
Disrupting the MAD2L2-Rev1 Complex Enhances Cell Death upon DNA Damage


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
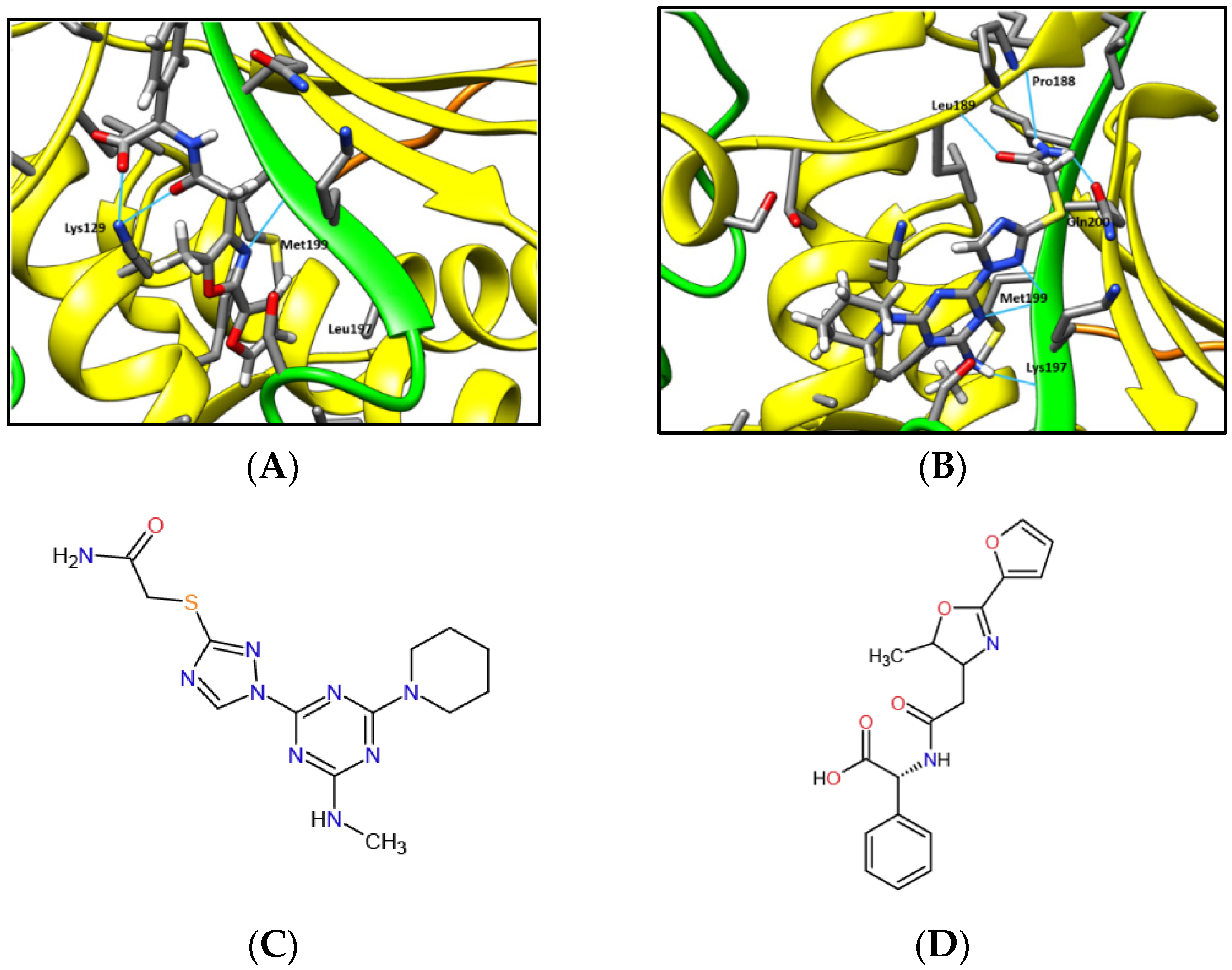
2.1. MAD2L2-MAD2L2-Rev1 Model and Docking Simulations
2.2. Both c#2 and c#3 Sensitize Cells to Cisplatin
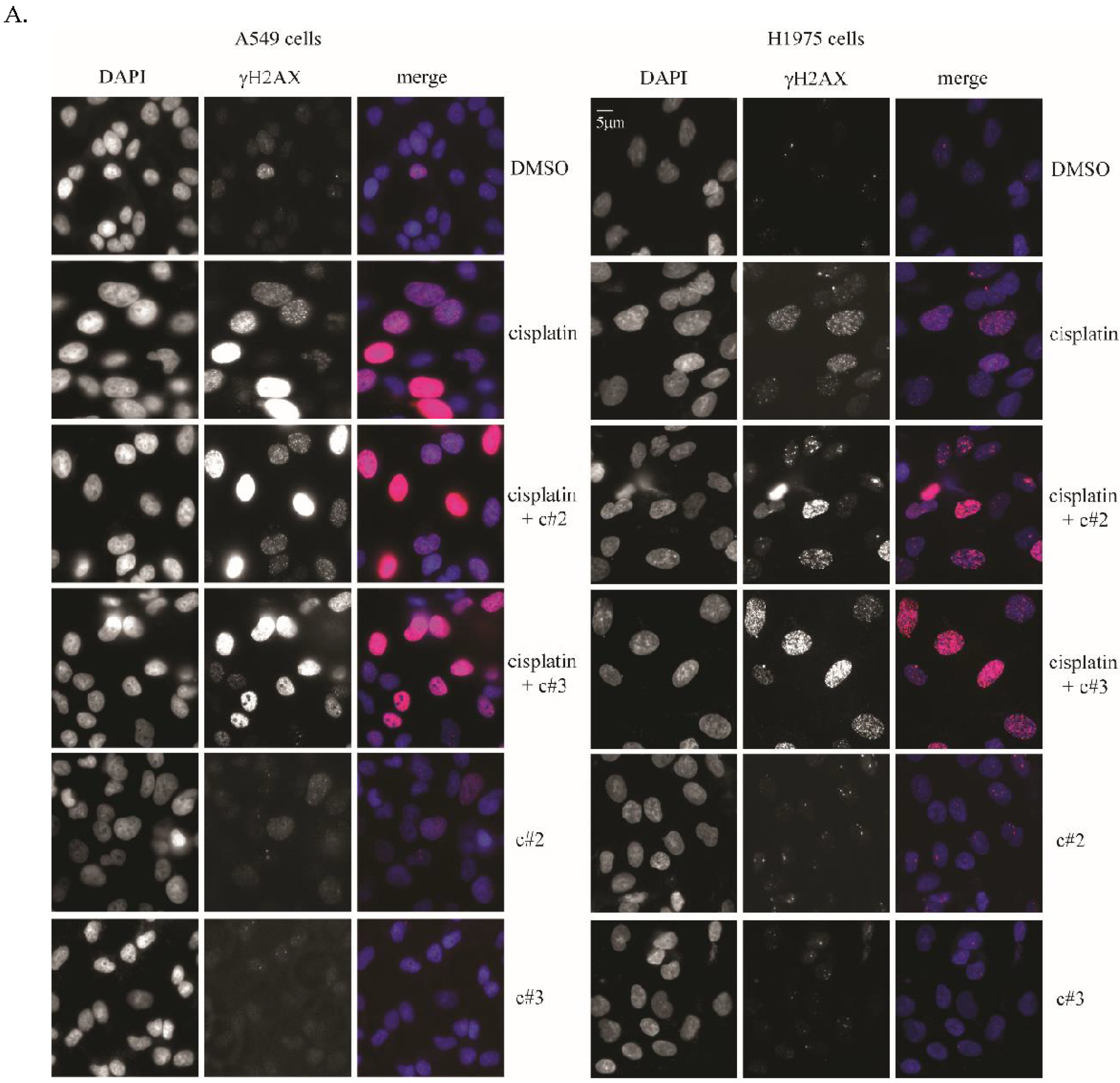
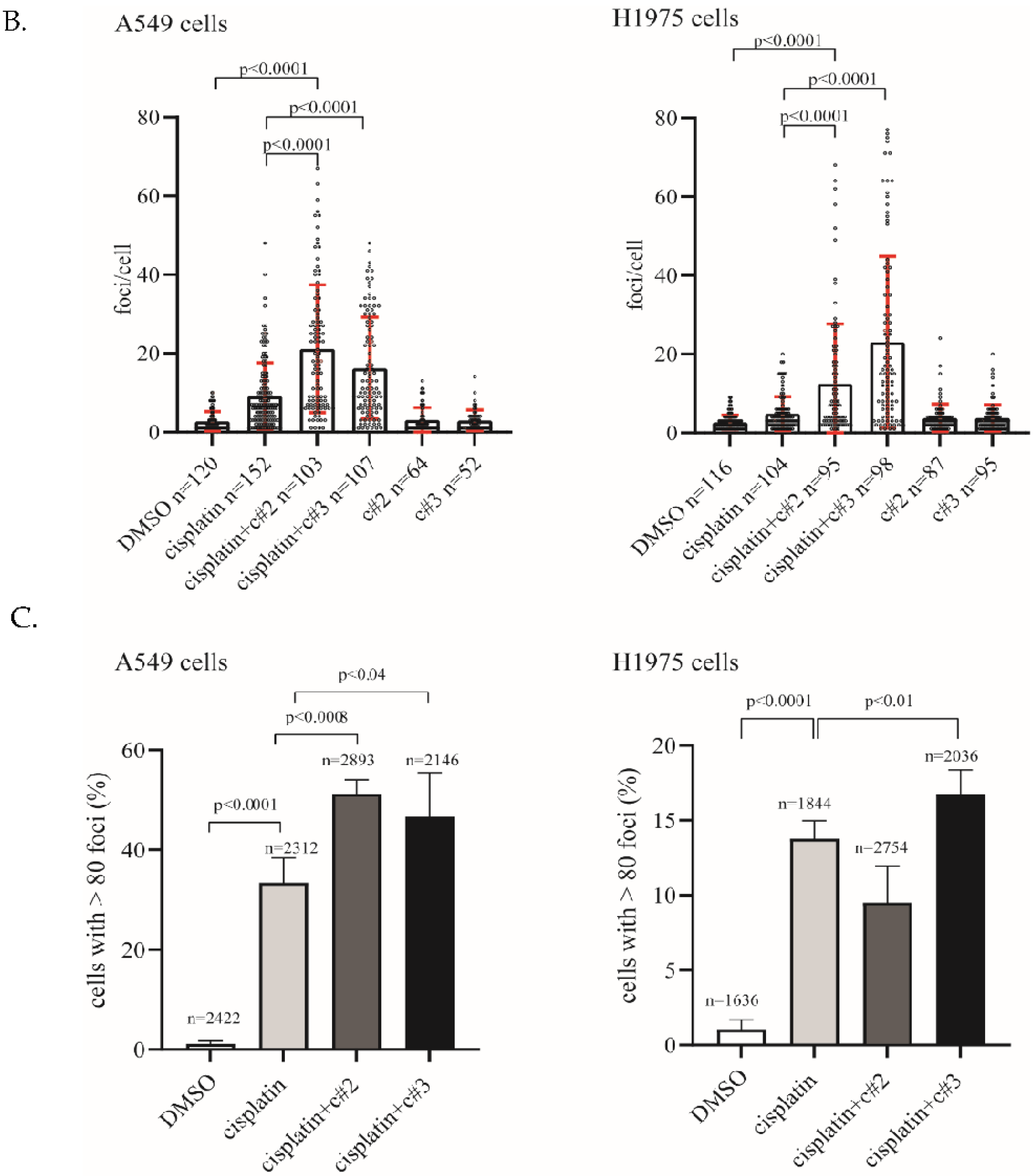
2.3. DNA Damage Is Elevated after Co-Treatment of Cisplatin Together with c#2 or c#3
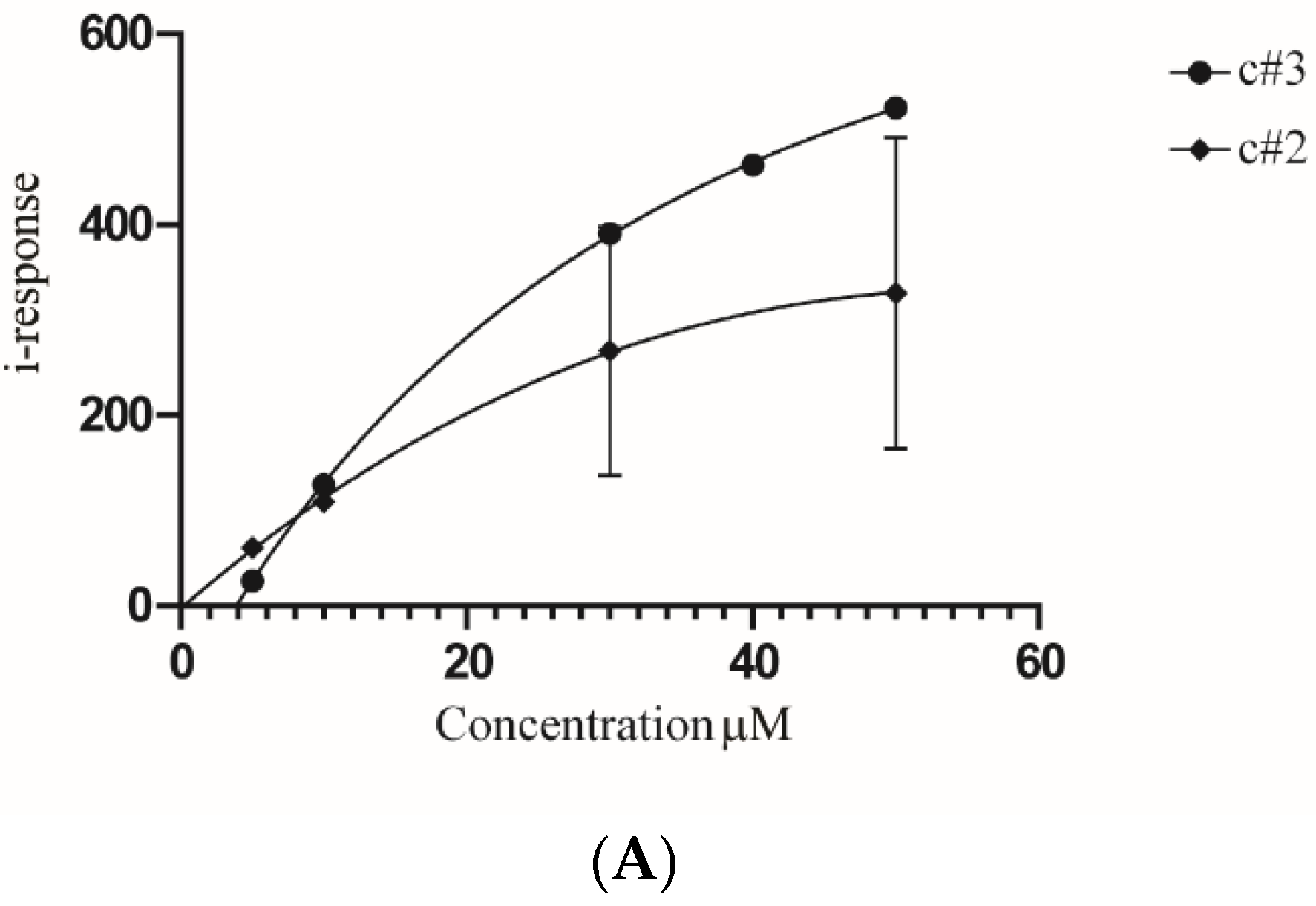
2.4. MAD2L2-Rev1 Interaction Is Reduced after Exposure to c#2 or c#3
3. Discussion
4. Materials and Methods
4.1. Generating MAD2L2-MAD2L2-Rev1 Model
4.2. Docking Simulations
4.3. Protein Induction, Purification and Field Effect Biosensing (FEB)
4.4. Cell Culture, Plasmids and Transfections
4.5. Colony Survival Assay
4.6. Western Blot and Co-Immunoprecipitation
4.7. Immunofluorescence
4.8. Cellular Thermal Shift Assay (CETSA)
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Lippard, S.J. Direct cellular responses to platinum-induced DNA damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chatterjee, N.; Hemann, M.T.; Walker, G.C. Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet. 2017, 13, e1006842. [Google Scholar] [CrossRef]
- Xie, K.; Doles, J.; Hemann, M.T.; Walkera, G.C. Error-prone translesion synthesis mediates acquired chemoresistance. Proc. Natl. Acad. Sci. USA 2010, 107, 20792–20797. [Google Scholar] [CrossRef]
- Ghosal, G.; Chen, J. DNA damage tolerance: A double-edged sword guarding the genome. Transl. Cancer Res. 2013, 2, 107–129. [Google Scholar] [CrossRef]
