
Proteolysis-Targeting Chimeras (PROTACs) in Cancer Therapy: Present and Future

Abstract
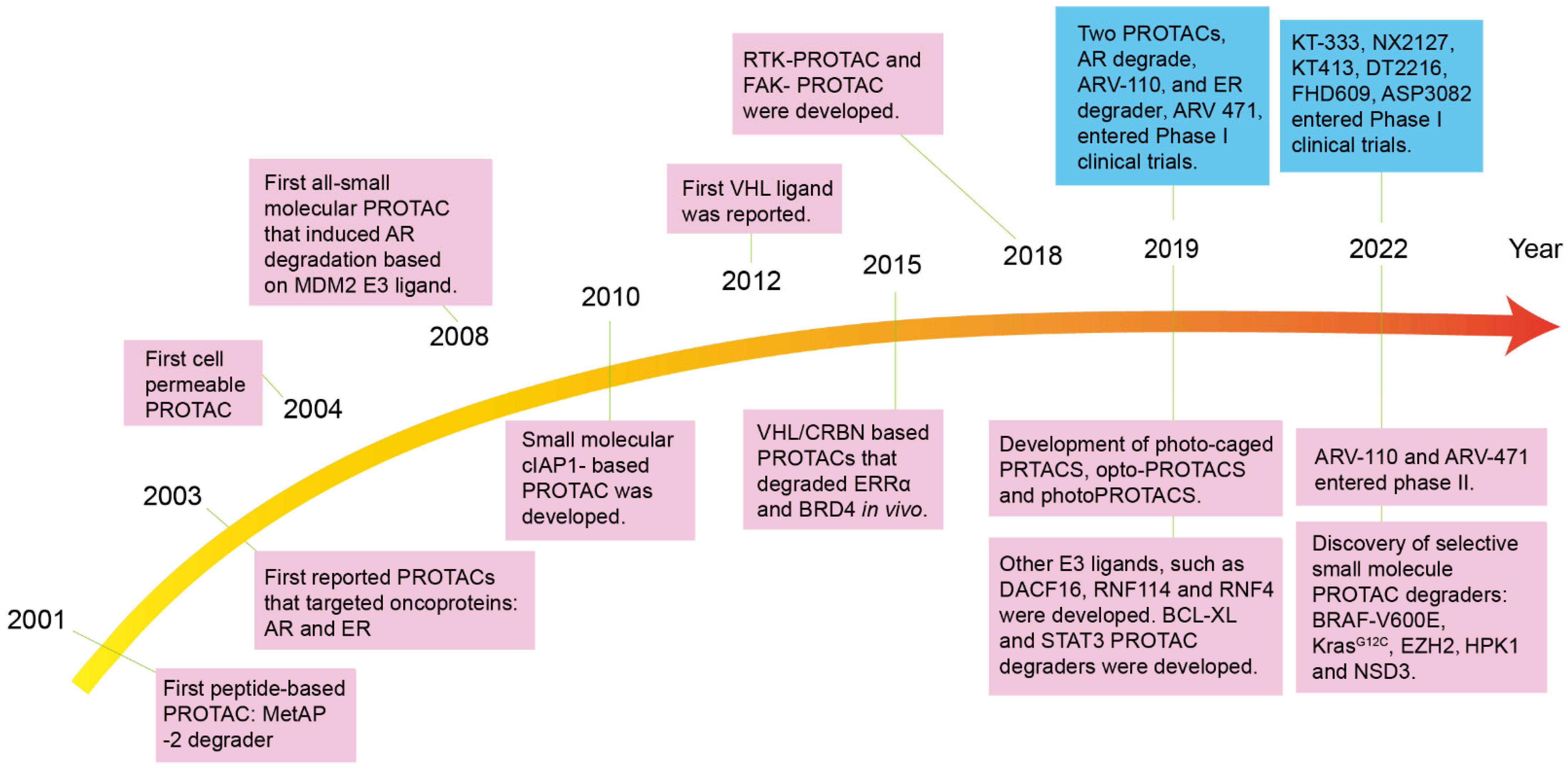
1. Introduction
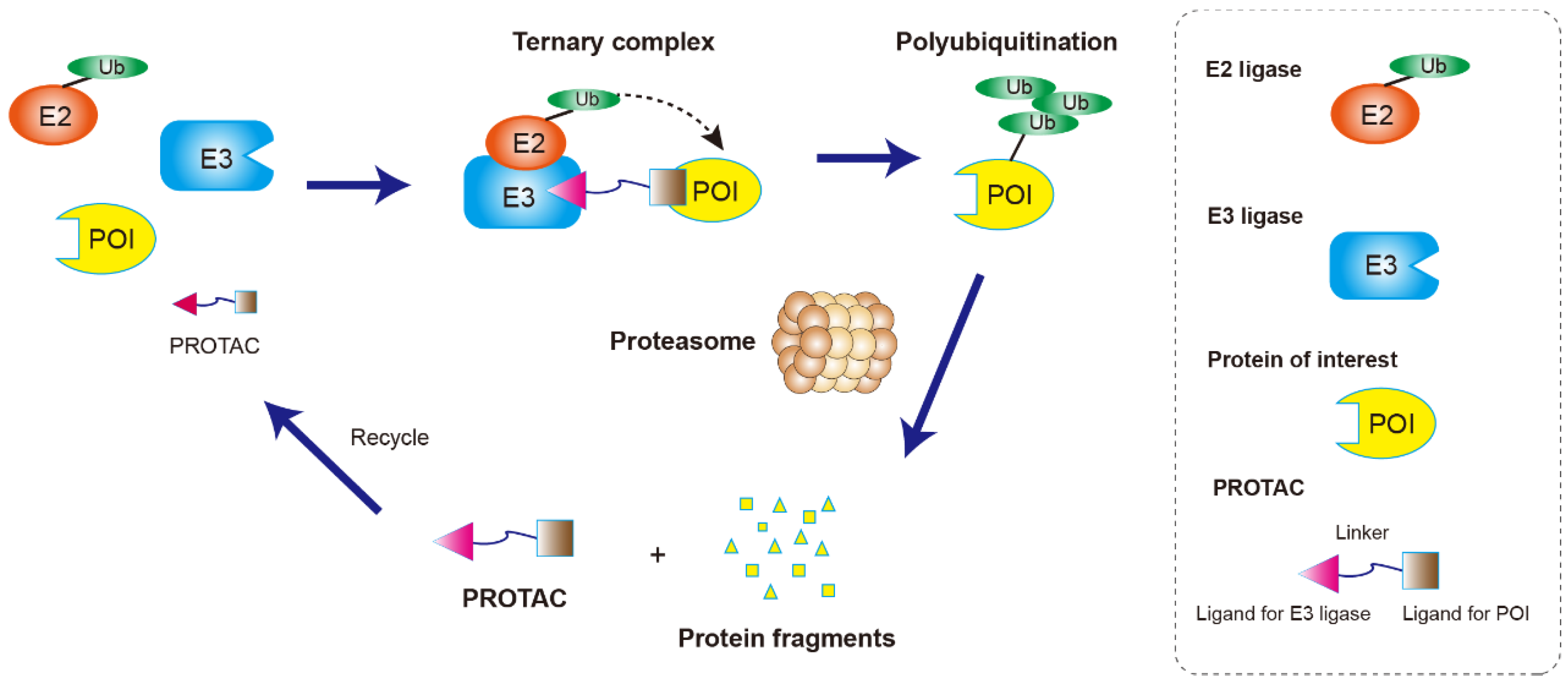
2. Structure Character and Mechanism of PROTACs
3. The Application of PROTACs in Anti-Cancer Drug Discovery and Development
3.1. Peptide-Based PROTAC
3.2. Small-Molecule-Based PROTACs
3.2.1. MDM2-Based PROTACs
3.2.2. VHL-Based PROTACs
3.2.3. CRBN-Based PROTACs
3.2.4. cIAP1-Based PROTACs
4. PROTACs in Clinical Trials
5. Challenges in PROTACs Study
6. Perspectives

Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Röth, S.; Fulcher, L.J.; Sapkota, G.P. Advances in targeted degradation of endogenous proteins. Cell. Mol. Life Sci. CMLS 2019, 76, 2761–2777. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Gao, X.; Vogelzang, N.J.; Garfield, M.H.; Taylor, I.; Moore, M.D.; Peck, R.A.; III, H.A.B. First-in-human phase I study of ARV-110, an androgen receptor (AR) PROTAC degrader in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) following enzalutamide (ENZ) and/or abiraterone (ABI). J. Clin. Oncol. 2020, 38 (Suppl. 15), 3500. [Google Scholar] [CrossRef]
- Snyder, L.B.; Flanagan, J.J.; Qian, Y.; Gough, S.M.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Chandler, J.; et al. Abstract 44: The discovery of ARV-471, an orally bioavailable estrogen receptor degrading PROTAC for the treatment of patients with breast cancer. Cancer Res. 2021, 81 (Suppl. 13), 44. [Google Scholar] [CrossRef]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Et Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Reviews. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- He, M.; Cao, C.; Ni, Z.; Liu, Y.; Song, P.; Hao, S.; He, Y.; Sun, X.; Rao, Y. PROTACs: Great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct. Target. Ther. 2022, 7, 181. [Google Scholar] [CrossRef]
- Sun, B.; Fiskus, W.; Qian, Y.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Saenz, D.T.; Mill, C.P.; et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 2018, 32, 343–352. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, H.Y.; Xi, X.X.; Liu, Y.J.; Xin, M.; Mao, S.; Zhang, J.J.; Lu, A.X.; Zhang, S.Q. Discovery of potent epidermal growth factor receptor (EGFR) degraders by proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem. 2020, 189, 112061. [Google Scholar] [CrossRef] [PubMed]
- Prozzillo, Y.; Fattorini, G.; Santopietro, M.V.; Suglia, L.; Ruggiero, A.; Ferreri, D.; Messina, G. Targeted Protein Degradation Tools: Overview and Future Perspectives. Biology 2020, 9, 421. [Google Scholar] [CrossRef]
