | Ligand ID | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| 3DNF | 30 December 2008 | A. aeolicus | Hanging drop | 0.1 M Tris-Hcl, 10% PEG 8000 | 8.0 | P 1 21 1 | 1.65 | Fe3 S4 | - | [36] |
| 4N7B | 20 November 2013 | P. falciparum | Sitting drop | 0.1 M Na citrate, 0.1 M (NH4)2SO4, 30% PEG 4000 | 5.6 | P 31 2 1 | 2.20 | Fe3 S4 | SO4 | [43] |
| 3F7T | 7 July 2009 | E. coli | Hanging drop | 1.6 M Potassium Phosphate | 8.0 | P 32 2 1 | 1.80 | Fe3 S4 | POP | [42] |
| 3KEF | 12 January 2010 | E. coli | Sitting drop | 0.1 M Bis-Tris, 0.2 M Li2SO4, 25% PEG 3350 | 6.5 | P 21 21 21 | 1.70 | Fe3 S4 | DMA | [37] |
| 3KEL | 1.80 | Fe3 S4 | POP | |||||||
| 3KE8 | 1.70 | Fe4 S4 | EIP | |||||||
| 3KE9 | 1.90 | Fe4 S4 | IPE | |||||||
| 3KEM | 7.0 | C 1 2 1 | 2.00 | Fe3 S4 | IPE | |||||
| 3T0G | 30 November 2011 | E. coli | Hanging drop | 0.1 M Bis-Tris, 0.2 M Li2SO4, 25% PEG3350 | 6.5 | P 21 21 21 | 2.10 | Fe3 S4 | H6P | [45] |
| 3T0F | 1.90 | Fe3 S4 | H6P | |||||||
| 3SZU | 1.40 | Fe3 S4 | H6P | |||||||
| 3SZL | 1.60 | Fe4 S4 | H6P | |||||||
| 3SZO | 1.60 | Fe4 S4 | H6P | |||||||
| 3URK | 5 September 2012 | E. coli | Sitting drop | 0.1 M Bis-Tris, 0.2 M (NH4)2SO4, 25% PEG3350 | 6.5 | P 21 21 21 | 1.50 | Fe4 S4 | 0CG | [46] |
| 3UTC | 1.90 | Fe4 S4 | 0JX | |||||||
| 3UTD | 1.70 | Fe3 S4 | OCJ | |||||||
| 3UV3 | 1.60 | Fe4 S4 | 0CM | |||||||
| 3UV6 | 1.70 | Fe4 S4 | 0CH | |||||||
| 3UV7 | 1.60 | Fe4 S4 | 0CN | |||||||
| 3UWM | 1.80 | Fe4 S4 | 0K2/0JX | |||||||
| 3ZGL | 9 January 2013 | E. coli | Hanging drop | 0.1M Bis-Tris, 0.2 M Li2SO4, 24% PEG 3350 | 6.5 | C 1 2 1 | 1.68 | Fe4 S4 | 10E | [47] |
| 3ZGN | 1.95 | Fe4 S4 | 10G | |||||||
| 4H4C | 23 January 2013 | E. coli | Sitting drop | 0.1 M Bis-Tris, 0.2 M (NH4)2SO4, 25% PEG3350 | 6.5 | P 21 21 21 | 1.80 | Fe4 S4 | 10D | [48] |
| 4H4D | 1.35 | Fe4 S4 | 10E | |||||||
| 4H4E | 1.70 | Fe4 S4 | 10G | |||||||
| 4EB3 | 6 February 2013 | E. coli | Sitting drop | 0.1 M Bis-Tris, 0.2 M (NH4)2SO4, 25% PEG3350 | 6.5 | P 21 21 21 | 1.90 | Fe4 S4 | 0O3 | [49] |
| 4MUY | 11 June 2014 | E. coli | Sitting drop | 0.1 M Bis-Tris, 0.2 M (NH4)2SO4, 25% PEG3350 | 6.5 | P 21 21 21 | 1.80 | Fe3 S4 | 2E5 | [50] |
| 4MV0 | 1.90 | Fe3 S4 | 2E6 | |||||||
| 4MV5 | 18 June 2014 | 1.90 | Fe3 S4 | 2E7 | ||||||
| 4MUX | 1.70 | Fe4 S4 | 2E4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jobelius, H.; Bianchino, G.I.; Borel, F.; Chaignon, P.; Seemann, M. The Reductive Dehydroxylation Catalyzed by IspH, a Source of Inspiration for the Development of Novel Anti-Infectives. Molecules 2022, 27, 708. https://doi.org/10.3390/molecules27030708
Jobelius H, Bianchino GI, Borel F, Chaignon P, Seemann M. The Reductive Dehydroxylation Catalyzed by IspH, a Source of Inspiration for the Development of Novel Anti-Infectives. Molecules. 2022; 27(3):708. https://doi.org/10.3390/molecules27030708
Chicago/Turabian StyleJobelius, Hannah, Gabriella Ines Bianchino, Franck Borel, Philippe Chaignon, and Myriam Seemann. 2022. "The Reductive Dehydroxylation Catalyzed by IspH, a Source of Inspiration for the Development of Novel Anti-Infectives" Molecules 27, no. 3: 708. https://doi.org/10.3390/molecules27030708
APA StyleJobelius, H., Bianchino, G. I., Borel, F., Chaignon, P., & Seemann, M. (2022). The Reductive Dehydroxylation Catalyzed by IspH, a Source of Inspiration for the Development of Novel Anti-Infectives. Molecules, 27(3), 708. https://doi.org/10.3390/molecules27030708