- Chang, D.J.; Cimprich, K.A. DNA damage tolerance: When it’s OK to make mistakes. Nat. Chem. Biol. 2009, 5, 82–90. [Google Scholar] [CrossRef]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef]
- Jansen, J.G.; Tsaalbi-Shtylik, A.; de Wind, N. Roles of mutagenic translesion synthesis in mammalian genome stability, health and disease. DNA Repair 2015, 29, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.A.; Korzhnev, D.M. The Rev1-Polζ Translesion Synthesis Mutasome: Structure, Interactions and Inhibition, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; Volume 45, ISBN 9780128173961. [Google Scholar]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.A.; Vassel, F.-M.; Chatterjee, N.; D’Souza, S.; Li, Y.; Hao, B.; Hemann, M.T.; Walker, G.C.; Korzhnev, D.M. Rev7 dimerization is important for assembly and function of the Rev1/Polζ translesion synthesis complex. Proc. Natl. Acad. Sci. USA 2018, 115, E8191–E8200. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, S.D.; Kokoska, R.J.; Chilkova, O.; Welch, C.M.; Johansson, E.; Burgers, P.M.J.; Kunkel, T.A. Enzymatic switching for efficient and accurate translesion DNA replication. Nucleic Acids Res. 2004, 32, 4665–4675. [Google Scholar] [CrossRef][Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokol, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 1999, 399, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Bluteau, D.; Masliah-Planchon, J.; Clairmont, C.; Rousseau, A.; Ceccaldi, R.; D’Enghien, C.D.; Bluteau, O.; Cuccuini, W.; Gachet, S.; de Latour, R.P.; et al. Biallelic inactivation ofREV7 is associated with Fanconi anemia. J. Clin. Investig. 