- Dai, M.; Radhakrishnan, S.; Li, R.; Tan, R.; Yan, K.; Fan, G.; Liu, M. Targeted Protein Degradation: An Important Tool for Drug Discovery for “Undruggable” Tumor Transcription Factors. Technol. Cancer Res. Treat. 2022, 21, 15330338221095950. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Burgess, K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem. Commun. (Camb. Engl.) 2019, 55, 2704–2707. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Yoon, H.; Xiong, Y.; Dixon-Clarke, S.E.; Nowak, R.P.; Fischer, E.S. Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nat. Struct. Mol. Biol. 2020, 27, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef]
- Jin, J.; Wu, Y.; Chen, J.; Shen, Y.; Zhang, L.; Zhang, H.; Chen, L.; Yuan, H.; Chen, H.; Zhang, W.; et al. The peptide PROTAC modality: A novel strategy for targeted protein ubiquitination. Theranostics 2020, 10, 10141–10153. [Google Scholar] [CrossRef]
- Rodriguez-Gonzalez, A.; Cyrus, K.; Salcius, M.; Kim, K.; Crews, C.M.; Deshaies, R.J.; Sakamoto, K.M. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene 2008, 27, 7201–7211. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Verma, R.; Ransick, A.; Stein, B.; Crews, C.M.; Deshaies, R.J. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell. Proteom. MCP 2003, 2, 1350–1358. [Google Scholar] [CrossRef]
- Hines, J.; Gough, J.D.; Corson, T.W.; Crews, C.M. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc. Natl. Acad. Sci. USA 2013, 110, 8942–8947. [Google Scholar] [CrossRef]
- Toure, M.; Crews, C.M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem. (Int. Ed. Engl.) 2016, 55, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorganic Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, J.S., Jr.; Fonseca, F.N.; Koldobskiy, M.; Mandal, A.; Deshaies, R.; Sakamoto, K.; Crews, C.M. Chemical genetic control of protein levels: Selective in vivo targeted degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef] [PubMed]
- Paiva, S.L.; Crews, C.M. Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Montrose, K.; Krissansen, G.W. Design of a PROTAC that antagonizes and destroys the cancer-forming X-protein of the hepatitis B virus. Biochem. Biophys. Res. Commun. 2014, 453, 735–740. [Google Scholar] [CrossRef]
- Bargagna-Mohan, P.; Baek, S.H.; Lee, H.; Kim, K.; Mohan, R. Use of PROTACS as molecular probes of angiogenesis. Bioorganic Med. Chem. Lett. 2005, 15, 2724–2727. [Google Scholar] [CrossRef]
