
Synthesis, Antimicrobial, Anti-Virulence and Anticancer Evaluation of New 5(4H)-Oxazolone-Based Sulfonamides

 , , , , ,
, , , , ,  and
and
Abstract
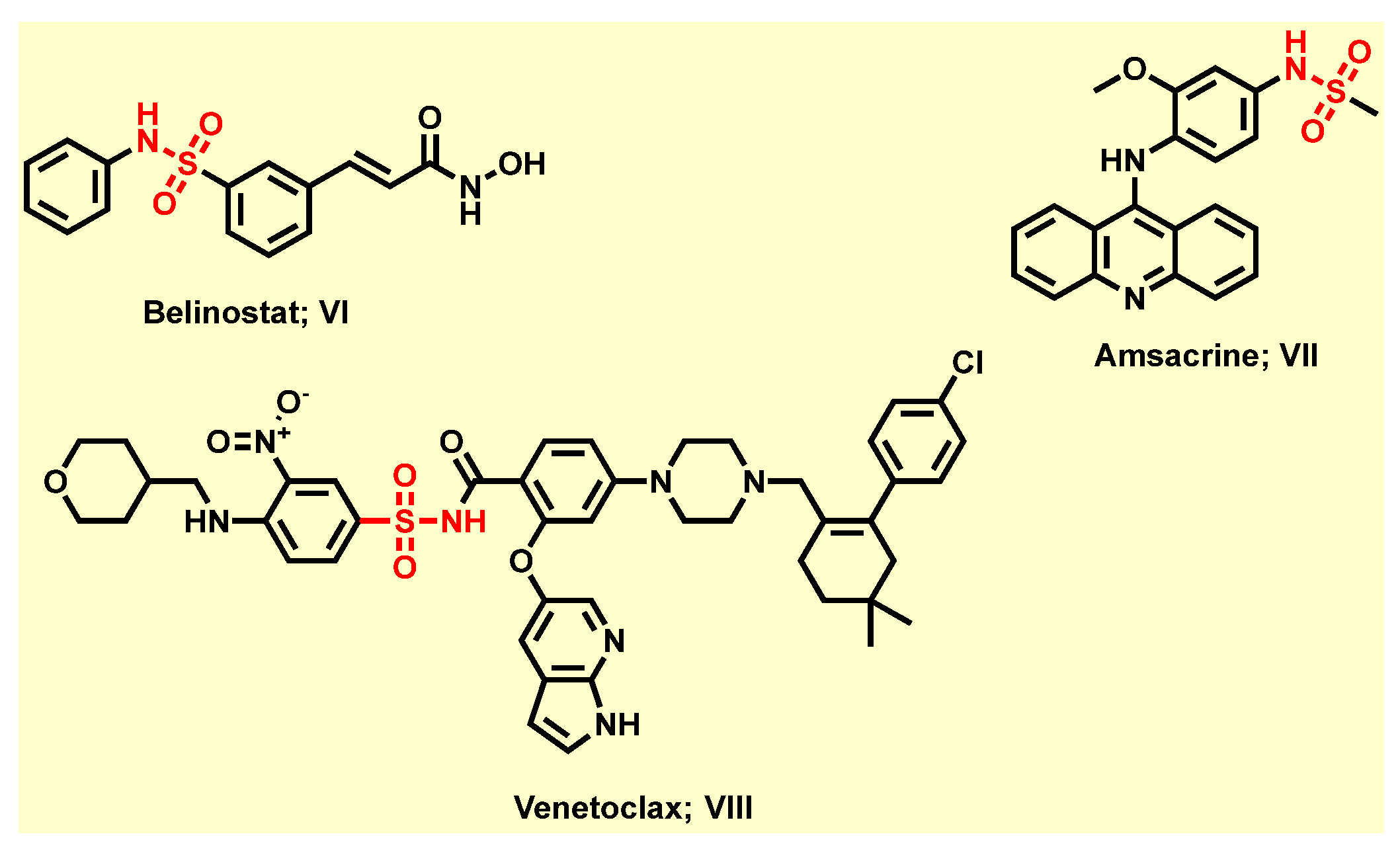
:1. Introduction
2. Results and Discussion
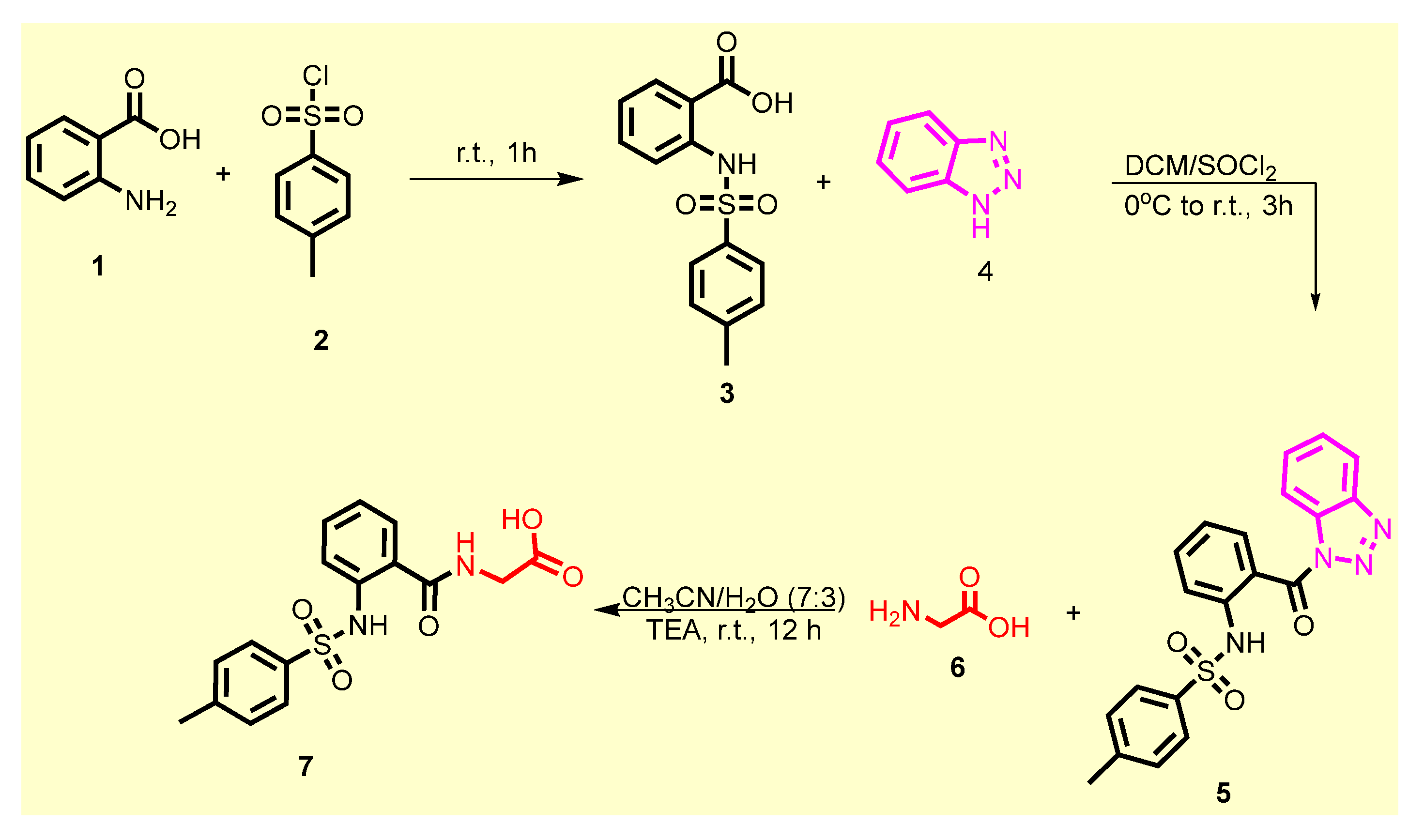
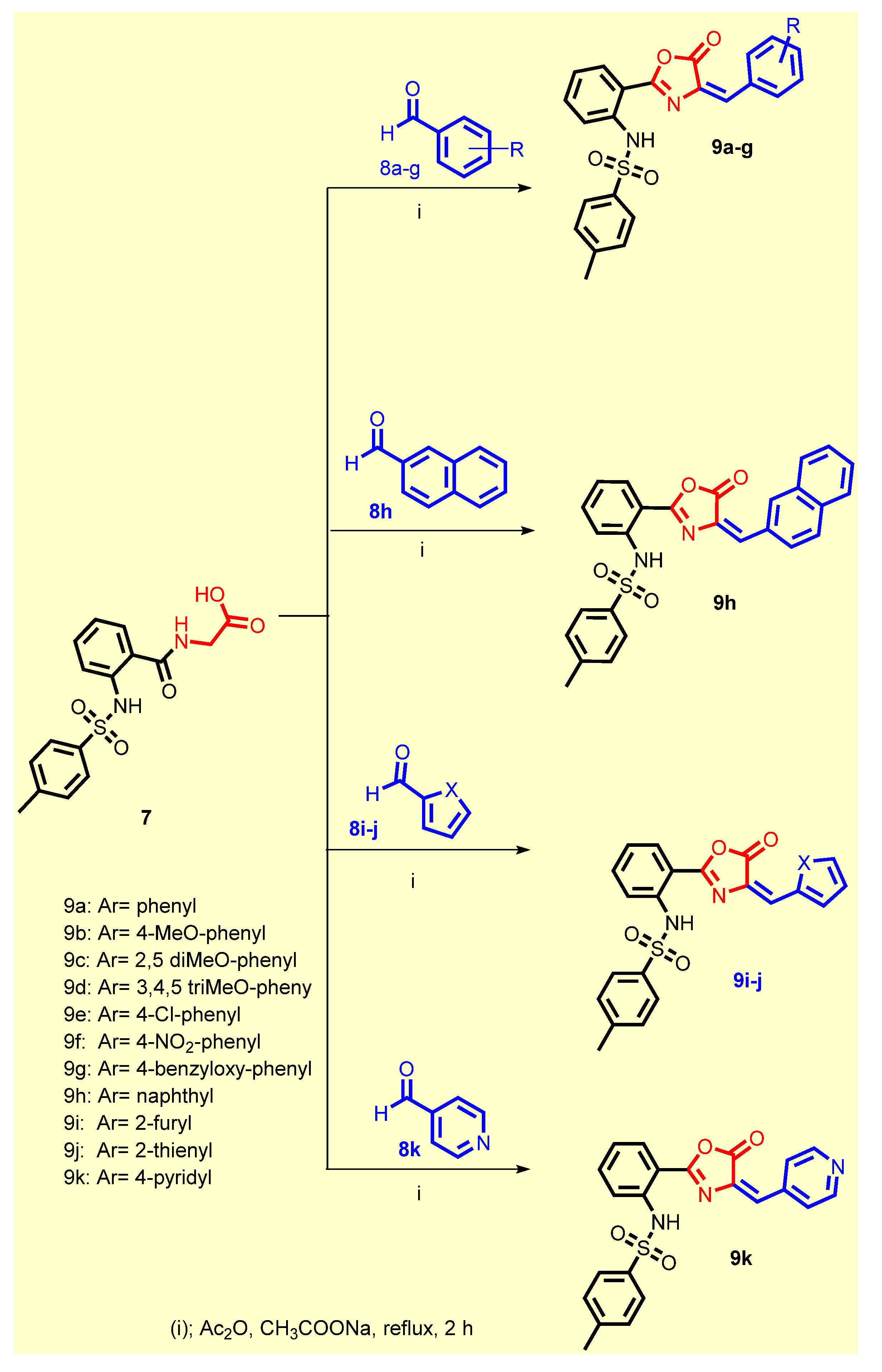
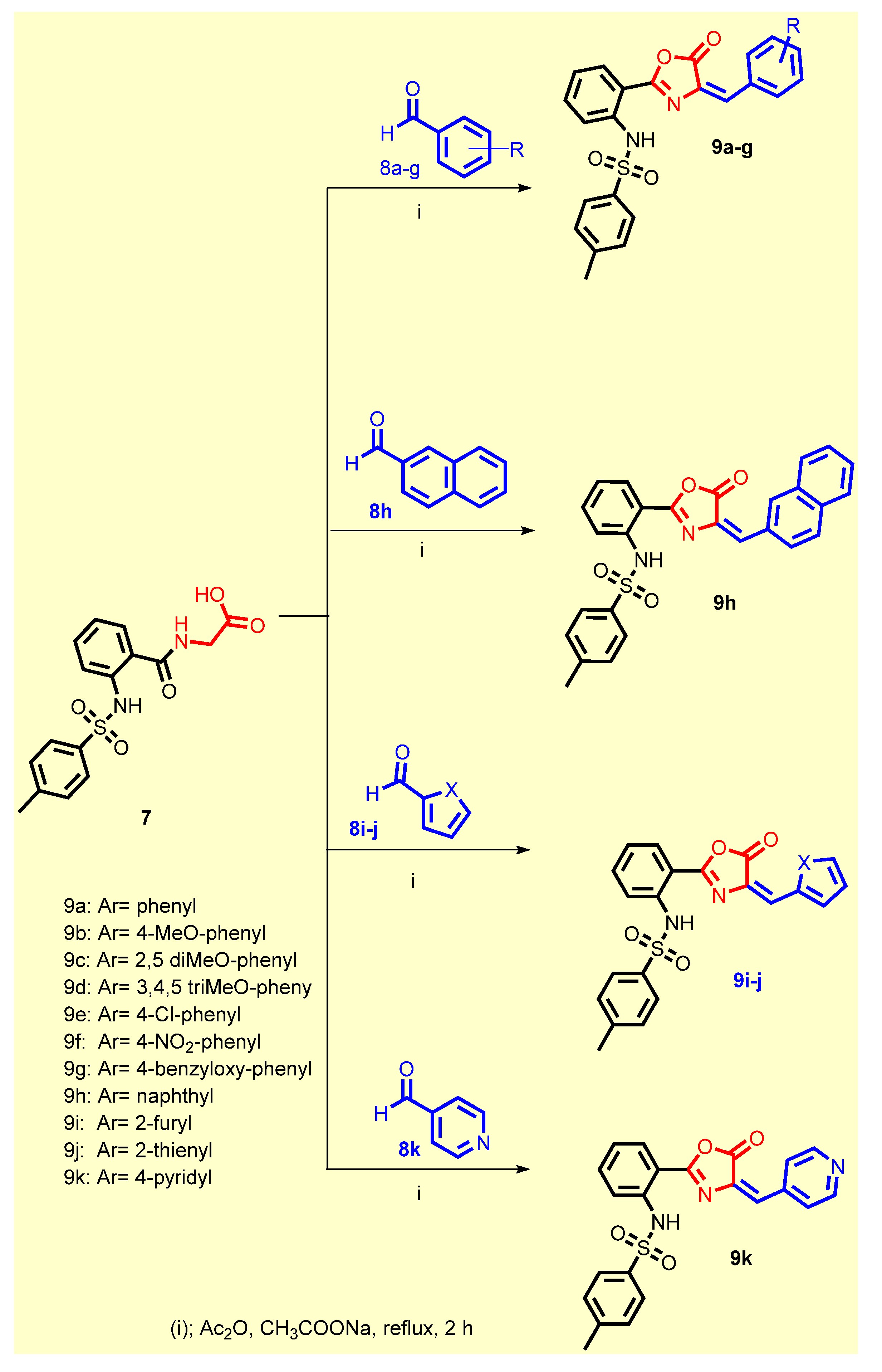
2.1. Chemistry
2.2. Biological Screening
2.2.1. In Vitro Antimicrobial Activity
Minimum Inhibitory Concentrations (MICs) of Synthesized Compounds against Different Gram-Positive and -Negative Bacteria
Antifungal Activity of Synthesized Compounds
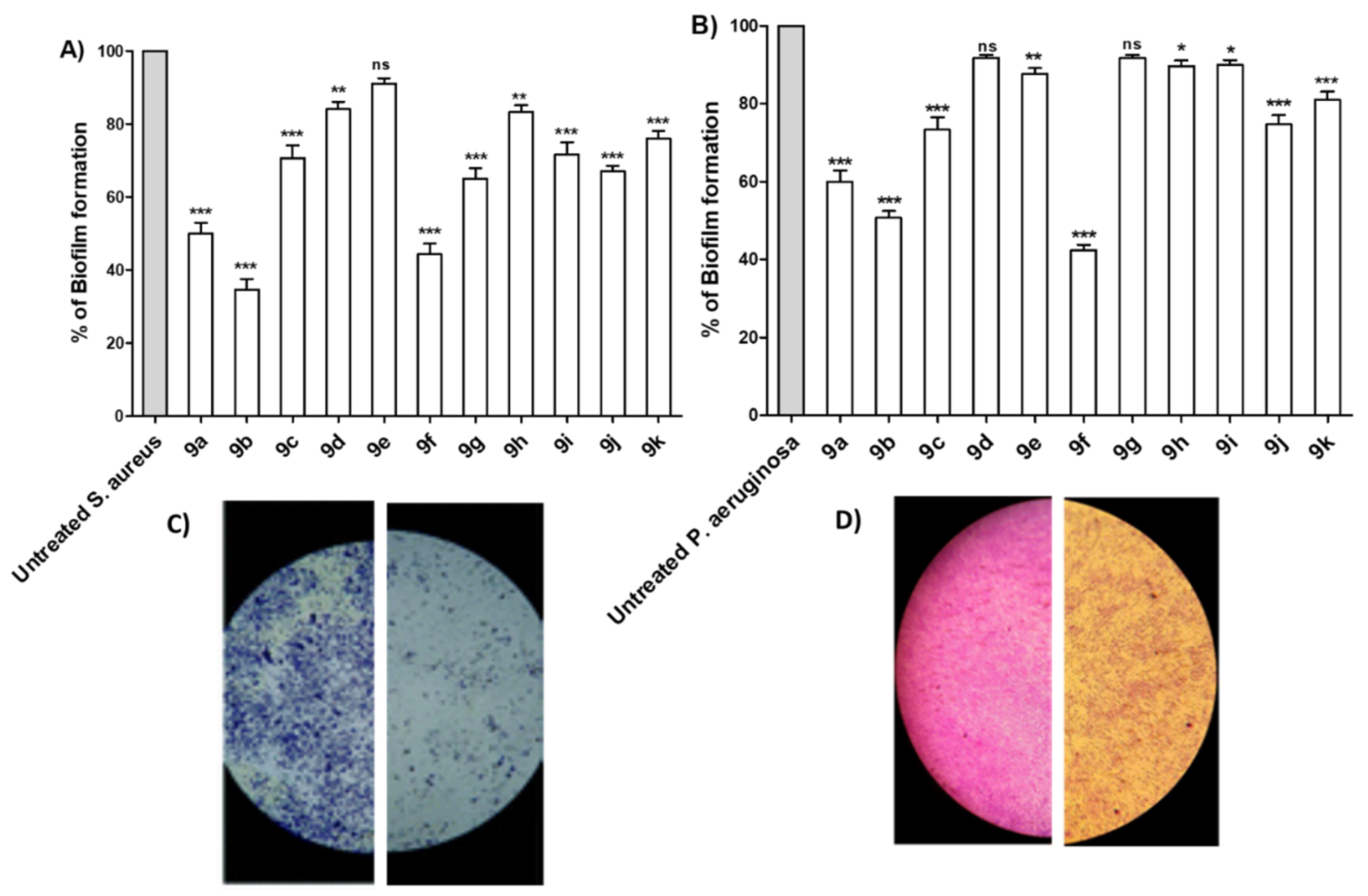
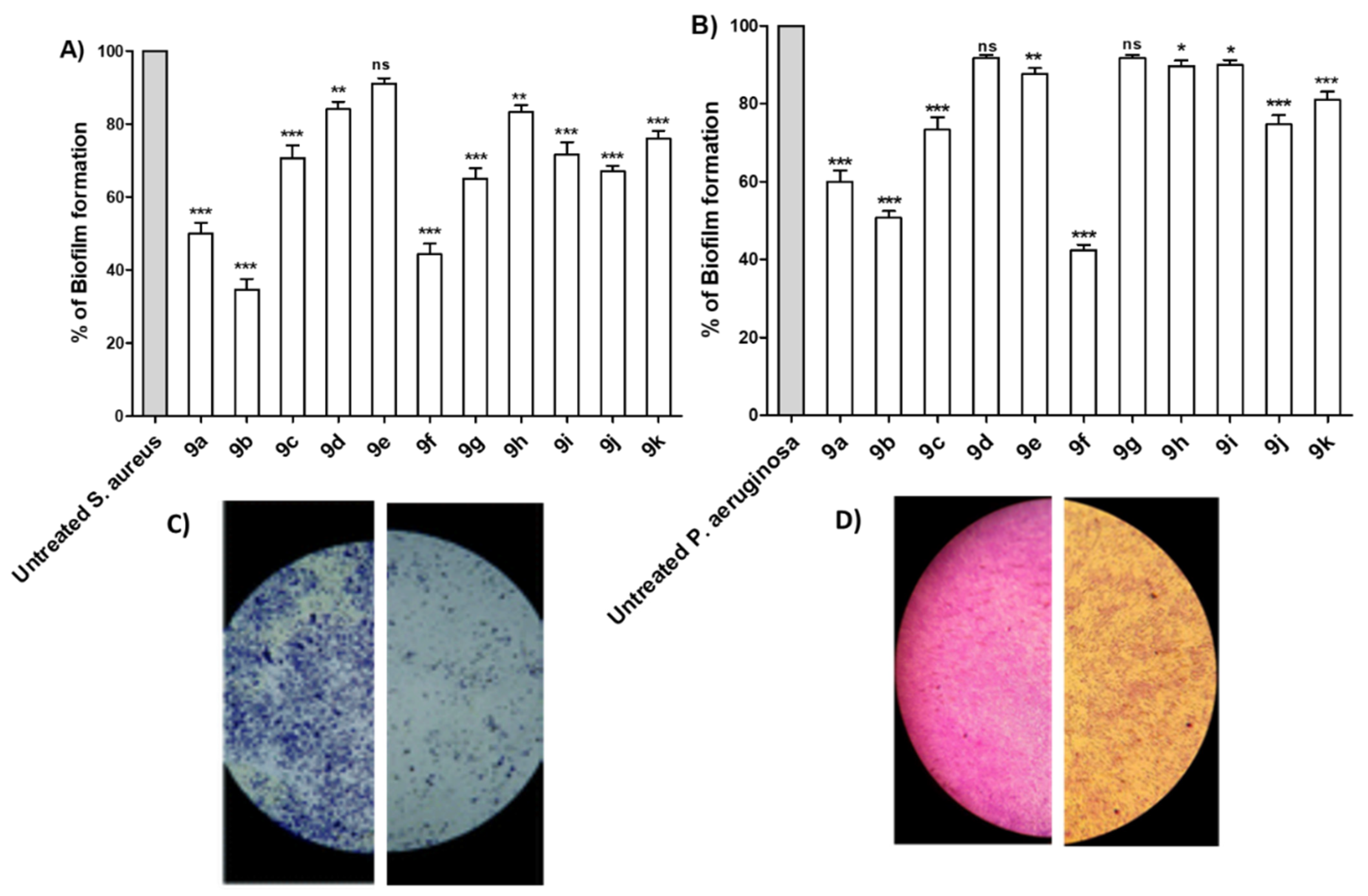
Antibiofilm Activity of Synthesized Compounds
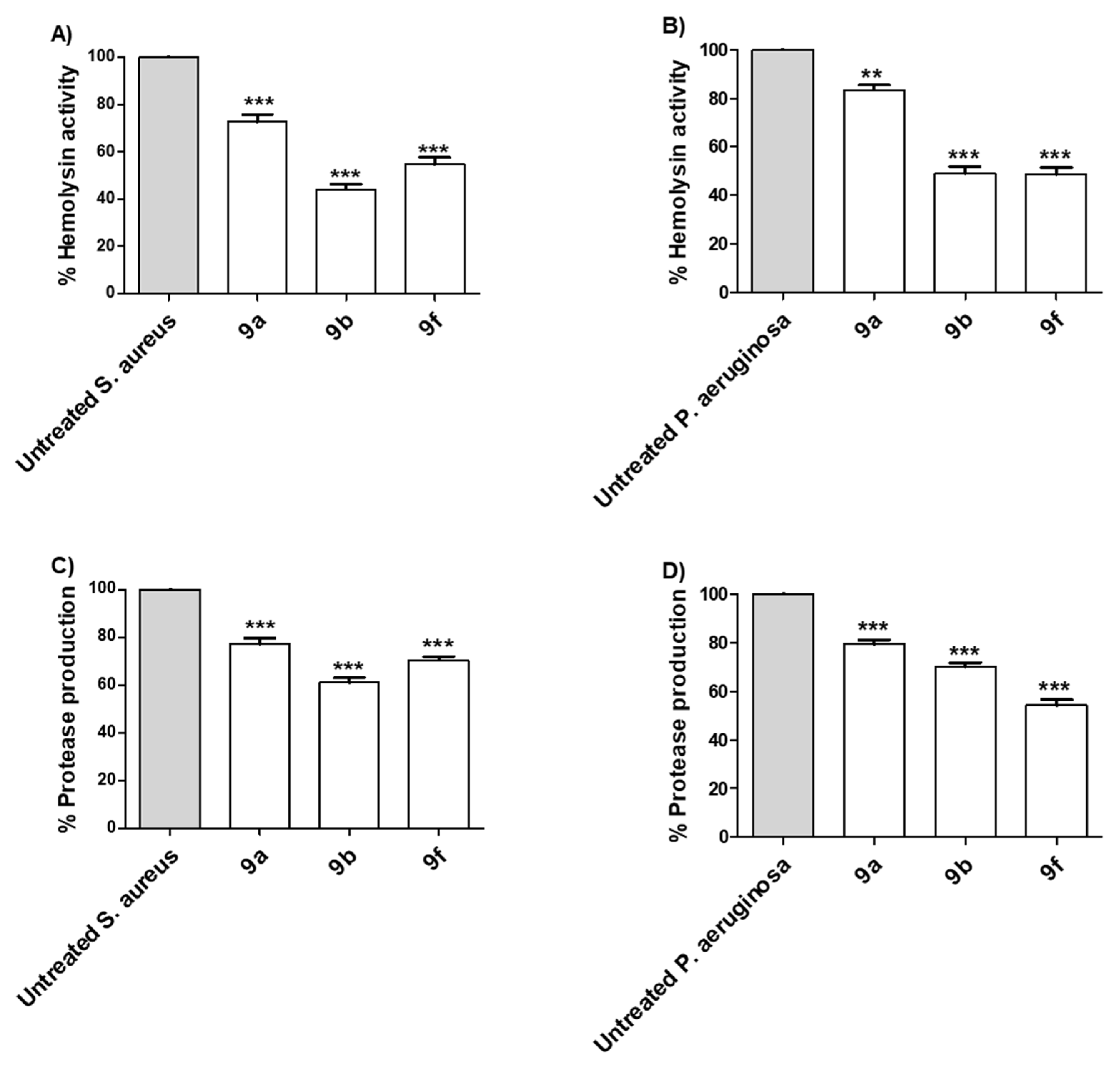
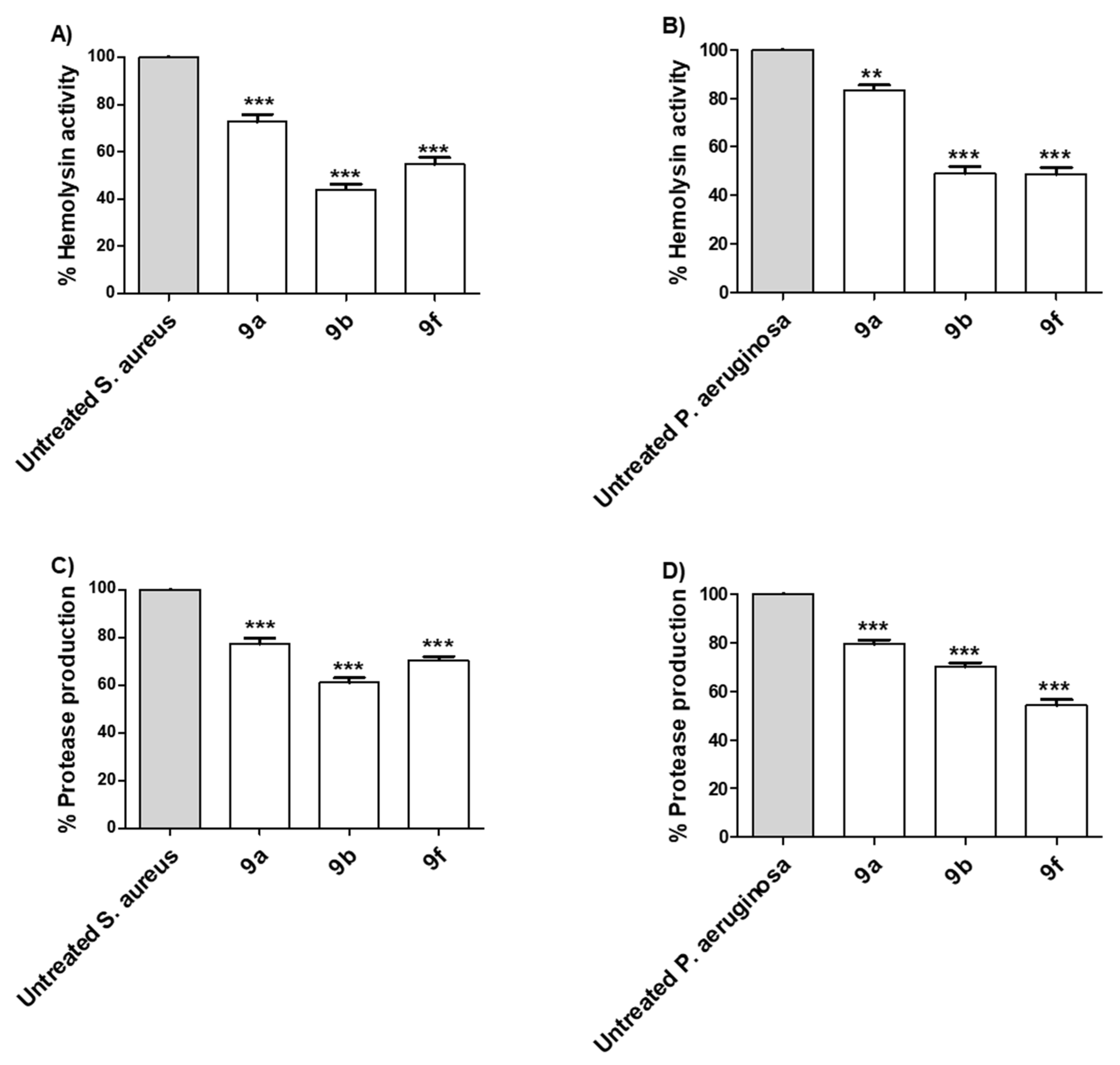
Tested Compounds Diminish the Production of Bacterial Virulence Extracellular Enzymes
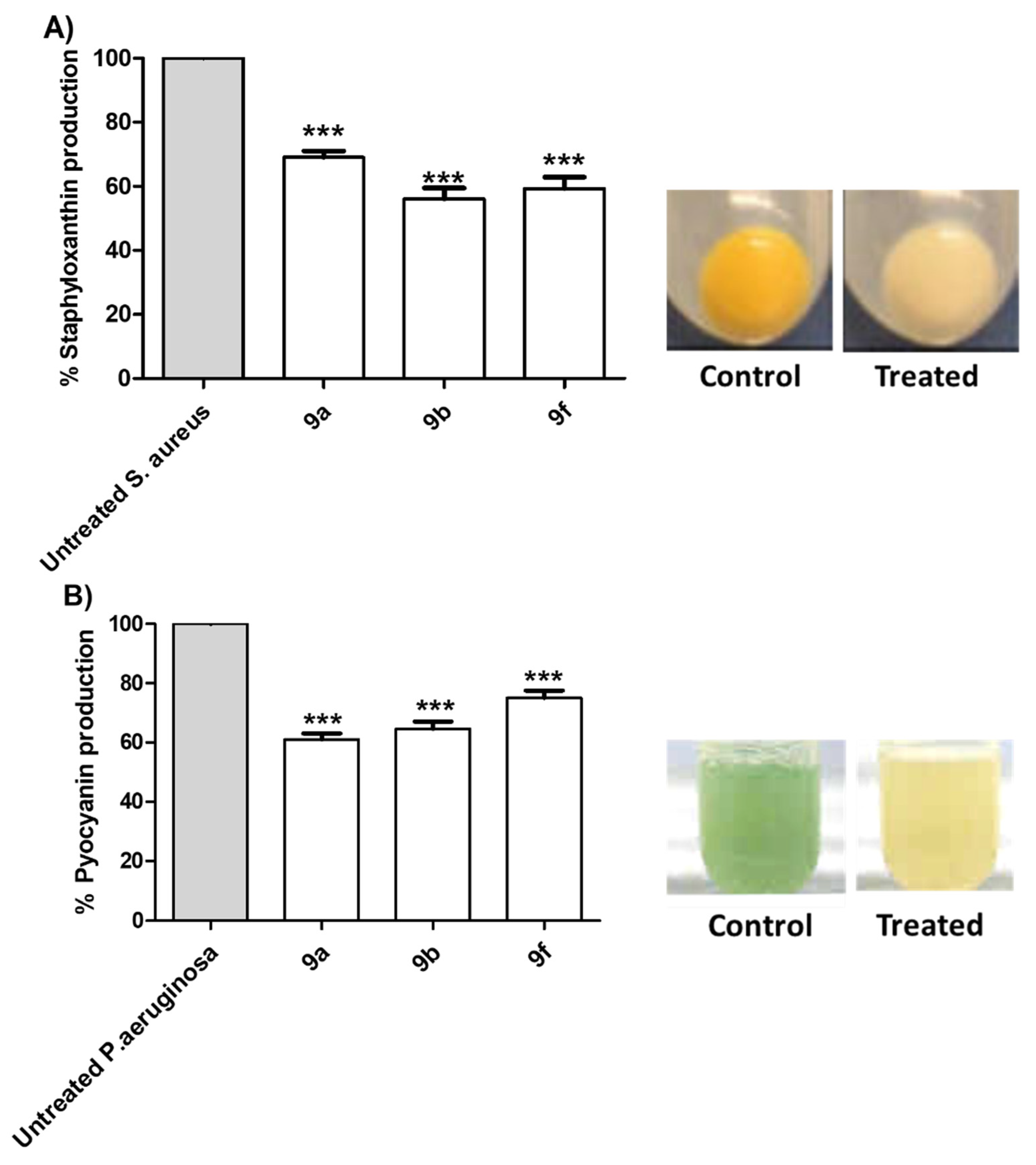
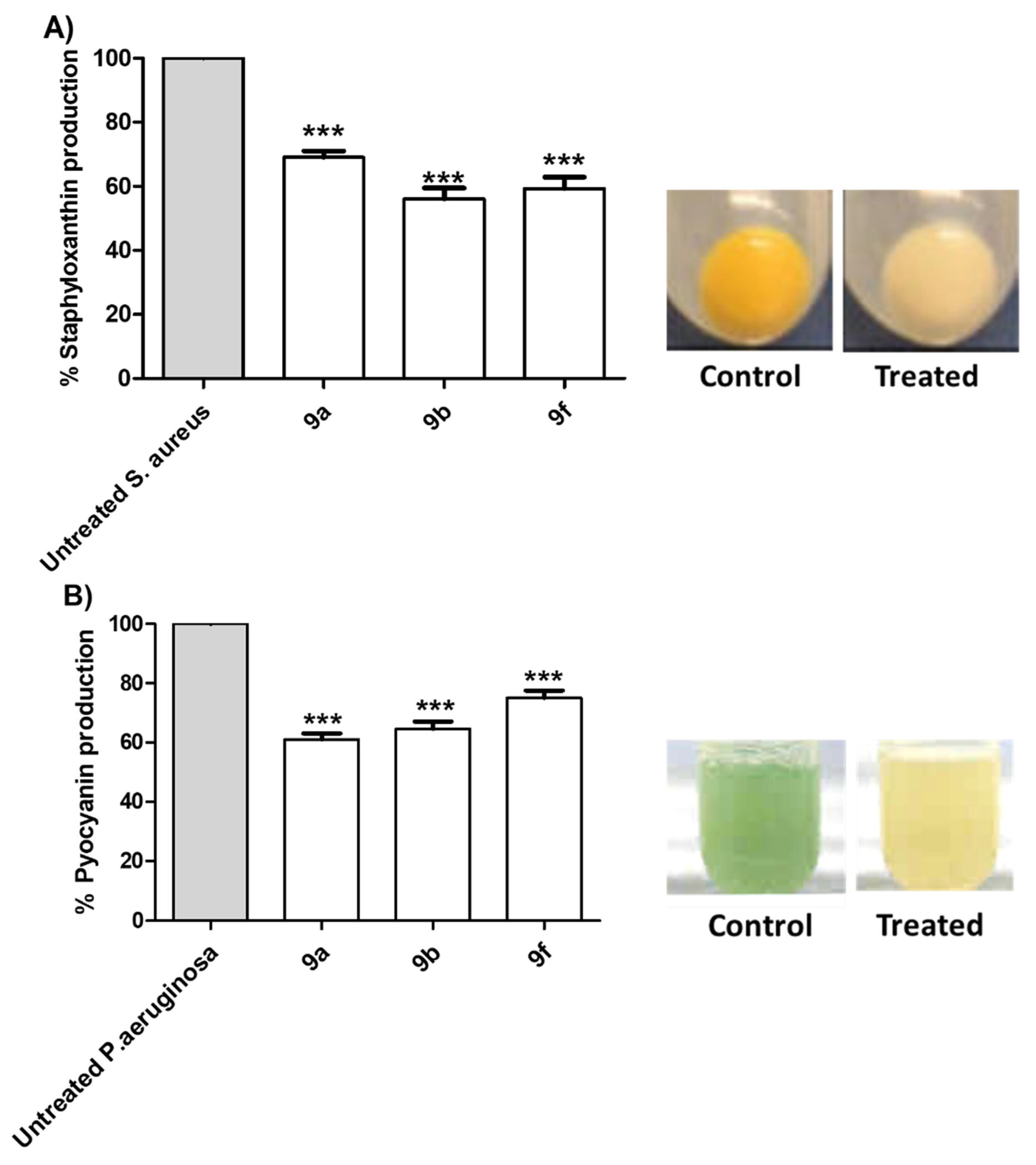