2017, 127, 1117. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wojtaszek, J.L.; Chatterjee, N.; Najeeb, J.; Ramos, A.; Lee, M.; Bian, K.; Xue, J.Y.; Fenton, B.A.; Park, H.; Li, D.; et al. A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 2019, 178, 152–159. [Google Scholar] [CrossRef]
- Niimi, K.; Murakumo, Y.; Watanabe, N.; Kato, T.; Mii, S.; Enomoto, A.; Asai, M.; Asai, N.; Yamamoto, E.; Kajiyama, H.; et al. Suppression of REV7 enhances cisplatin sensitivity in ovarian clear cell carcinoma cells. Cancer Sci. 2014, 105, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Vassel, F.; Bian, K.; Walker, G.C.; Hemann, M.T. Rev7 loss alters cisplatin response and increases drug efficacy in chemotherapy-resistant lung cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 28922–28924. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Ichinoe, M.; Yoshida, K.; Nakazato, Y.; Saito, S.; Satoh, M.; Nakada, N.; Sanoyama, I.; Umezawa, A.; Numata, Y.; et al. Inactivation of REV7 enhances chemosensitivity and overcomes acquired chemoresistance in testicular germ cell tumors. Cancer Lett. 2020, 489, 100–110. [Google Scholar] [CrossRef]
- Wang, X.; Pernicone, N.; Pertz, L.; Hua, D.; Zhang, T.; Listovsky, T.; Xie, W. REV7 has a dynamic adaptor region to accommodate small GTPase RAN/Shigella IpaB ligands and its activity is regulated by RanGTP/GDP switch. J. Biol. Chem. 2019, 294, 15733–15742. [Google Scholar] [CrossRef]
