
Evaluation of Candidatus Liberibacter Asiaticus Efflux Pump Inhibition by Antimicrobial Peptides

 , , , and
, , , and
Abstract
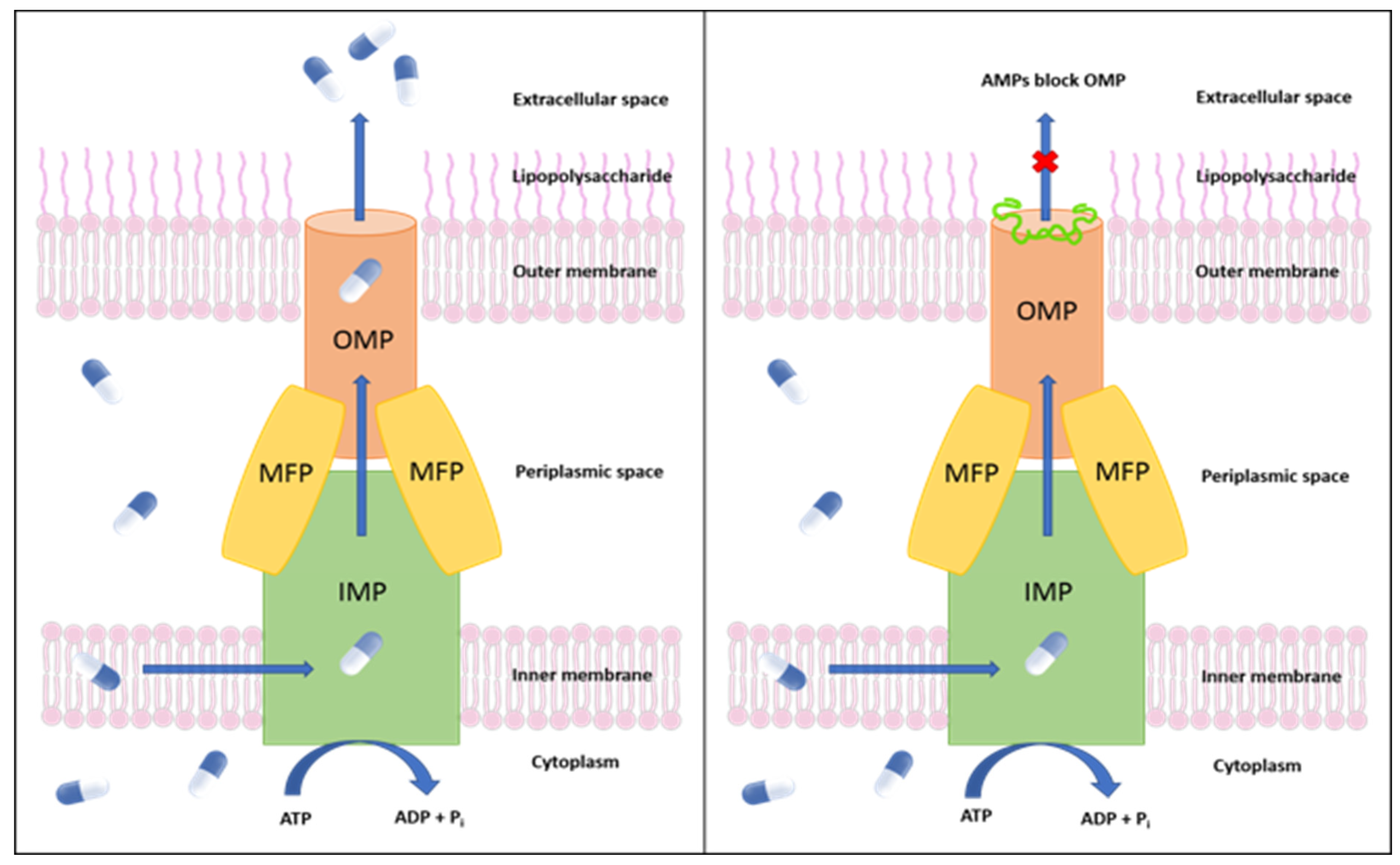
1. Introduction
2. Results and Discussion
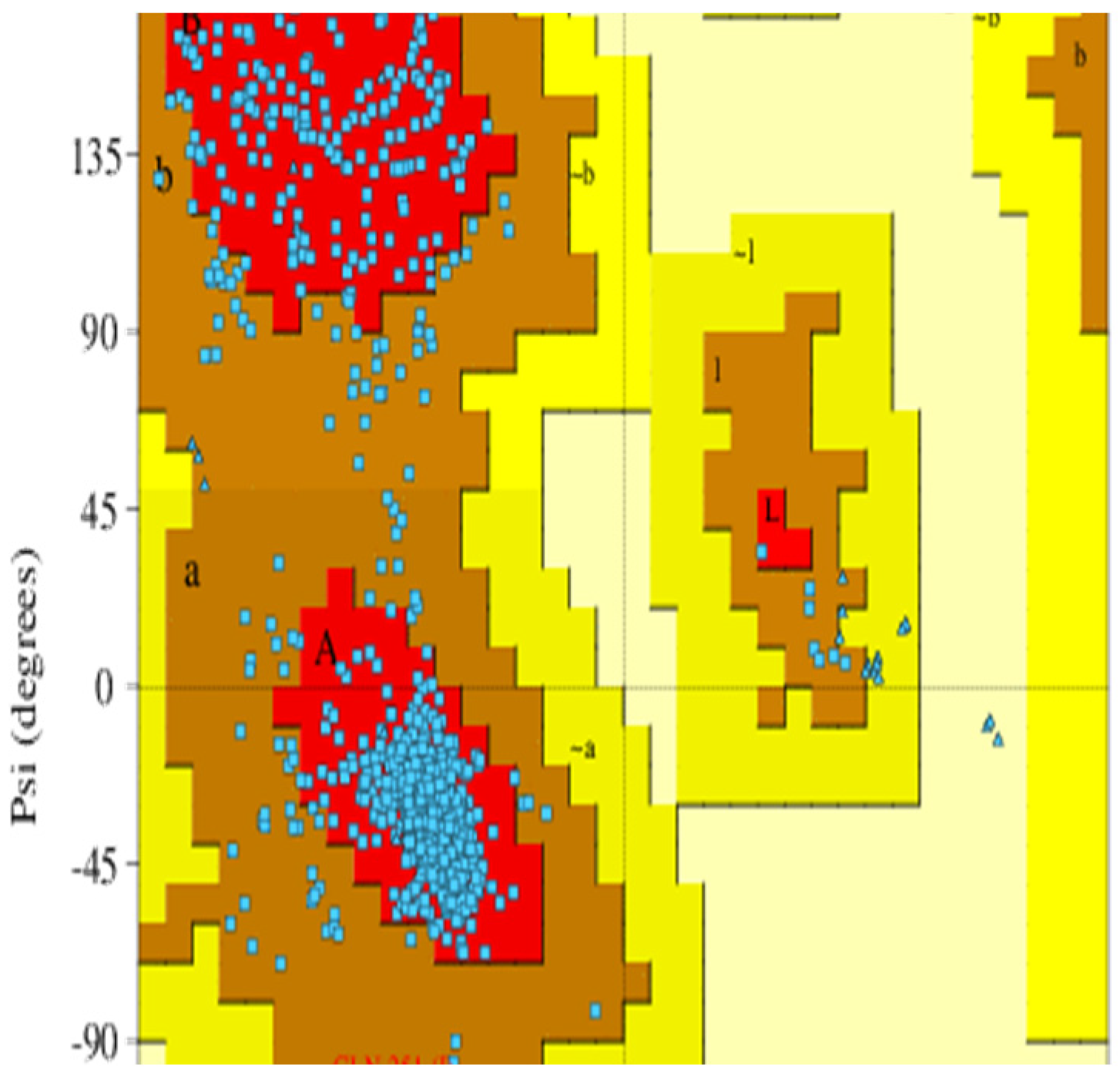
2.1. Protein Structure Modeling and Validation
2.2. Virtual Screening of AMPs
2.3. Molecular Docking Analysis (Glide)
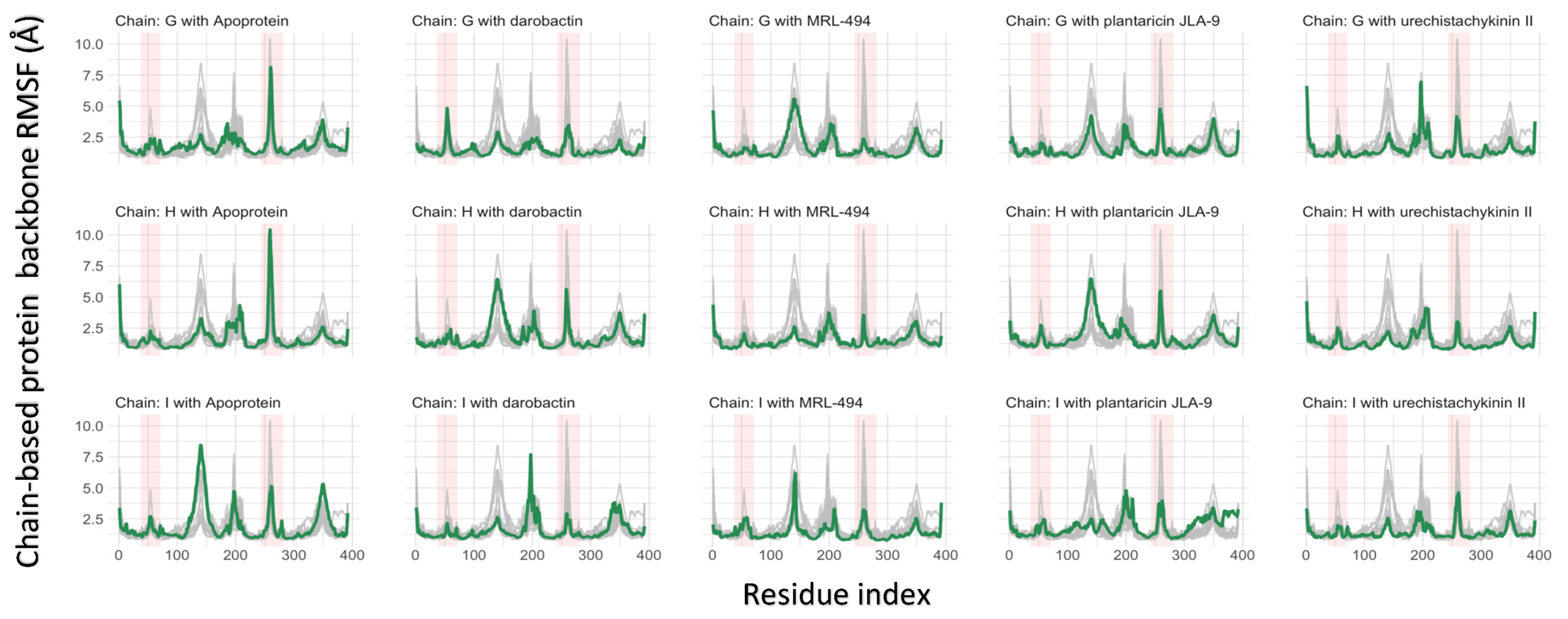
2.4. Molecular Dynamics Simulations
2.5. Prime MM-GBSA Analysis
2.6. Principal Component Analysis (PCA)
2.7. Biological Efficacy Assays
3. Materials and Methods
3.1. Sequence Analysis
3.2. Virtual Screening of AMPs
3.3. Molecular Docking
3.4. Molecular Dynamics Simulations
3.5. Calculation of Binding Free Energy
3.6. Principal Component Analysis
3.7. Peptide Synthesis
3.8. Biological Efficacy Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, N. The citrus huanglongbing crisis and potential solutions. Mol. Plant 2019, 12, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zhou, L.; Hall, D.G.; Li, W.; Doddapaneni, H.; Lin, H.; Liu, L.; Vahling, C.M.; Gabriel, D.W.; Williams, K.P. Complete genome sequence of citrus huanglongbing bacterium,‘Candidatus Liberibacter asiaticus’ obtained through metagenomics. Mol. Plant-Microbe Interact. 2009, 22, 1011–1020. [Google Scholar] [CrossRef]