Tested Compounds Diminish the Production of Bacterial Virulence Factors
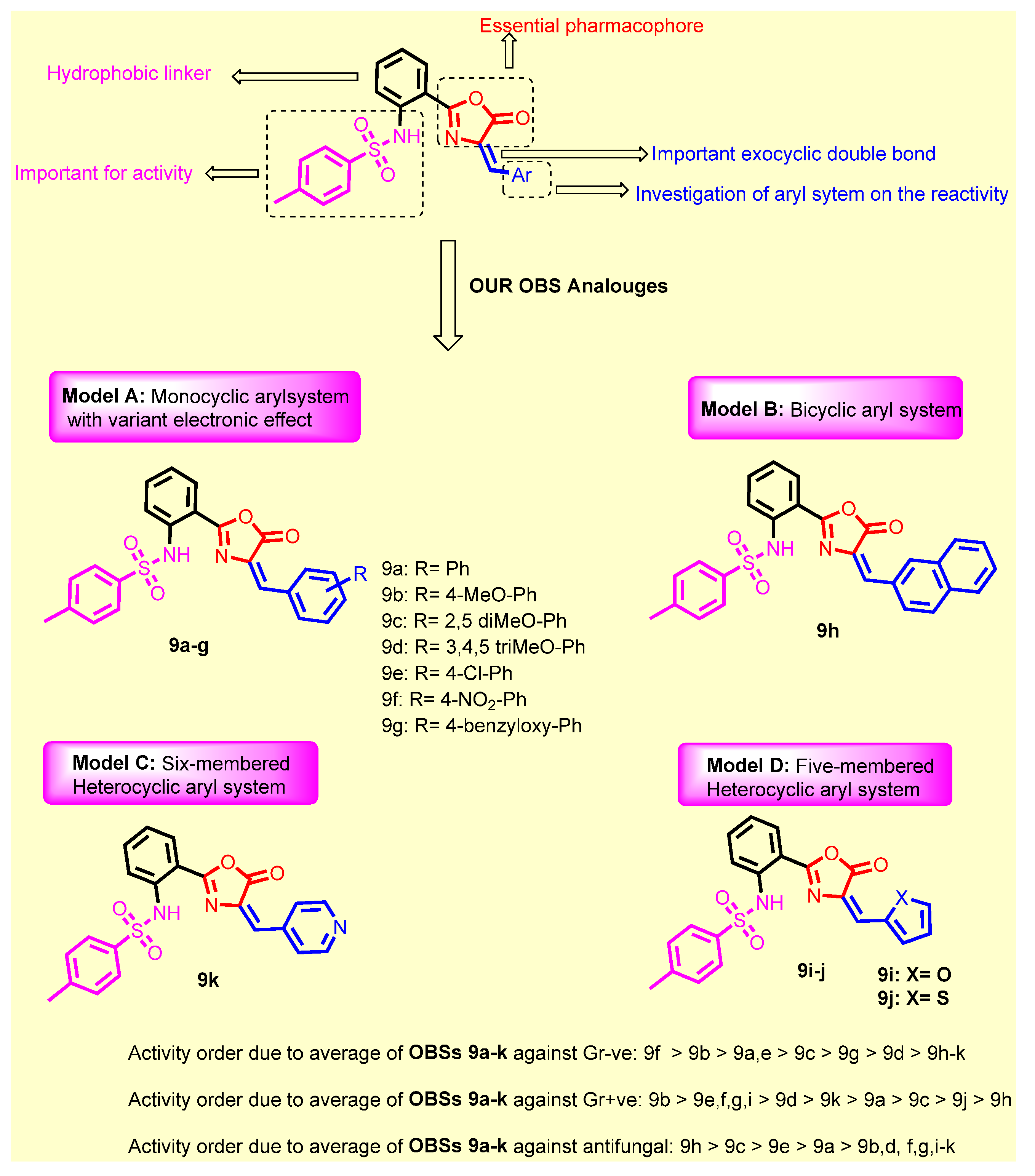
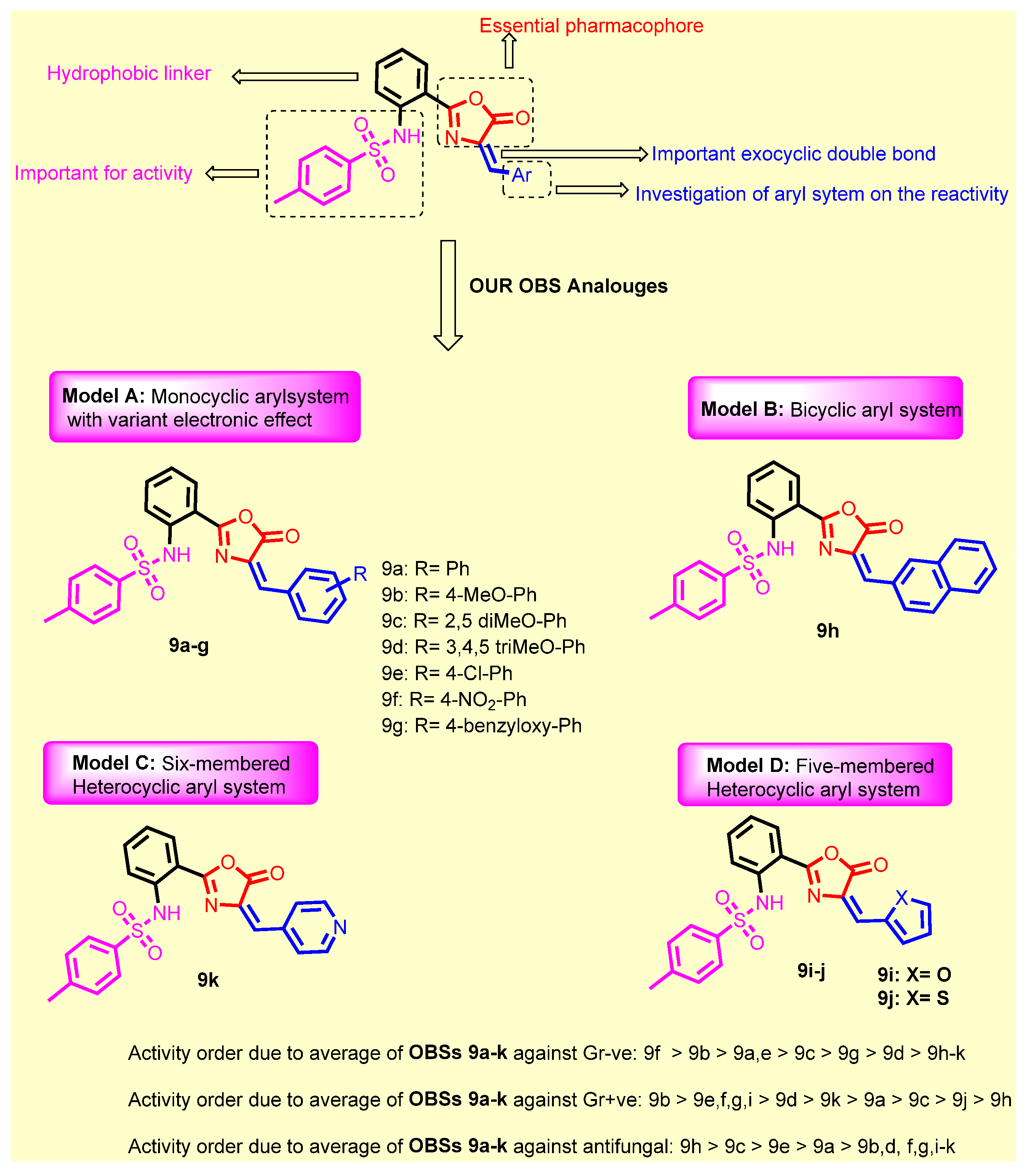
SAR Study
- Introduction of 4-methoxy group (9b) resulted in a 2-fold increase in the activity against Staphylococcus aureus, Staphylococcus epidermidis, Micrococcus spp. and Pseudomonas aeruginosae, while retaining the same activity as compound 9a against Klebsiella pneumonia, Salmonella typhimurium and Escherichia coli;
- Introduction of 2,5-dimethoxy group (9c) retained the same activity against Staphylococcus epidermidis and Micrococcus spp., and a 2-fold decrease against Staphylococcus aureus, Pseudomonas aeruginosae, Klebsiella pneumonia, Salmonella typhimurium and Escherichia coli;
- Replacement of hydrogen with trimethoxy groups (9d) resulted in the same activity against Staphylococcus aureus, improved activity (2-fold) against Staphylococcus epidermidis and Micrococcus spp., slightly