- Wang, K.; Dai, X.; Yu, A.; Feng, C.; Liu, K.; Huang, L. Peptide-based PROTAC degrader of FOXM1 suppresses cancer and decreases GLUT1 and PD-L1 expression. J. Exp. Clin. Cancer Res. CR 2022, 41, 289. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Wu, Y.; Xing, D. Developments of CRBN-based PROTACs as potential therapeutic agents. Eur. J. Med. Chem. 2021, 225, 113749. [Google Scholar] [CrossRef]
- Ohoka, N.; Shibata, N.; Hattori, T.; Naito, M. Protein Knockdown Technology: Application of Ubiquitin Ligase to Cancer Therapy. Curr. Cancer Drug Targets 2016, 16, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Et Biophys. Acta. Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; McGowan, P.M.; Crown, J.; O’Connor, D.; Gallagher, W.M. p53 as a target for the treatment of cancer. Cancer Treat. Rev. 2014, 40, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Hines, J.; Lartigue, S.; Dong, H.; Qian, Y.; Crews, C.M. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res. 2019, 79, 251–262. [Google Scholar] [CrossRef]
- Robinson, C.M.; Ohh, M. The multifaceted von Hippel-Lindau tumour suppressor protein. FEBS Lett. 2014, 588, 2704–2711. [Google Scholar] [CrossRef]
- Shmueli, M.D.; Levy-Kanfo, L.; Haj, E.; Schoenfeld, A.R.; Gazit, E.; Segal, D. Arginine refolds, stabilizes, and restores function of mutant pVHL proteins in animal model of the VHL cancer syndrome. Oncogene 2019, 38, 1038–1049. [Google Scholar] [CrossRef]
- Dale, B.; Cheng, M.; Park, K.S.; Kaniskan, H.; Xiong, Y.; Jin, J. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21, 638–654. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef]
- Buckley, D.L.; Gustafson, J.L.; Van Molle, I.; Roth, A.G.; Tae, H.S.; Gareiss, P.C.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1α. Angew. Chem. 2012, 51, 11463–11467. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. 2016, 55, 807–810. [Google Scholar] [CrossRef]
- Zhang, C.; Han, X.R.; Yang, X.; Jiang, B.; Liu, J.; Xiong, Y.; Jin, J. Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). Eur. J. Med. Chem. 2018, 151, 304–314. [Google Scholar] [CrossRef]
- Scheepstra, M.; Hekking, K.F.W.; van Hijfte, L.; Folmer, R.H.A. Bivalent Ligands for Protein Degradation in Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 160–176. [Google Scholar] [CrossRef]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. [Google Scholar] [CrossRef]