- Hara, K.; Taharazako, S.; Ikeda, M.; Fujita, H.; Mikami, Y.; Kikuchi, S.; Hishiki, A.; Yokoyama, H.; Ishikawa, Y.; Kanno, S.; et al. Dynamic feature of mitotic arrest deficient 2-like protein 2 (MAD2L2) and structural basis for its interaction with chromosome alignment maintaining phosphoprotein (CAMP). J. Biol. Chem. 2017, 292, 17658–17667. [Google Scholar] [CrossRef]
- Liang, L.; Feng, J.; Zuo, P.; Yang, J.; Lu, Y.; Yin, Y. Molecular basis for assembly of the shieldin complex and its implications for NHEJ. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of Octanol-Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- De Krijger, I.; Boersma, V.; Jacobs, J.J.L. Cell Biology REV7: Jack of many trades. Trends Cell Biol. 2021, 31, 686–701. [Google Scholar] [CrossRef]
- Boersma, V.; Moatti, N.; Segura-Bayona, S.; Peuscher, M.H.; van der Torre, J.; Wevers, B.A.; Orthwein, A.; Durocher, D.; Jacobs, J.J.L. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 2015, 521, 537–540. [Google Scholar] [CrossRef]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7–shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [PubMed]
- de Krijger, I.; Föhr, B.; Pérez, S.H.; Vincendeau, E.; Serrat, J.; Thouin, A.M.; Susvirkar, V.; Lescale, C.; Paniagua, I.; Hoekman, L.; et al. MAD2L2 dimerization and TRIP13 control shieldin activity in DNA repair. Nat. Commun. 2021, 12, 1–15. [Google Scholar]
- Xie, W.; Wang, S.; Wang, J.; De, M.J.; Xu, G.; Scaltriti, M. Molecular mechanisms of assembly and TRIP13-mediated remodeling of the human Shieldin complex. Proc. Natl. Acad. Sci. USA 2021, 118, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Clairmont, C.S.; D’andrea, A.D. REV7 directs DNA repair pathway choice. Trends Cell Biol. 2021, 31, 965–978. [Google Scholar] [CrossRef]