- Coletta-Filho, H.D.; Takita, M.A.; Targon, M.L.P.N.; Machado, M.A. Analysis of 16S rDNA sequences from citrus huanglongbing bacteria reveal a different “Ca. Liberibacter” strain associated with citrus disease in Sao Paulo. Plant Dis. 2005, 89, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Hodges, A.; Spreen, T. Economic Impacts of Citrus Greening (HLB) in Florida, 2006/07–2010/11. Available online: http://ufdcimages.uflib.ufl.edu/IR/00/00/56/15/00001/FE90300b.pdf (accessed on 3 March 2021).
- USDA. Citrus Greening Threatens America’s Citrus. Don’t Risk Citrus, Don’t Move Citrus. Available online: https://www.aphis.usda.gov/aphis/resources/pests-diseases/hungry-pests/the-threat/citrus-greening/citrus-greening-hp (accessed on 3 March 2021).
- Fletcher, J.; Berenbaum, M.; Gray, S.M.; Groves, R.; Scorza, R.; Triplett, L.; Trumble, J.; Yang, B. A Review of the Citrus Greening Research and Development Efforts Supported by the Citrus Research and Development Foundation: Fighting a Ravaging Disease; National Academy Press: Washington, DC, USA, 2018; pp. 1–270. [Google Scholar]