- Liu, J.; Xue, L.; Xu, X.; Luo, J.; Zhang, S. FAK-targeting PROTAC demonstrates enhanced antitumor activity against KRAS mutant non-small cell lung cancer. Exp. Cell Res. 2021, 408, 112868. [Google Scholar] [CrossRef]
- Wang, X.; Feng, S.; Fan, J.; Li, X.; Wen, Q.; Luo, N. New strategy for renal fibrosis: Targeting Smad3 proteins for ubiquitination and degradation. Biochem. Pharmacol. 2016, 116, 200–209. [Google Scholar] [CrossRef]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef] [PubMed]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [PubMed]
- Saenz, D.T.; Fiskus, W.; Qian, Y.; Manshouri, T.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Mill, C.P.; et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia 2017, 31, 1951–1961. [Google Scholar] [CrossRef]
- Gupta, P.; Zhang, G.N.; Barbuti, A.M.; Zhang, X.; Karadkhelkar, N.; Zhou, J.; Ding, K.; Pan, J.; Yoganathan, S.; Yang, D.H.; et al. Preclinical development of a novel BCR-ABL T315I inhibitor against chronic myeloid leukemia. Cancer Lett. 2020, 472, 132–141. [Google Scholar] [CrossRef]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Zhao, Q.; Ren, C.; Liu, L.; Chen, J.; Shao, Y.; Sun, N.; Sun, R.; Kong, Y.; Ding, X.; Zhang, X.; et al. Discovery of SIAIS178 as an Effective BCR-ABL Degrader by Recruiting Von Hippel-Lindau (VHL) E3 Ubiquitin Ligase. J. Med. Chem. 2019, 62, 9281–9298. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Qin, C.; Hu, Y.; Zhou, B.; Fernandez-Salas, E.; Yang, C.Y.; Liu, L.; McEachern, D.; Przybranowski, S.; Wang, M.; Stuckey, J.; et al. Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra-Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J. Med. Chem. 2018, 61, 6685–6704. [Google Scholar] [CrossRef]
- Zhang, H.; Li, G.; Zhang, Y.; Shi, J.; Yan, B.; Tang, H.; Chen, S.; Zhang, J.; Wen, P.; Wang, Z.; et al. Targeting BET Proteins with a PROTAC Molecule Elicits Potent Anticancer Activity in HCC Cells. Front. Oncol. 2019, 9, 1471. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTK(C481S)-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ding, N.; Song, Y.; Yang, Z.; Liu, W.; Zhu, J.; Rao, Y. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia 2019, 33, 2105–2110. [Google Scholar] [CrossRef]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27 (Suppl. 3), iii4–iii15. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Bauer, T.M.; de Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. New Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef]