- Dev, H.; Chiang, T.W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, C.; Friederichs, J.; Rosenberg, R.; Holzmann, B.; Siewert, J.R.; Janssen, K.P. Expression of the mitotic checkpoint gene MAD2L2 has prognostic significance in colon cancer. Int. J. Cancer 2007, 120, 207–211. [Google Scholar] [CrossRef]
- Pernicone, N.; Peretz, L.; Grinshpon, S.; Listovsky, T. MDA-MB-157 Cell Line Presents High Levels of MAD2L2 and Dysregulated Mitosis. Anticancer Res. 2020, 40, 5471–5480. [Google Scholar] [CrossRef]
- Chen, J.; Fang, G. MAD2B is an inhibitor of the anaphase-promoting complex. Genes Dev. 2001, 15, 1765–1770. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, C.M.; Salic, A.; Lee, E.; Kirschner, M.W. Inhibition of Cdh1-APC by the MAD2-related protein MAD2L2: A novel mechanism for regulating Cdh1. Genes Dev. 2001, 15, 1759–1764. [Google Scholar] [CrossRef]
- Listovsky, T.; Sale, J.E. Sequestration of cdh1 by mad2l2 prevents premature apc/c activation prior to anaphase onset. J. Cell Biol. 2013, 203, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.; Wu, Z.; Maher, V.M.; McCormick, J.J.; Xiao, W. Rev7/Mad2b plays a critical role in the assembly of a functional mitotic spindle. Cell Cycle 2015, 14, 3929–3938. [Google Scholar] [CrossRef] [PubMed]
- Itoh, G.; Kanno, S.I.; Uchida, K.S.K.; Chiba, S.; Sugino, S.; Watanabe, K.; Mizuno, K.; Yasui, A.; Hirota, T.; Tanaka, K. CAMP (C13orf8, ZNF828) is a novel regulator of kinetochore-microtubule attachment. EMBO J. 2011, 30, 130–144. [Google Scholar] [CrossRef]