decreased activity (2-fold) against Escherichia coli, and significantly decreased the antibacterial activity against Pseudomonas aeruginosae, Klebsiella pneumonia, and Salmonella typhimurium;
- Introduction of the weakly deactivating Cl group (9e) improved potency (2-fold increase in activity) against all Gram-positive organisms, Staphylococcus epidermidis, and Micrococcus spp., improved potency (2-fold increase) against Klebsiella pneumonia, resulted in the same activity against Pseudomonas aeruginosae and Salmonella typhimurium, and a 2-fold decrease in activity against Escherichia coli;
- Introduction of the strongly activating NO2 group (9f) improved potency (2-fold) against all strains except against Escherichia coli (2-fold decrease);
- Replacement of phenyl group with benzyloxy group (9g) led to the improvement of antibacterial activity against all Gram-positive organisms, the same antibacterial activity against the Gram-negative organism Escherichia coli, moderate antibacterial activity against Klebsiella pneumonia, and weak antibacterial activity against Pseudomonas aeruginosae and Salmonella typhimurium;
- Replacement of phenyl group with naphthyl one (9h) resulted in loss of activity against all bacterial strains, whereas it displayed the highest antifungal activity against Aspergillus niger and Candida albicans;
- Introduction of 2-furyl moiety (9i) led to a 2-fold increase against the three test Gram-positive strains Staphylococcus aureus, Staphylococcus epidermidis and Micrococcus spp.;
- Introduction of 2-thienyl group (9j) resulted in a decrease in activity by 4-fold against Staphylococcus aureus, and 2-fold against Staphylococcus epidermidis and Micrococcus spp.;
- Shifting from phenyl group to 2-pyridyl group (9k) increased the antibacterial activity by 2-fold against Micrococcus spp., while retaining the same activity against Staphylococcus aureus and Staphylococcus epidermidis.