- Kang, C.H.; Lee, D.H.; Lee, C.O.; Du Ha, J.; Park, C.H.; Hwang, J.Y. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem. Biophys. Res. Commun. 2018, 505, 542–547. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. New Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Yang, Z.; Gao, H.; Yang, H.; Zhu, S.; An, Z.; Wang, J.; Li, Q.; Chandarlapaty, S.; Deng, H.; et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 2019, 62, 7575–7582. [Google Scholar] [CrossRef] [PubMed]
- Papatzimas, J.W.; Gorobets, E.; Maity, R.; Muniyat, M.I.; MacCallum, J.L.; Neri, P.; Bahlis, N.J.; Derksen, D.J. From Inhibition to Degradation: Targeting the Antiapoptotic Protein Myeloid Cell Leukemia 1 (MCL1). J. Med. Chem. 2019, 62, 5522–5540. [Google Scholar] [CrossRef]
- Wang, Z.; He, N.; Guo, Z.; Niu, C.; Song, T.; Guo, Y.; Cao, K.; Wang, A.; Zhu, J.; Zhang, X.; et al. Proteolysis Targeting Chimeras for the Selective Degradation of Mcl-1/Bcl-2 Derived from Nonselective Target Binding Ligands. J. Med. Chem. 2019, 62, 8152–8163. [Google Scholar] [CrossRef]
- Li, W.; Gao, C.; Zhao, L.; Yuan, Z.; Chen, Y.; Jiang, Y. Phthalimide conjugations for the degradation of oncogenic PI3K. Eur. J. Med. Chem. 2018, 151, 237–247. [Google Scholar] [CrossRef] [PubMed]
- You, I.; Erickson, E.C.; Donovan, K.A.; Eleuteri, N.A.; Fischer, E.S.; Gray, N.S.; Toker, A. Discovery of an AKT Degrader with Prolonged Inhibition of Downstream Signaling. Cell Chem. Biol. 2020, 27, 66–73.e7. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorganic Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511.e17. [Google Scholar] [CrossRef]
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.M.; Büsselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis Int. J. Program. Cell Death 2017, 22, 898–919. [Google Scholar] [CrossRef]
- Feltham, R.; Bettjeman, B.; Budhidarmo, R.; Mace, P.D.; Shirley, S.; Condon, S.M.; Chunduru, S.K.; McKinlay, M.A.; Vaux, D.L.; Silke, J.; et al. Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J. Biol. Chem. 2011, 286, 17015–17028. [Google Scholar] [CrossRef]
- Khan, S.; He, Y.; Zhang, X.; Yuan, Y.; Pu, S.; Kong, Q.; Zheng, G.; Zhou, D. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 2020, 39, 4909–4924. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Ishikawa, M.; Kitaguchi, R.; Sato, S.; Naito, M.; Hashimoto, Y. Development of target protein-selective degradation inducer for protein knockdown. Bioorganic Med. Chem. 2011, 19, 3229–3241. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Xiang, Y.; Kang, X. Small-Molecule PROTACs for Cancer Immunotherapy. Molecules 2022, 27, 5439. [Google Scholar] [CrossRef]