- Chatterjee, N.; Whitman, M.A.; Harris, C.A.; Min, S.M.; Jonas, O.; Lien, E.C.; Luengo, A.; Heiden, M.G.V.; Hong, J.; Zhou, P.; et al. REV1 inhibitor JH-RE-06 enhances tumor cell response to chemotherapy by triggering senescence hallmarks. Proc. Natl. Acad. Sci. USA 2020, 117, 28918–28921. [Google Scholar] [CrossRef]
- Yoon, J.H.; Johnson, R.E.; Prakash, L.; Prakash, S. Implications of inhibition of Rev1 interaction with Y family DNA polymerases for cisplatin chemotherapy. Genes Dev. 2021, 35, 1256–1270. [Google Scholar] [CrossRef]
- Hara, K.; Hashimoto, H.; Murakumo, Y.; Kobayashi, S.; Kogame, T.; Unzai, S.; Akashi, S.; Takeda, S.; Shimizu, T.; Sato, M. Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase ζ and REV1. J. Biol. Chem. 2010, 285, 12299–12307. [Google Scholar] [CrossRef]
- Chaudhury, S.; Berrondo, M.; Weitzner, B.D.; Muthu, P.; Bergman, H.; Gray, J.J. Benchmarking and Analysis of Protein Docking Performance in Rosetta v3.2. PLoS ONE 2011, 6, e22477. [Google Scholar] [CrossRef]
- Lyskov, S.; Gray, J.J. The RosettaDock server for local protein-protein docking. Nucleic Acids Res. 2008, 36, W233–W238. [Google Scholar] [CrossRef]
- Lyskov, S.; Chou, F.C.; Conchúir, S.Ó.; Der, B.S.; Drew, K.; Kuroda, D.; Xu, J.; Weitzner, B.D.; Renfrew, P.D.; Sripakdeevong, P.; et al. Serverification of Molecular Modeling Applications: The Rosetta Online Server That Includes Everyone (ROSIE). PLoS ONE 2013, 8, e63906. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Shoichet, B.K.; Kuntz, I.D.; Bodian, D.L. Molecular docking using shape descriptors. J. Comput. Chem. 1992, 13, 380–397. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Qvit, N.; Kornfeld, O.S.; Mochly-Rosen, D. Engineered Substrate-Specific Delta PKC Antagonists to Enhance Cardiac Therapeutics. Angew. Chemie-Int. Ed. 2016, 55, 15672–15679. [Google Scholar] [CrossRef] [PubMed]