2.2.2. The Antitumor Activity of the Tested Compounds
Cell Viability Assay
Tested Compounds Suppress Cellular Proliferation of Cancer Cell Lines
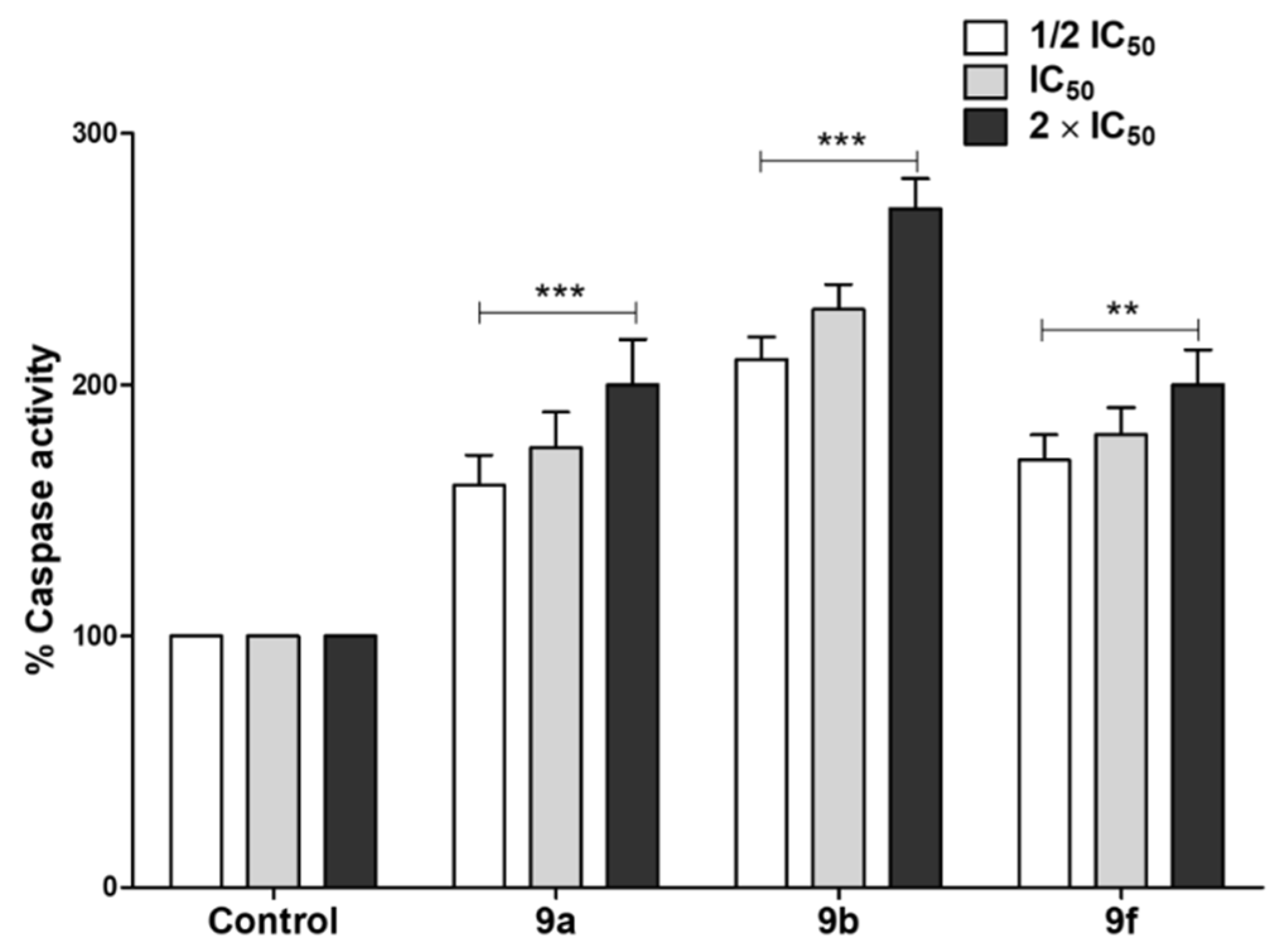
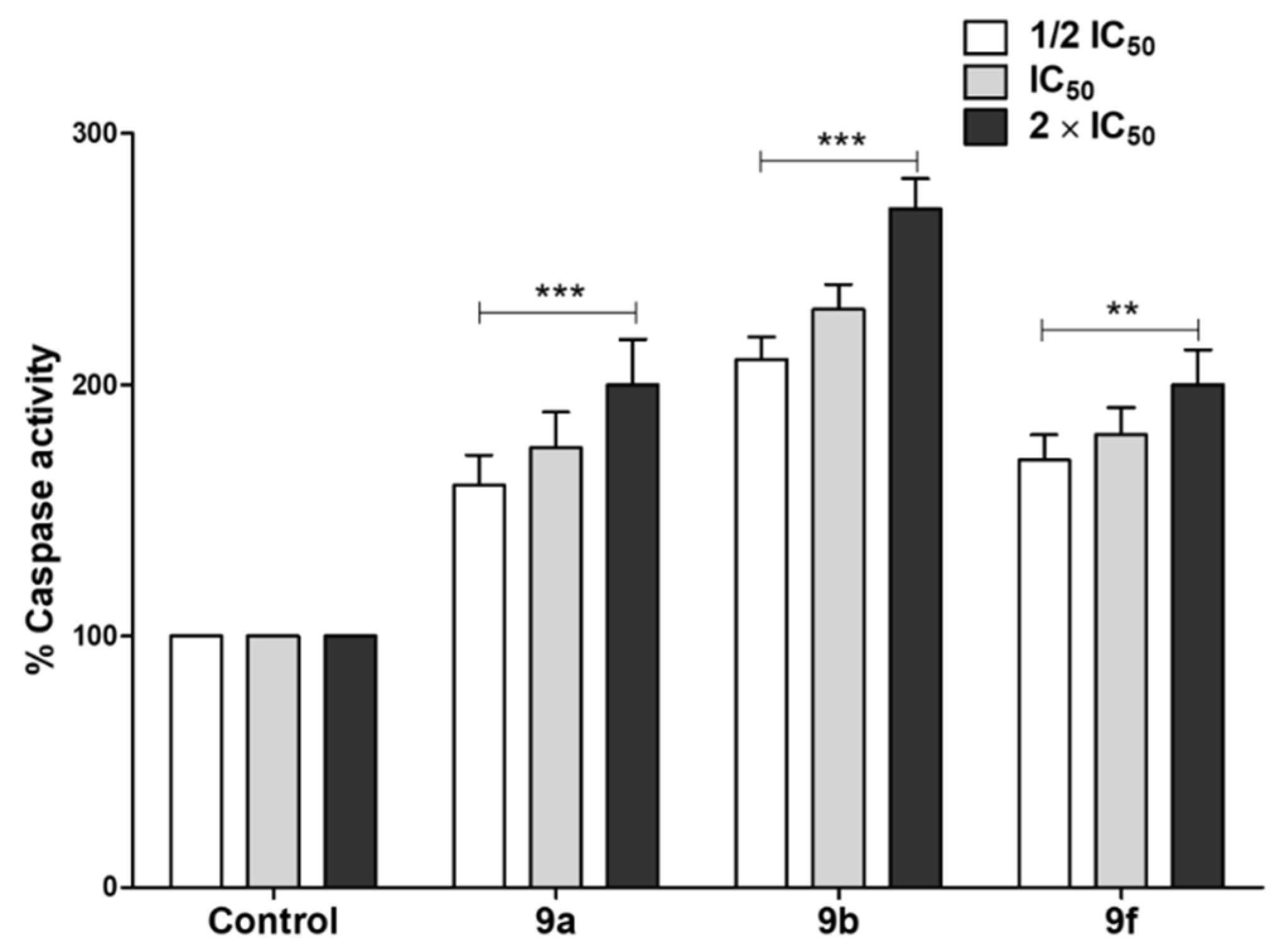
Tested Compounds Can Induce Apoptotic Cell Death
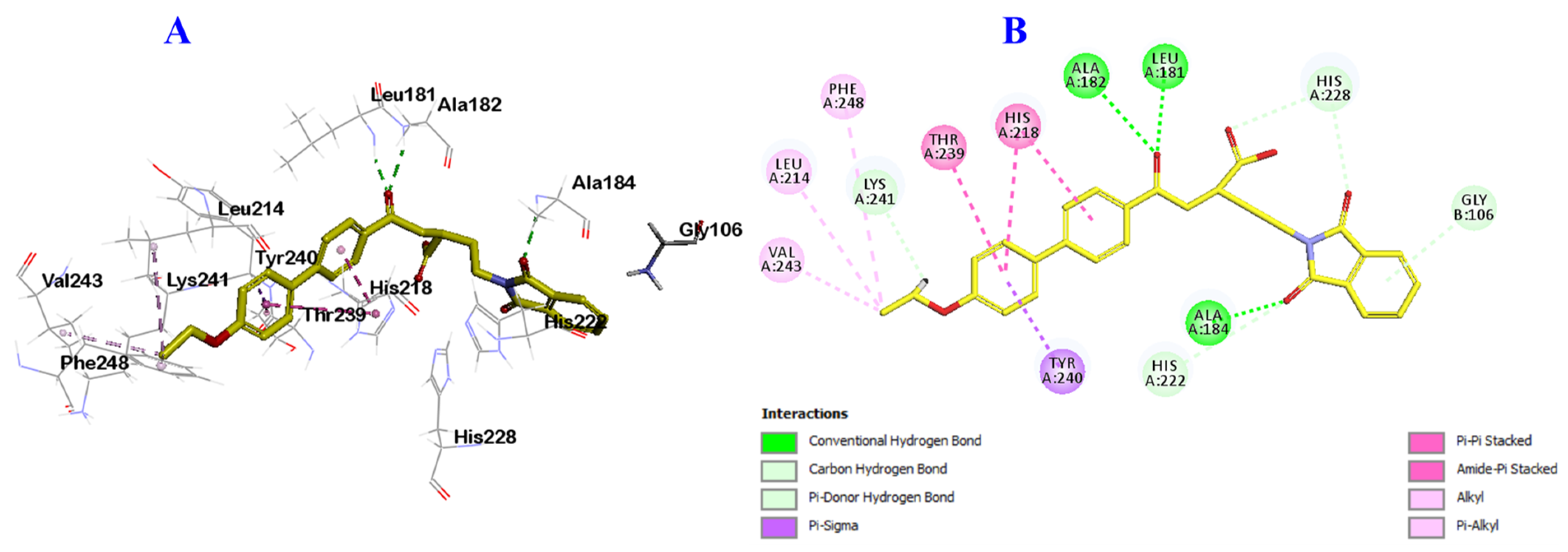
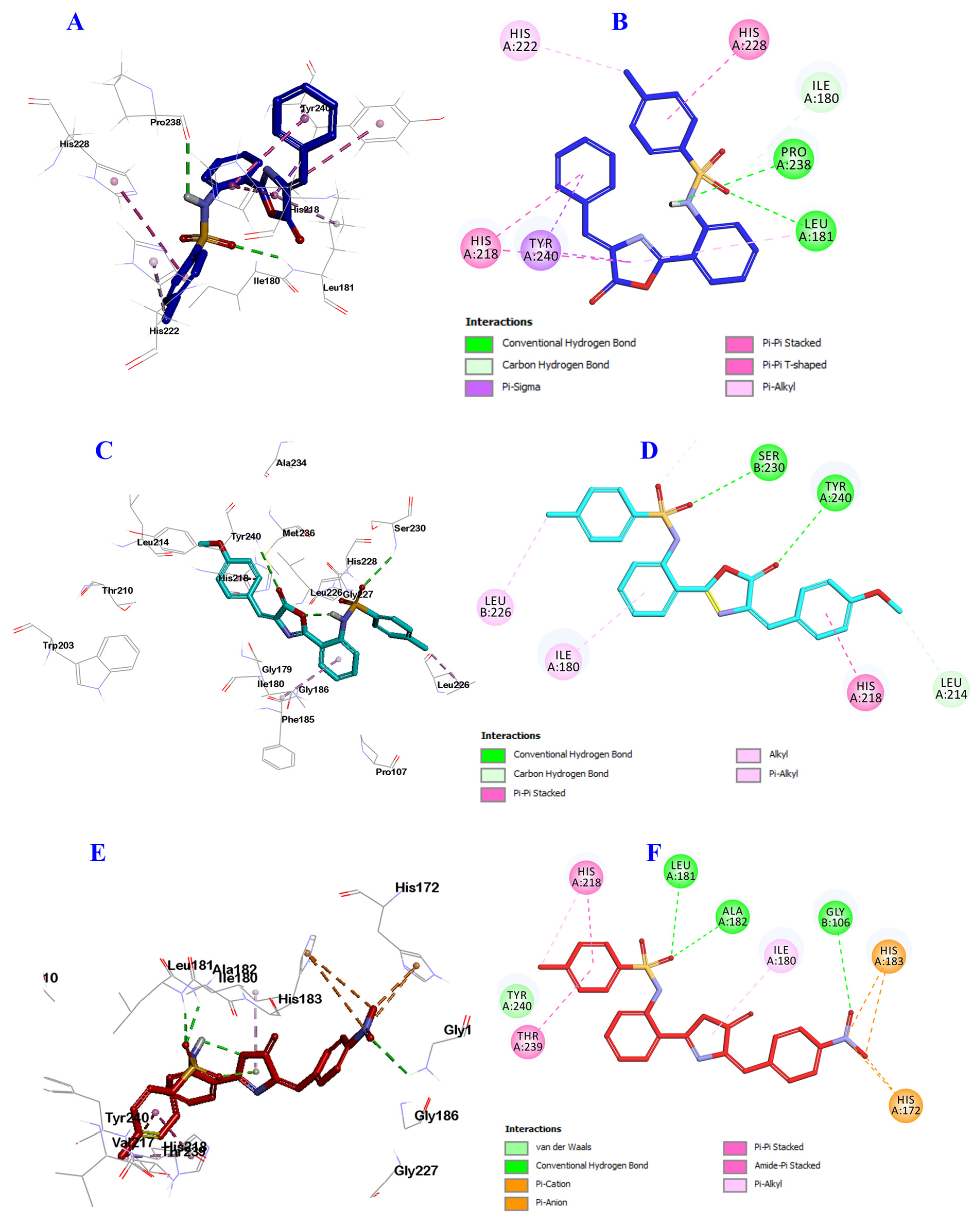
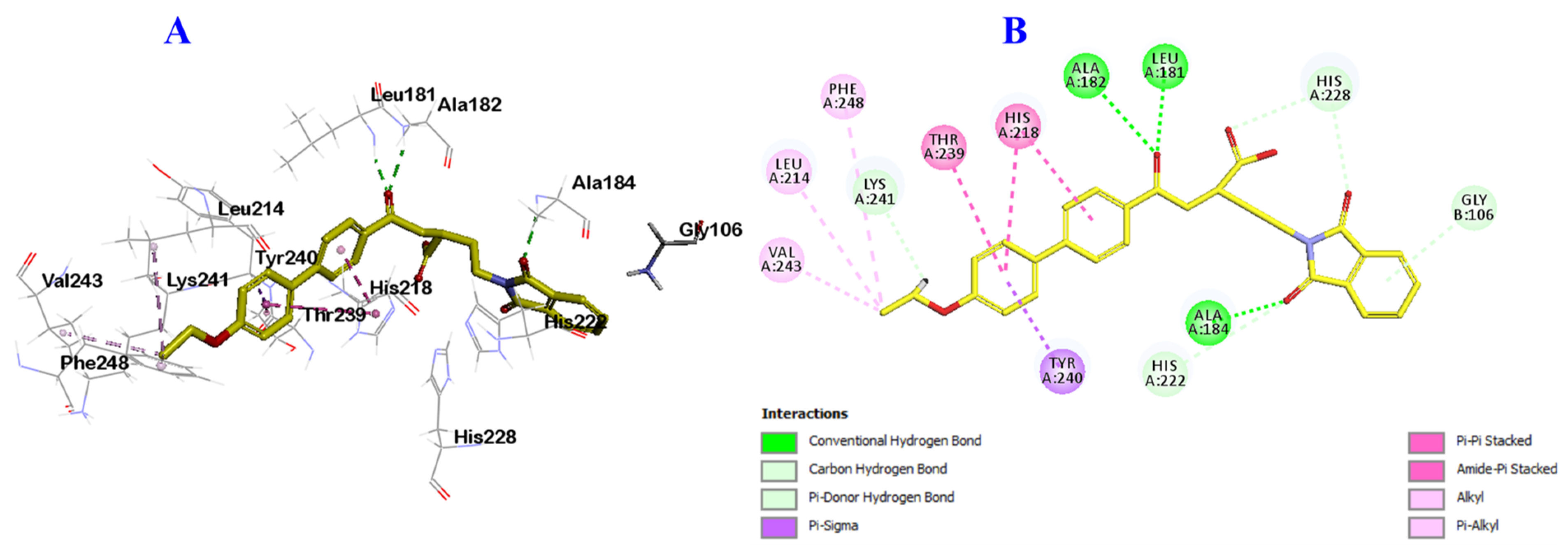
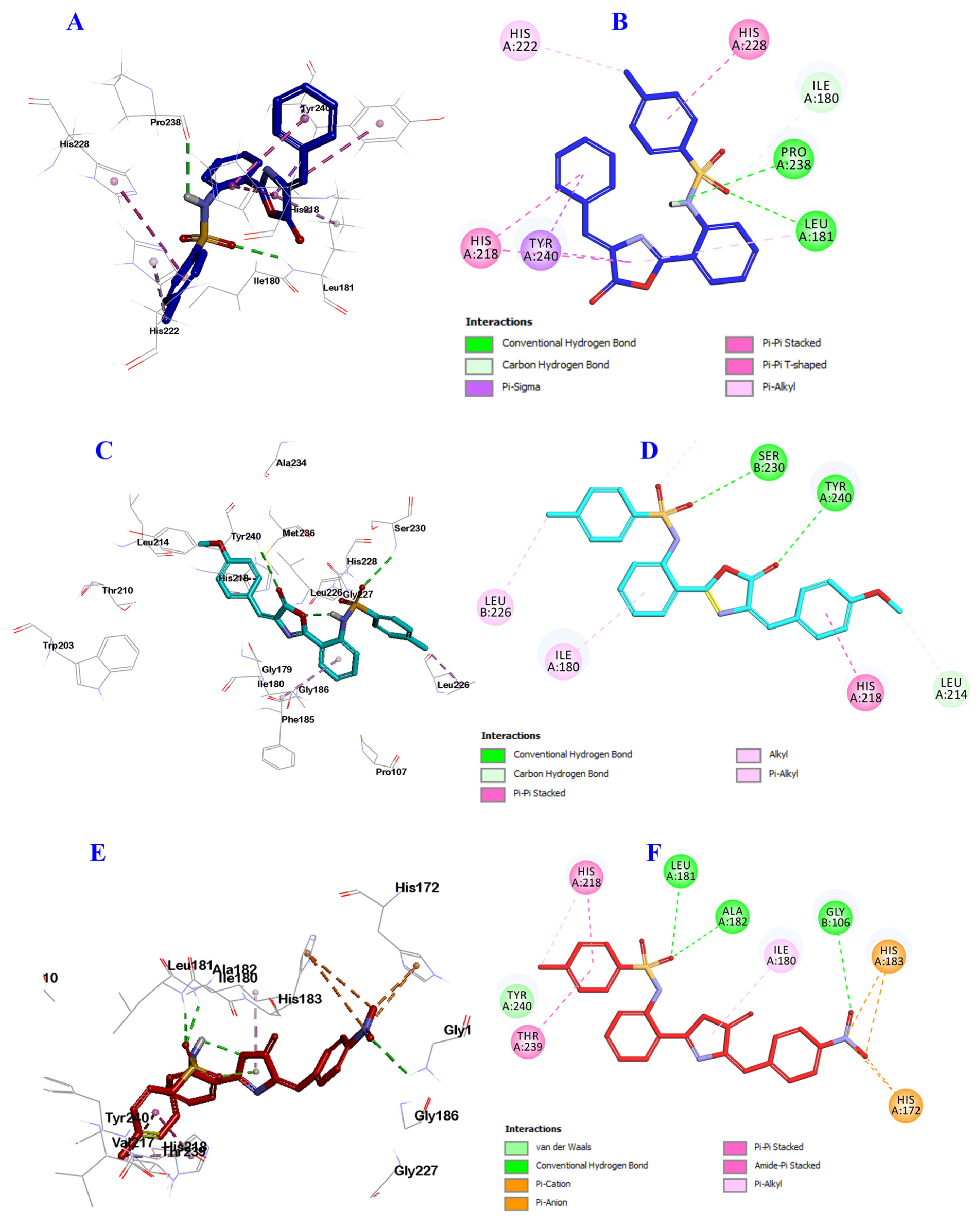
2.3. Docking Study into Pseudomonas aeruginosa QS Receptors
3. Materials and Methods
3.1. Chemistry
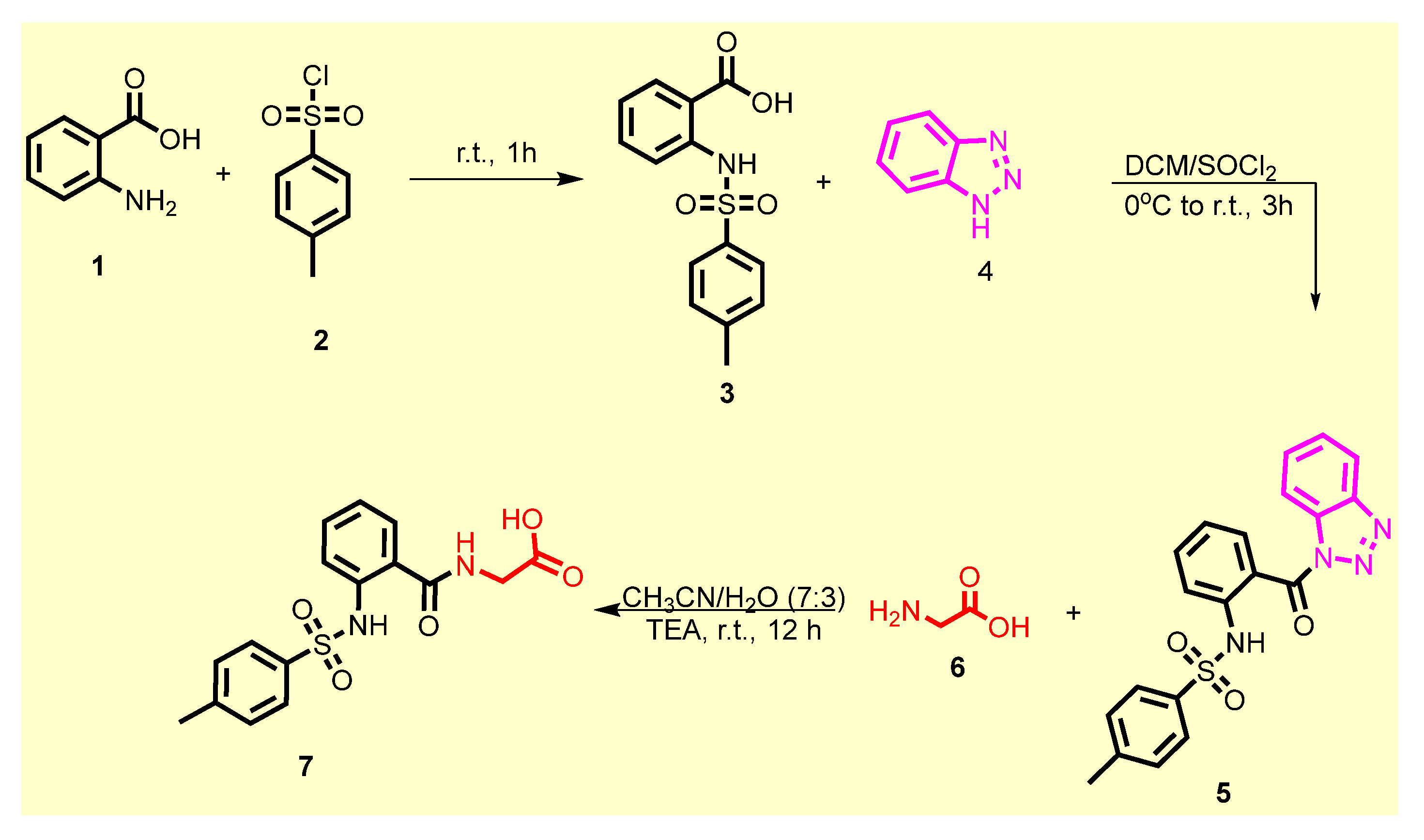
3.1.1. General Procedure for the Synthesis of 2-((4-Methylphenyl)sulfonamido)benzoic acid 3
3.1.2. General Procedure for the Synthesis of N-(2-(1H-Benzo[d][1,2,3]triazole -1-carbonyl) phenyl)-4-methylbenzenesulfonamide 5
3.1.3. General Procedure for the Synthesis of (2-((4-Methylphenyl)sulfonamido) benzoyl)glycine 7
3.1.4. General Procedure for the Synthesis of 9a–j
3.1.5. (E)-N-(2-(4-Benzylidene-5-oxo-4,5-dihydrooxazol-2-yl)phenyl)-4-methylbenzenesulfonamide 9a
3.1.6. (E)-N-(2-(4-(4-Methoxybenzylidene)-5-oxo-4,5-dihydrooxazol-2-yl)phenyl)-4-methylbenzenesulfonamide 9b
3.1.7. (E)-N-(2-(4-(2,5-diMethoxybenzylidene)-5-oxo-4,5-dihydrooxazol-2-yl)phenyl)-4-methylbenzenesulfonamide 9c
3.1.8. (E)-4-Methyl-N-(2-(5-oxo-4-(3,4,5-trimethoxybenzylidene)-4,5-dihydrooxazol-2-yl)phenyl)be-nzenesulfonamide 9d
3.1.9. (E)-N-(2-(4-(4-Chlorobenzylidene)-5-oxo-4,5-dihydrooxazol-2-yl)phenyl)-4-methylbenzenesulfonamide 9e
3.1.10. (E)-4-Methyl-N-(2-(4-(4-nitrobenzylidene)-5-oxo-4,5-dihydrooxazol-2-yl)phenyl)benzenesulfonamide 9f
3.1.11. (E)-N-(2-(4-(4-(Benzyloxy)benzylidene)-5-oxo-4,5-dihydrooxazol-2-yl)phenyl)-4-methylbenzenesulfonamide 9g
3.1.12. (E)-4-Methyl-N-(2-(4-(naphthalen-1-ylmethylene)-5-oxo-4,5-dihydrooxazol-2-yl)phenyl)benzenesulfonamide 9h
3.1.13. (E)-N-(2-(4-(Furan-2-ylmethylene)-5-oxo-4,5-dihydrooxazol-2-yl)phenyl)-4-methylbenzenesulfonamide 9i
3.1.14. (E)-4-Methyl-N-(2-(5-oxo-4-(thiophen-2-ylmethylene)-4,5-dihydrooxazol-2-yl)phenyl)benzenesulfonamide 9j
3.1.15. (E)-4-Methyl-N-(2-(5-oxo-4-(pyridin-4-ylmethylene)-4,5-dihydrooxazol-2-yl)phenyl)benzenesulfonamide 9k
3.2. Biological Activity
3.2.1. Evaluation of Antimicrobial and Anti-Virulence Activities
Determination of Minimum Inhibitory Concentration (MIC)
Excluding the Effect of Compounds on Bacterial Growth
Assay of Biofilm Formation
Assay of Protease Production
Assay of Hemolytic Activity
Quantification of Staphyloxanthin Pigment
Quantification of Pyocyanin Pigment
3.2.2. Evaluation of Antitumor Activities of Synthesized Compounds
Effect of Synthesized Compounds on Cellular Proliferation
Evaluation of Caspase-3/7 Activity
3.3. In Silico Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. P T Peer-Rev. J. Formul. Manag. 2015, 40, 277–283. [Google Scholar]
- Ibrahim, T.S.; Almalki, A.J.; Moustafa, A.H.; Allam, R.M.; Abuo-Rahma, G.E.-D.A.; El Subbagh, H.I.; Mohamed, M.F.A. Novel 1,2,4-oxadiazole-chalcone/oxime hybrids as potential antibacterial DNA gyrase inhibitors: Design, synthesis, ADMET prediction and molecular docking study. Bioorg. Chem. 2021, 111, 104885. [Google Scholar] [CrossRef] [PubMed]
- Hofny, H.A.; Mohamed, M.F.A.; Gomaa, H.A.M.; Abdel-Aziz, S.A.; Youssif, B.G.M.; El-koussi, N.A.; Aboraia, A.S. Design, synthesis, and antibacterial evaluation of new quinoline-1,3,4-oxadiazole and quinoline-1,2,4-triazole hybrids as potential inhibitors of DNA gyrase and topoisomerase IV. Bioorg. Chem. 2021, 112, 104920. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Misba, L.; Khan, A.U. Antibiotics versus biofilm: An emerging battleground in microbial communities. Antimicrob. Resist. Infect. Control 2019, 8, 1–10. [Google Scholar] [CrossRef]
- Uruén, C.; Chopo-Escuin, G.; Tommassen, J.; Mainar-Jaime, R.C.; Arenas, J. Biofilms as promoters of bacterial antibiotic resistance and tolerance. Antibiotics 2021, 10, 3. [Google Scholar] [CrossRef]
- Martins, P.; Jesus, J.; Santos, S.; Raposo, L.R.; Roma-Rodrigues, C.; Baptista, P.V.; Fernandes, A.R. Heterocyclic Anticancer Compounds: Recent Advances and the Paradigm Shift towards the Use of Nanomedicine’s Tool Box. Molecules 2015, 20, 16852–16891. [Google Scholar] [CrossRef]
- Eftekhari-Sis, B.; Zirak, M.; Akbari, A. Arylglyoxals in Synthesis of Heterocyclic Compounds. Chem. Rev. 2013, 113, 2958–3043. [Google Scholar] [CrossRef]
- Wan, Y.; Fang, G.; Chen, H.; Deng, X.; Tang, Z. Sulfonamide derivatives as potential anti-cancer agents and their SARs elucidation. Eur. J. Med. Chem. 2021, 226, 113837. [Google Scholar] [CrossRef]
- Fisk, J.S.; Mosey, R.A.; Tepe, J.J. The diverse chemistry of oxazol-5-(4H)-ones. Chem. Soc. Rev. 2007, 36, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.A.B.; Martinho, J.M.G.; Afonso, C.A.M. Synthesis of a Biologically Active Oxazol-5-(4H)-one via an Erlenmeyer–Plöchl Reaction. J. Chem. Educ. 2015, 92, 1543–1546. [Google Scholar] [CrossRef]
- Tandon, M.; Coffen, D.L.; Gallant, P.; Keith, D.; Ashwell, M.A. Potent and selective inhibitors of bacterial methionyl tRNA synthetase derived from an oxazolone–dipeptide scaffold. Bioorg. Med. Chem. Lett. 2004, 14, 1909–1911. [Google Scholar] [CrossRef]
- Mesaik, M.A.; Rahat, S.; Khan, K.M.; Zia, U.; Choudhary, M.I.; Murad, S.; Ismail, Z.; Attaur, R.; Ahmad, A. Synthesis and immunomodulatory properties of selected oxazolone derivatives. Bioorg. Med. Chem. 2004, 12, 2049–2057. [Google Scholar] [CrossRef]
- Hegazy, W.A.H.; Henaway, M. Hepatitis C virus pathogenesis: Serum IL-33 level indicates liver damage. Afr. J. Microbiol. Res. 2015, 9, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Mariappan, G.; Saha, B.P.; Datta, S.; Kumar, D.; Haldar, P.K. Design, synthesis and antidiabetic evaluation of oxazolone derivatives. J. Chem. Sci. 2011, 123, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Pinto, I.L.; West, A.; Debouck, C.M.; DiLella, A.G.; Gorniak, J.G.; O’Donnell, K.C.; O’Shannessy, D.J.; Patel, A.; Jarvest, R.L. Novel, selective mechanism-based inhibitors of the herpes proteases. Bioorg. Med. Chem. Lett. 1996, 6, 2467–2472. [Google Scholar] [CrossRef]