- Hughes, S.J.; Testa, A.; Thompson, N.; Churcher, I. The rise and rise of protein degradation: Opportunities and challenges ahead. Drug Discov. Today 2021, 26, 2889–2897. [Google Scholar] [CrossRef] [PubMed]
- The PROTAC DT2216 Targets Cancer by Promoting BCL-XL Degradation. Cancer Discov. 2020, 10, 174. [CrossRef]
- Qi, S.M.; Dong, J.; Xu, Z.Y.; Cheng, X.D.; Zhang, W.D.; Qin, J.J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 2021, 12, 692574. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Huang, W.; Zheng, X.; Liyan, C.; Zhang, Z.; Wang, J.; Shen, Z. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: Recent progress and future challenges. Eur. J. Med. Chem. 2021, 210, 112981. [Google Scholar] [CrossRef]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef]
- Hanzl, A.; Winter, G.E. Targeted protein degradation: Current and future challenges. Curr. Opin. Chem. Biol. 2020, 56, 35–41. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)–Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Dai, M.Y.; Shi, Y.Y.; Wang, A.J.; Liu, X.L.; Liu, M.; Cai, H.B. High-potency PD-1/PD-L1 degradation induced by Peptide-PROTAC in human cancer cells. Cell Death Dis. 2022, 13, 924. [Google Scholar] [CrossRef] [PubMed]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, P.; Samarasinghe, K.T.G.; Crews, C.M.; Carreira, E.M. Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Cent. Sci. 2019, 5, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-Induced Protein Degradation with Photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef]
- Alabi, S.B.; Crews, C.M. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Degrader | Target | E3 ligase | Indications | NCT Numbers (If Applicable) | Phase | Company | ROA | Start Year |
|---|---|---|---|---|---|---|---|---|
| CC-94676 | AR | CRBN | Prostate cancer | NCT04428788 | Phase Ⅰ | Bristol Myers Squibb | Oral | 2020.6 |
| HP518 | AR | Undisclosed | Metastatic castration-resistant prostate cancer | NCT05252364 | Phase Ⅰ | Hinova Pharmaceuticals | Oral | 2021.12 |
| ARV-766 | AR | Undisclosed | Prostate cancer | NCT05067140 | Phase Ⅰ | Arvinas | Oral | 2021.9 |
| AC176 | AR | Undisclosed | Prostate cancer | NCT05241613 | Phase Ⅰ | Accutar Biotech | Oral | 2022.1 |
| ARV-110 | AR | CRBN | Prostate cancer | NCT03888612(U.S.) NCT05177042(Canada) | Phase Ⅱ | Arvinas | Oral | 2019.3(U.S.) 2021.11(Canada) |
| DT2216 | BCL-XL | VHL | Liquid and solid tumors | NCT04886622 | Phase Ⅰ | Dialectic Therapeutics | I.V. | 2021.4 |