- Blaustein, R.A.; Lorca, G.L.; Teplitski, M. Challenges for managing Candidatus Liberibacter spp.(huanglongbing disease pathogen): Current control measures and future directions. Phytopathology 2018, 108, 424–435. [Google Scholar] [CrossRef]
- Li, S.; Wu, F.; Duan, Y.; Singerman, A.; Guan, Z. Citrus Greening: Management Strategies and Their Economic Impact. HortScience 2020, 1, 1–9. [Google Scholar] [CrossRef]
- Jain, M.; Cai, L.; Fleites, L.; Munoz-Bodnar, A.; Davis, M.; Gabriel, D. Liberibacter crescens is a cultured surrogate for functional genomics of uncultured pathogenic ‘Candidatus Liberibacter’spp. and is naturally competent for transformation. Phytopathology 2019, 109, 1811–1819. [Google Scholar] [CrossRef]
- Ammar, E.-D.; Alessandro, R.; Shatters, R.G., Jr.; Hall, D.G. Behavioral, ultrastructural and chemical studies on the honeydew and waxy secretions by nymphs and adults of the Asian citrus psyllid Diaphorina citri (Hemiptera: Psyllidae). PLoS ONE 2013, 8, e64938. [Google Scholar] [CrossRef] [PubMed]
- Kishk, A.; Anber, H.A.; AbdEl-Raof, T.K.; El-Sherbeni, A.H.D.; Hamed, S.; Gowda, S.; Killiny, N. RNA interference of carboxyesterases causes nymph mortality in the Asian citrus psyllid, Diaphorina citri. Arch. Insect Biochem. Physiol. 2017, 94, e21377. [Google Scholar] [CrossRef]
- Yu, X.; Killiny, N. RNA interference-mediated control of Asian citrus psyllid, the vector of the huanglongbing bacterial pathogen. Trop. Plant Pathol. 2020, 45, 298–305. [Google Scholar] [CrossRef]
- Li, J.; Pang, Z.; Duan, S.; Lee, D.; Kolbasov, V.G.; Wang, N. The in planta effective concentration of oxytetracycline against ‘Candidatus Liberibacter asiaticus’ for suppression of citrus huanglongbing. Phytopathology 2019, 109, 2046–2054. [Google Scholar] [CrossRef]
- Yang, C.; Powell, C.A.; Duan, Y.; Shatters, R.; Zhang, M. Antimicrobial nanoemulsion formulation with improved penetration of foliar spray through citrus leaf cuticles to control citrus huanglongbing. PLoS ONE 2015, 10, e0133826. [Google Scholar] [CrossRef] [PubMed]
- EPA. Final Registration Decision for the New Use of the Active Ingredient Oxytetracycline Hydrochloride on Citrus Crop Group 10–10; Environmental Protection Agency: Washington, DC, USA, 2018.