- Taile, V.; Hatzade, K.; Gaidhane, P.; Ingle, V. Synthesis and Biological Activity of 4-(4-Hydroxybenzylidene)-2-(substituted styryl) oxazol-5-ones and Their o-glucosides. Turk. J. Chem. 2009, 33, 295–305. [Google Scholar]
- Jat, L.; Mishra, R.; Pathak, D. Synthesis and anticancer activity of 4-Benzylidene-2-phenyloxazol-5 (4H)-one derivatives. Int. J. Pharm. Pharm. Sci. 2012, 4, 378–380. [Google Scholar]
- Hassanein, H.H.; Khalifa, M.M.; El-Samaloty, O.N.; El-Rahim, M.A.; Taha, R.A.; Magda; Ismail, M.F. Synthesis and biological evaluation of novel imidazolone derivatives as potential COX-2 inhibitors. Arch. Pharmacal. Res. 2008, 31, 562. [Google Scholar] [CrossRef] [PubMed]
- Witvrouw, M.; Pannecouque, C.; De Clercq, E.; Fernández-Alvarez, E.; Marco, J.L. Inhibition of Human Immunodeficiency Virus Type (HIV-1) Replication by some Diversely Functionalized Spirocyclopropyl Derivatives. Arch. Der Pharm. Int. J. Pharm. Med. Chem. 1999, 332, 163–166. [Google Scholar] [CrossRef]
- Tsukumo, Y.; Harada, D.; Manabe, H. Pharmacological Characterization of Itch-Associated Response Induced by Repeated Application of Oxazolone in Mice. J. Pharmacol. Sci. 2010, 113, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.M.; Mughal, U.R.; Khan, M.T.H.; Zia, U.; Perveen, S.; Iqbal Choudhary, M. Oxazolones: New tyrosinase inhibitors; synthesis and their structure–activity relationships. Bioorg. Med. Chem. 2006, 14, 6027–6033. [Google Scholar] [CrossRef]
- Hamidian, H.; Azizi, S. Synthesis of novel compounds containing morpholine and 5(4H)-oxazolone rings as potent tyrosinase inhibitors. Bioorg. Med. Chem. 2015, 23, 7089–7094. [Google Scholar] [CrossRef]
- Kiptoo, P.K.; Paudel, K.S.; Hammell, D.C.; Hamad, M.O.; Crooks, P.A.; Stinchcomb, A.L. In vivo evaluation of a transdermal codrug of 6-β-naltrexol linked to hydroxybupropion in hairless guinea pigs. Eur. J. Pharm. Sci. 2008, 33, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Tandel, R.; Mammen, D. Synthesis and Study of Some Compounds Containing Oxazolone Ring, Showing Biological Activity. Indian J. Chem. 2008, 47, 932–937. [Google Scholar] [CrossRef]
- Sánchez, C.; Méndez, C.; Salas, J.A. Indolocarbazole natural products: Occurrence, biosynthesis, and biological activity. Nat. Prod. Rep. 2006, 23, 1007–1045. [Google Scholar] [CrossRef] [PubMed]
- Rix, U.; Zheng, J.; Remsing Rix, L.L.; Greenwell, L.; Yang, K.; Rohr, J. The dynamic structure of jadomycin B and the amino acid incorporation step of its biosynthesis. J. Am. Chem. Soc. 2004, 126, 4496–4497. [Google Scholar] [CrossRef]
- Wookey, A.; Turner, P.J.; Greenhalgh, J.M.; Eastwood, M.; Clarke, J.; Sefton, C. AZD2563, a novel oxazolidinone: Definition of antibacterial spectrum, assessment of bactericidal potential and the impact of miscellaneous factors on activity in vitro. Clin. Microbiol. Infect. 2004, 10, 247–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, M.; Levine, L. Peptide Synthesis via Active Esters. IV. Racemization and Ring-Opening Reactions of Opitcally Active Oxazolones. J. Am. Chem. Soc. 1964, 86, 2918–2922. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Alsaid, M.S.; El-Gaby, M.S.A.; Safwat, N.A.; Elaasser, M.M.; Soliman, A.M. Biological evaluation of some new N-(2,6-dimethoxypyrimidinyl) thioureido benzenesulfonamide derivatives as potential antimicrobial and anticancer agents. Eur. J. Med. Chem. 2016, 124, 299–310. [Google Scholar] [CrossRef]
- Alaoui, S.; Dufies, M.; Driowya, M.; Demange, L.; Bougrin, K.; Robert, G.; Auberger, P.; Pagès, G.; Benhida, R. Synthesis and anti-cancer activities of new sulfonamides 4-substituted-triazolyl nucleosides. Bioorg. Med. Chem. Lett. 2017, 27, 1989–1992. [Google Scholar] [CrossRef]
- Durgun, M.; Turkmen, H.; Zengin, G.; Zengin, H.; Koyunsever, M.; Koyuncu, I. Synthesis, characterization, in vitro cytotoxicity and antimicrobial investigation and evaluation of physicochemical properties of novel 4-(2-methylacetamide)benzenesulfonamide derivatives. Bioorg. Chem. 2017, 70, 163–172. [Google Scholar] [CrossRef]
- Dai, H.-X.; Stepan, A.F.; Plummer, M.S.; Zhang, Y.-H.; Yu, J.-Q. Divergent C–H Functionalizations Directed by Sulfonamide Pharmacophores: Late-Stage Diversification as a Tool for Drug Discovery. J. Am. Chem. Soc. 2011, 133, 7222–7228. [Google Scholar] [CrossRef] [PubMed]
- Gul, H.I.; Tugrak, M.; Sakagami, H.; Taslimi, P.; Gulcin, I.; Supuran, C.T. Synthesis and bioactivity studies on new 4-(3-(4-Substitutedphenyl)-3a,4-dihydro-3H-indeno[1,2-c]pyrazol-2-yl) benzenesulfonamides. J. Enzym. Inhib. Med. Chem. 2016, 31, 1619–1624. [Google Scholar] [CrossRef] [Green Version]
- Lal, J.; Gupta, S.K.; Thavaselvam, D.; Agarwal, D.D. Biological activity, design, synthesis and structure activity relationship of some novel derivatives of curcumin containing sulfonamides. Eur. J. Med. Chem. 2013, 64, 579–588. [Google Scholar] [CrossRef]
- Lu, X.-Y.; Wang, Z.-C.; Ren, S.-Z.; Shen, F.-Q.; Man, R.-J.; Zhu, H.-L. Coumarin sulfonamides derivatives as potent and selective COX-2 inhibitors with efficacy in suppressing cancer proliferation and metastasis. Bioorg. Med. Chem. Lett. 2016, 26, 3491–3498. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Casini, A.; Scozzafava, A. Protease inhibitors of the sulfonamide type: Anticancer, antiinflammatory, and antiviral agents. Med. Res. Rev. 2003, 23, 535–558. [Google Scholar] [CrossRef]
- Scozzafava, A.; Owa, T.; Mastrolorenzo, A.; Supuran, C.T. Anticancer and antiviral sulfonamides. Curr. Med. Chem. 2003, 10, 925–953. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Guo, Y.; Ma, X.; Zhu, R.; Tian, C.; Zhang, Z.; Wang, X.; Ma, Z.; Liu, J. Design, synthesis and pharmacological evaluation of (E)-3,4-dihydroxy styryl sulfonamides derivatives as multifunctional neuroprotective agents against oxidative and inflammatory injury. Bioorg. Med. Chem. 2013, 21, 5589–5597. [Google Scholar] [CrossRef]