| FHD-609 | BRD9 | Undisclosed | Synovial sarcoma | NCT04965753 | Phase Ⅰ | Foghorn Therapeutics | I.V. | 2021.6 |
| CFT8634 | BRD9 | CRBN | Synovial sarcoma | NCT05355753 | Phase Ⅰ/Ⅱ | C4 Therapeutics | Oral | 2022.4 |
| NX-5948 | BTK | CRBN | B cell malignancies and autoimmune disease | NCT05131022 | Phase Ⅰ | Nurix Therapeutics | Oral | 2021.11 |
| NX-2127 | BTK | CRBN | B cell malignancies | NCT04830137 | Phase Ⅰ | Nurix Therapeutics | Oral | 2021.3 |
| BGB-16673 | BTK | Undisclosed | B cell malignancy and lymphoma | NCT05294731(China) NCT05006716(U.S.) | Phase Ⅰ | BeiGene | Oral | 2022.2(China) 2021.8(U.S.) |
| HSK29116 | BTK | Undisclosed | Relapsed/refractory B cell malignancies | NCT04861779 | Phase Ⅰ | Haisco | Oral | 2021.8 |
| CFT8919 | EGFR-L858R | CRBN | Non-small-cell-lung cancer | IND-e | C4 Therapeutics | Oral | ||
| ARV-471 | ER | CRBN | Breast cancer | NCT04072952(U.S.) NCT05463952(Japan) NCT05501769(U.S.) | Phase Ⅱ | Arvinas/Pfizer | Oral | 2019.8(U.S.) 2022.7(Japan) 2022.8(U.S.) |
| AC682 | ER | CRBN | Breast cancer | NCT05080842(U.S.) NCT05489679(China) | Phase Ⅰ | Accutar Biotech | Oral | 2021.9(U.S.) 2022.7(China) |
| KT-413 | IRAK4 | CRBN | Diffuse large B cell lymphoma (MYD88-mutant) | NCT05233033 | Phase Ⅰ | Kymera | I.V. | 2022.1 |
| ASP3082 | KRAS G12D | Undisclosed | Solid tumor | NCT05382559 | Phase Ⅰ | Astellas Pharma | Intravenous infusion | 2022 |
| KT-333 | STAT3 | Undisclosed | Liquid and solid tumors | NCT05225584 | Phase Ⅰ | Kymera | Undisclosed | 2021.12 |
| CG001419 | TRK | CRBN | Cancer and other indications | IND-e | Cullgen | Oral |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Liu, M.; Yang, Z.; Li, J.; Gao, Y.; Tan, R. Proteolysis-Targeting Chimeras (PROTACs) in Cancer Therapy: Present and Future. Molecules 2022, 27, 8828. https://doi.org/10.3390/molecules27248828
Li R, Liu M, Yang Z, Li J, Gao Y, Tan R. Proteolysis-Targeting Chimeras (PROTACs) in Cancer Therapy: Present and Future. Molecules. 2022; 27(24):8828. https://doi.org/10.3390/molecules27248828
Chicago/Turabian StyleLi, Rui, Miao Liu, Zhenya Yang, Jiao Li, Yuxin Gao, and Ruirong Tan. 2022. "Proteolysis-Targeting Chimeras (PROTACs) in Cancer Therapy: Present and Future" Molecules 27, no. 24: 8828. https://doi.org/10.3390/molecules27248828
APA StyleLi, R., Liu, M., Yang, Z., Li, J., Gao, Y., & Tan, R. (2022). Proteolysis-Targeting Chimeras (PROTACs) in Cancer Therapy: Present and Future. Molecules, 27(24), 8828. https://doi.org/10.3390/molecules27248828