- EPA. Pesticide Emergency Exemptions: Agency Decisions and State and Federal Agency Crisis Declarations; Environmental Protection Agency: Washington, DC, USA, 2020.
- McKenna, M. Antibiotics set to flood Florida’s troubled orange orchards. Nature 2019, 567, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Miyata, S.-i.; Inoue, H.; Iwanami, T. Unique features of a Japanese ‘Candidatus Liberibacter asiaticus’ strain revealed by whole genome sequencing. PLoS ONE 2014, 9, e106109. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Iwanami, T.; Fujikawa, T. Alterations of Candidatus Liberibacter asiaticus-associated microbiota decrease survival of Ca. L. asiaticus in in vitro assays. Front. Microbiol. 2018, 9, 3089. [Google Scholar] [CrossRef]
- Manyi-Loh, C.; Iwanami, T.; Fujikawa, T. Antibiotic Use in Agriculture and Its Consequential Resistance in Environmental Sources: Potential Public Health Implications. Molecules 2018, 23, 795. [Google Scholar] [CrossRef]
- Dall, C. Lawmakers Urge EPA to Rethink Use of Antibiotics on Citrus Trees. 2019. Available online: https://www.cidrap.umn.edu/news-perspective/2019/08/lawmakers-urge-epa-rethink-use-antibiotics-citrus-trees (accessed on 3 March 2021).
- Center for Biological Diversity. Trump Administration Approves Antibiotic Residue on Citrus Fruit. Medically Important Antibiotic Oxytetracycline Allowed on Oranges, Tangerines. 2018. Available online: https://www.biologicaldiversity.org/news/press_releases/2018/oxytetracycline-12-06-2018.php (accessed on 3 March 2021).
- Center for Biological Diversity. EPA Extends ‘Emergency’ Antibiotic Use on California, Florida Citrus Crops. Approval of Streptomycin as Pesticide Sidesteps Full Safety Assessment. 2020. Available online: https://biologicaldiversity.org/w/news/press-releases/epa-extends-emergency-antibiotic-use-on-california-florida-citrus-crops-2020-02-11/ (accessed on 3 March 2021).