- Qin, H.-L.; Zhang, Z.-W.; Lekkala, R.; Alsulami, H.; Rakesh, K.P. Chalcone hybrids as privileged scaffolds in antimalarial drug discovery: A key review. Eur. J. Med. Chem. 2020, 193, 112215. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Zhao, C.; Rakesh, K.P.; Ravidar, L.; Fang, W.-Y.; Qin, H.-L. Pharmaceutical and medicinal significance of sulfur (SVI)-Containing motifs for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 162, 679–734. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.L.; Chen, L.; Lanning, M.E.; Fletcher, S. Expanding the Cancer Arsenal with Targeted Therapies: Disarmament of the Antiapoptotic Bcl-2 Proteins by Small Molecules. J. Med. Chem. 2017, 60, 821–838. [Google Scholar] [CrossRef]
- Ballatore, C.; Huryn, D.M.; Smith, A.B., III. Carboxylic Acid (Bio)Isosteres in Drug Design. ChemMedChem 2013, 8, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Ammazzalorso, A.; De Filippis, B.; Giampietro, L.; Amoroso, R. N-acylsulfonamides: Synthetic routes and biological potential in medicinal chemistry. Chem. Biol. Drug Des. 2017, 90, 1094–1105. [Google Scholar] [CrossRef]
- Parsonnet, J. Bacterial infection as a cause of cancer. Environ. Health Perspect. 1995, 103 (Suppl. S8), 263–268. [Google Scholar] [CrossRef] [Green Version]
- Aldawsari, M.F.; Khafagy, E.S.; Saqr, A.A.; Alalaiwe, A.; Abbas, H.A.; Shaldam, M.A.; Hegazy, W.A.H.; Goda, R.M. Tackling Virulence of Pseudomonas aeruginosa by the Natural Furanone Sotolon. Antibiotics 2021, 10, 871. [Google Scholar] [CrossRef]
- Hegazy, W.A.H.; Khayat, M.T.; Ibrahim, T.S.; Nassar, M.S.; Bakhrebah, M.A.; Abdulaal, W.H.; Alhakamy, N.A.; Bendary, M.M. Repurposing Anti-diabetic Drugs to Cripple Quorum Sensing in Pseudomonas aeruginosa. Microorganisms 2020, 8, 1285. [Google Scholar] [CrossRef] [PubMed]
- Saqr, A.A.; Aldawsari, M.F.; Khafagy, E.S.; Shaldam, M.A.; Hegazy, W.A.H.; Abbas, H.A. A Novel Use of Allopurinol as A Quorum-Sensing Inhibitor in Pseudomonas aeruginosa. Antibiotics 2021, 10, 1385. [Google Scholar] [CrossRef] [PubMed]
- Abbas, H.A.; Hegazy, W.A.H. Repurposing anti-diabetic drug “Sitagliptin” as a novel virulence attenuating agent in Serratia marcescens. PLoS ONE 2020, 15, e0231625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegazy, W.A.H.; Rajab, A.A.H.; Abu Lila, A.S.; Abbas, H.A. Anti-diabetics and antimicrobials: Harmony of mutual interplay. World J. Diabetes 2021, 12, 1832–1855. [Google Scholar] [CrossRef] [PubMed]
- Khayyat, A.N.; Abbas, H.A.; Khayat, M.T.; Shaldam, M.A.; Askoura, M.; Asfour, H.Z.; Khafagy, E.S.; Abu Lila, A.S.; Allam, A.N.; Hegazy, W.A.H. Secnidazole Is a Promising Imidazole Mitigator of Serratia marcescens Virulence. Microorganisms 2021, 9, 2333. [Google Scholar] [CrossRef]
- Khayyat, A.N.; Abbas, H.A.; Mohamed, M.F.A.; Asfour, H.Z.; Khayat, M.T.; Ibrahim, T.S.; Youns, M.; Khafagy, E.-S.; Abu Lila, A.S.; Safo, M.K.; et al. Not Only Antimicrobial: Metronidazole Mitigates the Virulence of Proteus mirabilis Isolated from Macerated Diabetic Foot Ulcer. Appl. Sci. 2021, 11, 6847. [Google Scholar] [CrossRef]
- Khayyat, A.N.; Hegazy, W.A.H.; Shaldam, M.A.; Mosbah, R.; Almalki, A.J.; Ibrahim, T.S.; Khayat, M.T.; Khafagy, E.S.; Soliman, W.E.; Abbas, H.A. Xylitol Inhibits Growth and Blocks Virulence in Serratia marcescens. Microorganisms 2021, 9, 1083. [Google Scholar] [CrossRef]
- Hegazy, W.A.H.; Khayat, M.T.; Ibrahim, T.S.; Youns, M.; Mosbah, R.; Soliman, W.E. Repurposing of antidiabetics as Serratia marcescens virulence inhibitors. Braz. J. Microbiol. 2021, 52, 627–638. [Google Scholar] [CrossRef]
- Abbas, H.A.; Hegazy, W.A.H. Targeting the virulence factors of Serratia marcescens by ambroxol. Roum. Arch. Microbiol. Immunol. 2017, 76, 27–32. [Google Scholar]
- Askoura, M.; Youns, M.; Hegazy, W.A.H. Investigating the influence of iron on Campylobacter jejuni transcriptome in response to acid stress. Microb. Pathog. 2020, 138, 103777. [Google Scholar] [CrossRef]
- Askoura, M.; Hegazy, W.A.H. Ciprofloxacin interferes with Salmonella Typhimurium intracellular survival and host virulence through repression of Salmonella pathogenicity island-2 (SPI-2) genes expression. Pathog. Dis. 2020, 78, ftaa011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Suo, Y.; Zhang, D.; Jin, F.; Zhao, H.; Shi, C. Genetic and Virulent Difference Between Pigmented and Non-pigmented Staphylococcus aureus. Front. Microbiol. 2018, 9, 598. [Google Scholar] [CrossRef]
- Youns, M.; Askoura, M.; Abbas, H.A.; Attia, G.H.; Khayyat, A.N.; Goda, R.M.; Almalki, A.J.; Khafagy, E.S.; Hegazy, W.A.H. Celastrol Modulates Multiple Signaling Pathways to Inhibit Proliferation of Pancreatic Cancer via DDIT3 and ATF3 Up-Regulation and RRM2 and MCM4 Down-Regulation. Onco Targets 2021, 14, 3849–3860. [Google Scholar] [CrossRef] [PubMed]
- Bendary, M.M.; Ibrahim, D.; Mosbah, R.A.; Mosallam, F.; Hegazy, W.A.H.; Awad, N.F.S.; Alshareef, W.A.; Alomar, S.Y.; Zaitone, S.A.; Abd El-Hamid, M.I. Thymol Nanoemulsion: A New Therapeutic Option for Extensively Drug Resistant Foodborne Pathogens. Antibiotics 2020, 10, 25. [Google Scholar] [CrossRef]
- Morales, R.; Perrier, S.; Florent, J.-M.; Beltra, J.; Dufour, S.; De Mendez, I.; Manceau, P.; Tertre, A.; Moreau, F.; Compere, D.; et al. Crystal Structures of Novel Non-peptidic, Non-zinc Chelating Inhibitors Bound to MMP-12. J. Mol. Biol. 2004, 341, 1063–1076. [Google Scholar] [CrossRef]
- Kamal, A.; Reddy, J.S.; Bharathi, E.V.; Dastagiri, D. Base-free monosulfonylation of amines using tosyl or mesyl chloride in water. Tetrahedron Lett. 2008, 49, 348–353. [Google Scholar] [CrossRef]
- Vishwa, B.; Moin, A.; Gowda, D.V.; Rizvi, S.M.D.; Hegazy, W.A.H.; Abu Lila, A.S.; Khafagy, E.S.; Allam, A.N. Pulmonary Targeting of Inhalable Moxifloxacin Microspheres for Effective Management of Tuberculosis. Pharmaceutics 2021, 13, 79. [Google Scholar] [CrossRef]
- Agha, K.A.; Abo-Dya, N.E.; Ibrahim, T.S.; Abdel-Aal, E.H.; Hegazy, W.A.H. Benzotriazole-Mediated Synthesis and Antibacterial Activity of Novel N-Acylcephalexins. Sci. Pharm. 2016, 84, 484–496. [Google Scholar] [CrossRef] [Green Version]