- Qvit, N.; Disatnik, M.H.; Sho, E.; Mochly-Rosen, D. Selective Phosphorylation Inhibitor of Delta Protein Kinase C-Pyruvate Dehydrogenase Kinase Protein-Protein Interactions: Application for Myocardial Injury in Vivo. J. Am. Chem. Soc. 2016, 138, 7626–7635. [Google Scholar] [CrossRef] [PubMed]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundbäck, T.; Nordlund, P.; Molina, D.M. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef] [PubMed]
- Molina, D.M.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pernicone, N.; Elias, M.; Onn, I.; Tobi, D.; Listovsky, T. Disrupting the MAD2L2-Rev1 Complex Enhances Cell Death upon DNA Damage. Molecules 2022, 27, 636. https://doi.org/10.3390/molecules27030636
Pernicone N, Elias M, Onn I, Tobi D, Listovsky T. Disrupting the MAD2L2-Rev1 Complex Enhances Cell Death upon DNA Damage. Molecules. 2022; 27(3):636. https://doi.org/10.3390/molecules27030636
Chicago/Turabian StylePernicone, Nomi, Maria Elias, Itay Onn, Dror Tobi, and Tamar Listovsky. 2022. "Disrupting the MAD2L2-Rev1 Complex Enhances Cell Death upon DNA Damage" Molecules 27, no. 3: 636. https://doi.org/10.3390/molecules27030636
APA StylePernicone, N., Elias, M., Onn, I., Tobi, D., & Listovsky, T. (2022). Disrupting the MAD2L2-Rev1 Complex Enhances Cell Death upon DNA Damage. Molecules, 27(3), 636. https://doi.org/10.3390/molecules27030636