- Sharff, A.; Fanutti, C.; Shi, J.; Calladine, C.; Luisi, B. The role of the TolC family in protein transport and multidrug efflux. From stereochemical certainty to mechanistic hypothesis. Eur. J. Biochem. 2001, 268, 5011–5026. [Google Scholar] [CrossRef]
- Clark, K.; Franco, J.Y.; Schwizer, S.; Pang, Z.Q.; Hawara, E.; Liebrand, T.W.H.; Pagliaccia, D.; Zeng, L.P.; Gurung, F.B.; Wang, P.C.; et al. An effector from the Huanglongbing-associated pathogen targets citrus proteases. Nat. Commun. 2018, 9, 1718. [Google Scholar] [CrossRef]
- Ghosh, D.K.; Motghare, M.; Gowda, S. Citrus greening: Overview of the most severe disease of citrus. Adv. Agric. Res. Technol. J. 2018, 2, 83–100. [Google Scholar]
- Wang, N.; Pierson, E.A.; Setubal, J.C.; Xu, J.; Levy, J.G.; Zhang, Y.Z.; Li, J.Y.; Rangel, L.T.; Martins, J. The Candidatus Liberibacter-Host Interface: Insights into Pathogenesis Mechanisms and Disease Control. Annu. Rev. Phytopathol. 2017, 55, 451–482. [Google Scholar] [CrossRef]
- Costa, T.R.; Felisberto-Rodrigues, C.; Meir, A.; Prevost, M.S.; Redzej, A.; Trokter, M.; Waksman, G. Secretion systems in Gram-negative bacteria: Structural and mechanistic insights. Nat. Rev. Microbiol. 2015, 13, 343–359. [Google Scholar] [CrossRef]
- da Graça, J.V.; Douhan, G.W.; Halbert, S.E.; Keremane, M.L.; Lee, R.F.; Vidalakis, G.; Zhao, H. Huanglongbing: An overview of a complex pathosystem ravaging the world’s citrus. J. Integr. Plant Biol. 2016, 58, 373–387. [Google Scholar] [CrossRef]
- Morgan, J.L.; Acheson, J.F.; Zimmer, J. Structure of a Type-1 Secretion System ABC Transporter. Structure 2017, 25, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Sugio, A.; MacLean, A.M.; Kingdom, H.N.; Grieve, V.M.; Manimekalai, R.; Hogenhout, S.A. Diverse Targets of Phytoplasma Effectors: From Plant Development to Defense Against Insects. Annu. Rev. Phytopathol. 2011, 49, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.Q.; Zhang, L.; Coaker, G.; Ma, W.B.; He, S.Y.; Wang, N. Citrus CsACD2 Is a Target of Candidatus Liberibacter Asiaticus in Huanglongbing Disease. Plant Physiol. 2020, 184, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919. [Google Scholar]
- Mannoor, M.S.; Zhang, S.Y.; Link, A.J.; McAlpine, M.C. Electrical detection of pathogenic bacteria via immobilized antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2010, 107, 19207–19212. [Google Scholar] [CrossRef]
- Nawrot, R.; Barylski, J.; Nowicki, G.; Broniarczyk, J.; Buchwald, W.; Goździcka-Józefiak, A. Plant antimicrobial peptides. Folia Microbiol. 2014, 59, 181–196. [Google Scholar] [CrossRef]
- Guerra-Lupian, M.A.; Ruiz-Medrano, R.; Ramirez-Pool, J.A.; Ramirez-Ortega, F.A.; Lopez-Buenfil, J.A.; Loeza-Kuk, E.; Morales-Galvan, O.; Chavarin-Palacio, C.; Hinojosa-Moya, J.; Xoconostle-Cazares, B. Localized expression of antimicrobial proteins mitigates huanglongbing symptoms in Mexican lime. J. Biotechnol. 2018, 285, 74–83. [Google Scholar] [CrossRef]
- Guzman, J.V.; Basu, S.; Rabara, R.; Huynh, L.; Basu, G.; Nguyen, H.; Shaw, J.; Shi, Q.; Zhang, S.; Stover, E.; et al. Liposome delivery system of antimicrobial peptides against Huanglongbing. Biophys. J. Phytopathol. 2018, 114, 266a. [Google Scholar] [CrossRef]