- Hegazy, W.A.H.; Abbas, H.A. Evaluation of the role of SsaV ‘Salmonella pathogenicity island-2 dependent type III secretion system components on the virulence behavior of Salmonella enterica serovar Typhimurium. Afr. J. Biotechnol. 2017, 16, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Berlutti, F.; Frioni, A.; Natalizi, T.; Pantanella, F.; Valenti, P. Influence of sub-inhibitory antibiotics and flow condition on Staphylococcus aureus ATCC 6538 biofilm development and biofilm growth rate: BioTimer assay as a study model. J. Antibiot. 2014, 67, 763–769. [Google Scholar] [CrossRef]
- El-Hamid, A.; Marwa, I.; Y El-Naenaeey, E.S.; Hegazy, W.A.H.; Mosbah, R.A.; Nassar, M.S.; Bakhrebah, M.A.; Abdulaal, W.H.; Alhakamy, N.A.; Bendary, M.M. Promising Antibiofilm Agents: Recent Breakthrough against Biofilm Producing Methicillin-Resistant Staphylococcus aureus. Antibiotics 2020, 9, 667. [Google Scholar] [CrossRef]
- Askoura, M.; Almalki, A.J.; Lila, A.S.A.; Almansour, K.; Alshammari, F.; Khafagy, E.-S.; Ibrahim, T.S.; Hegazy, W.A.H. Alteration of Salmonella enterica Virulence and Host Pathogenesis through Targeting sdiA by Using the CRISPR-Cas9 System. Microorganisms 2021, 9, 2564. [Google Scholar] [CrossRef] [PubMed]
- Aldawsari, M.F.; Alalaiwe, A.; Khafagy, E.S.; Al Saqr, A.; Alshahrani, S.M.; Alsulays, B.B.; Alshehri, S.; Abu Lila, A.S.; Danish Rizvi, S.M.; Hegazy, W.A.H. Efficacy of SPG-ODN 1826 Nanovehicles in Inducing M1 Phenotype through TLR-9 Activation in Murine Alveolar J774A.1 Cells: Plausible Nano-Immunotherapy for Lung Carcinoma. Int. J. Mol. Sci. 2021, 22, 6833. [Google Scholar] [CrossRef] [PubMed]
- Youns, M.; Hegazy, W.A.H. The Natural Flavonoid Fisetin Inhibits Cellular Proliferation of Hepatic, Colorectal, and Pancreatic Cancer Cells through Modulation of Multiple Signaling Pathways. PLoS ONE 2017, 12, e0169335. [Google Scholar] [CrossRef] [PubMed]
- Al Saqr, A.; Khafagy, E.S.; Alalaiwe, A.; Aldawsari, M.F.; Alshahrani, S.M.; Anwer, M.K.; Khan, S.; Lila, A.S.A.; Arab, H.H.; Hegazy, W.A.H. Synthesis of Gold Nanoparticles by Using Green Machinery: Characterization and In Vitro Toxicity. Nanomaterials 2021, 11, 808. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.S.; Moustafa, A.H.; Almalki, A.J.; Allam, R.M.; Althagafi, A.; Md, S.; Mohamed, M.F.A. Novel chalcone/aryl carboximidamide hybrids as potent anti-inflammatory via inhibition of prostaglandin E2 and inducible NO synthase activities: Design, synthesis, molecular docking studies and ADMET prediction. J. Enzym. Inhib. Med. Chem. 2021, 36, 1067–1078. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Gram-Positive Bacteria | Gram-Negative Bacteria | Fungi | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Staphylococcus aureus ATCC 6538 | Staphylococcus epidermidis ATCC 12228 | Micrococcus spp. ATCC 10240 | Pseudomonas aeruginosae ATCC 47085 | Klebsiella pneumoniae ATCC 27736 | Salmonella typhimurium ATCC 14028 | Escherichia coli ATCC 10536 | Aspergillus niger ATCC 16404 | Candida albicans ATCC 10231 | |
| 9a | 4 | 4 | 4 | 8 | 4 | 4 | 2 | 32 | 16 |
| 9b | 2 | 1 | 1 | 4 | 4 | 4 | 2 | >32 | >32 |
| 9c | 8 | 4 | 4 | 16 | 8 | 8 | 4 | 8 | 4 |
| 9d | 4 | 2 | 2 | 32 | 16 | 16 | 4 | >32 | >32 |
| 9e | 2 | 2 | 2 | 8 | 2 | 4 | 4 | 16 | 8 |
| 9f | 2 | 2 | 2 | 4 | 2 | 2 | 4 | >32 | >32 |
| 9g | 2 | 2 | 2 | 16 | 8 | 16 | 2 | >32 | >32 |
| 9h | >32 | >32 | >32 | >32 | >32 | >32 | >32 | 4 | 2 |
| 9i | 2 | 2 | 2 | >32 | >32 | >32 | >32 | >32 | >32 |
| 9j | 16 | 8 | 8 | >32 | >32 | >32 | >32 | >32 | >32 |
| 9k | 4 | 4 | 2 | >32 | >32 | >32 | 16 | >32 | >32 |
| Amoxycillin | 0.5 | 0.5 | 0.25 | 2 | 1 | 1 | 0.5 | - | - |
| Cefotaxime | 0.5 | 0.25 | 0.25 | 1 | 0.5 | 1 | 0.125 | - | - |
| Sulphamethoxazole | 2 | 2 | 1 | 4 | 2 | 2 | 1 | - | - |
| Nystatin | - | - | - | - | - | - | - | 4 | 4 |
| Compounds | IC50 (µg/mL) | |||
|---|---|---|---|---|
| HepG-2 | Panc-1 | BxPC-3 | HPDE | |
| 9a | 10.96 ± 2.39 | 11.95 ± 0.76 | 14.39 ± 0.73 | >50 mg/mL |
| 9b | 8.53 ± 1.10 | 13.63 ± 1.16 | 14.88 ± 4.19 | >50 mg/mL |
| 9c | 19.08 ± 3.23 | 15.13 ± 4.13 | 21.47 ± 1.08 | >50 mg/mL |
| 9d | 22.71 ± 2.30 | 25.32 ± 1.91 | 18.05 ± 3.56 | >50 mg/mL |
| 9e | 15.04 ± 2.89 | 12.15 ± 1.29 | 13.28 ± 1.57 | >50 mg/mL |
| 9f | 6.39 ± 1.25 | 12.60 ± 1.20 | 14.18 ± 1.87 | >50 mg/mL |
| 9g | 32.53 ± 1.45 | 29.26 ± 4.23 | 25.70 ± 4.89 | >50 mg/mL |
| 9h | 17.34 ± 0.96 | 14.72 ± 2.87 | 12.60 ± 0.62 | >50 mg/mL |
| 9i | 27.017 ± 5.32 | 16.13 ± 1.820 | 21.83 ± 2.98 | >50 mg/mL |
| 9j | 25.54 ± 0.144 | 16.04 ± 3.18 | 21.42 ± 2.15 | >50 mg/mL |
| 9k | 33.11 ± 5.12 | 19.878 ± 3.35 | 7.27 ± 1.49 | >50 mg/mL |
| Doxorubicin | 5.11 ± 0.98 | 6.90 ± 0.93 | 7.31 ± 1.12 | >50 mg/mL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almalki, A.J.; Ibrahim, T.S.; Taher, E.S.; Mohamed, M.F.A.; Youns, M.; Hegazy, W.A.H.; Al-Mahmoudy, A.M.M. Synthesis, Antimicrobial, Anti-Virulence and Anticancer Evaluation of New 5(4H)-Oxazolone-Based Sulfonamides. Molecules 2022, 27, 671. https://doi.org/10.3390/molecules27030671
Almalki AJ, Ibrahim TS, Taher ES, Mohamed MFA, Youns M, Hegazy WAH, Al-Mahmoudy AMM. Synthesis, Antimicrobial, Anti-Virulence and Anticancer Evaluation of New 5(4H)-Oxazolone-Based Sulfonamides. Molecules. 2022; 27(3):671. https://doi.org/10.3390/molecules27030671
Chicago/Turabian StyleAlmalki, Ahmad J., Tarek S. Ibrahim, Ehab S. Taher, Mamdouh F. A. Mohamed, Mahmoud Youns, Wael A. H. Hegazy, and Amany M. M. Al-Mahmoudy. 2022. "Synthesis, Antimicrobial, Anti-Virulence and Anticancer Evaluation of New 5(4H)-Oxazolone-Based Sulfonamides" Molecules 27, no. 3: 671. https://doi.org/10.3390/molecules27030671
APA StyleAlmalki, A. J., Ibrahim, T. S., Taher, E. S., Mohamed, M. F. A., Youns, M., Hegazy, W. A. H., & Al-Mahmoudy, A. M. M. (2022). Synthesis, Antimicrobial, Anti-Virulence and Anticancer Evaluation of New 5(4H)-Oxazolone-Based Sulfonamides. Molecules, 27(3), 671. https://doi.org/10.3390/molecules27030671