- Irigoyen, S.; Ramasamy, M.; Pant, S.; Niraula, P.; Bedre, R.; Gurung, M.; Rossi, D.; Laughlin, C.; Gorman, Z.; Achor, D.; et al. Plant hairy roots enable high throughput identification of antimicrobials against Candidatus Liberibacter spp. Nat. Commun. 2020, 11, 5802. [Google Scholar] [CrossRef]
- Van Gijsegem, F.; Genin, S.; Boucher, C. Conservation of secretion pathways for pathogenicity determinants of plant and animal bacteria. Trends Microbiol. 1993, 1, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Enguene, Y.V.N.; Phan, G.; Garnier, C.; Ducruix, A.; Prange, T.; Broutin, I. Xenon for tunnelling analysis of the efflux pump component OprN. PLoS ONE 2017, 12, e0184045. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck—A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.T.; Chou, T.H.; Su, C.C.; Bolla, J.R.; Kumar, N.; Radhakrishnan, A.; Long, F.; Delmar, J.A.; Do, S.V.; Rajashankar, K.R.; et al. Crystal Structure of the Open State of the Neisseria gonorrhoeae MtrE Outer Membrane Channel. PLoS ONE 2014, 9, e97475. [Google Scholar] [CrossRef]
- Kulathila, R.; Indic, M.; van den Berg, B. Crystal Structure of Escherichia coli CusC, the Outer Membrane Component of a Heavy Metal Efflux Pump. PLoS ONE 2011, 6, e15610. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Lim, Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef]
- Simmaco, M.; Mignogna, G.; Canofeni, S.; Miele, R.; Mangoni, M.L.; Barra, D. Temporins, antimicrobial peptides from the European red frog Rana temporaria. Eur. J. Biochem. 1996, 242, 788–792. [Google Scholar] [CrossRef]
- Ikeda, T.; Minakata, H.; Nomoto, K.; Kubota, I.; Muneoka, Y. 2 Novel Tachykinin-Related Neuropeptides in the Echiuroid Worm, Urechis-Unicinctus. Biochem. Biophys. Res. Commun. 1993, 192, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y. A new antibiotic’colistin’produced by spore-forming soil bacteria. J. Antibiot. 1950, 3, 457–458. [Google Scholar]
- Nation, R.L.; Li, J. Colistin in the 21st century. Curr. Opin. Infect. Dis. 2009, 22, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.M.; Han, J.Z.; Bie, X.M.; Lu, Z.X.; Zhang, C.; Lv, F.X. Purification and Characterization of Plantaricin JLA-9: A Novel Bacteriocin against Bacillus spp. Produced by Lactobacillus plantarum JLA-9 from Suan-Tsai, a Traditional Chinese Fermented Cabbage. J. Agric. Food Chem. 2016, 64, 2754–2764. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Lushnikova, T.; Golla, R.M.; Wang, X.Q.; Wang, G.S. Design and surface immobilization of short anti-biofilm peptides. Acta Biomater. 2017, 49, 316–328. [Google Scholar] [CrossRef]
- Zarena, D.; Mishra, B.; Lushnikoya, T.; Wang, F.Y.; Wang, G.S. The pi Configuration of the WWW Motif of a Short Trp-Rich Peptide Is Critical for Targeting Bacterial Membranes, Disrupting Preformed Biofilms, and Killing Methicillin-Resistant Staphylococcus aureus. Biochemistry 2017, 56, 4039–4043. [Google Scholar] [CrossRef]
- Imai, Y.; Meyer, K.J.; Iinishi, A.; Favre-Godal, Q.; Green, R.; Manuse, S.; Caboni, M.; Mori, M.; Niles, S.; Ghiglieri, M.; et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576, 459. [Google Scholar] [CrossRef]
- Hart, E.M.; Mitchell, A.M.; Konovalova, A.; Grabowicz, M.; Sheng, J.; Han, X.Q.; Rodriguez-Rivera, F.P.; Schwaid, A.G.; Malinverni, J.C.; Balibar, C.J.; et al. A small-molecule inhibitor of BamA impervious to efflux and the outer membrane permeability barrier. Proc. Natl. Acad. Sci. USA 2019, 116, 21748–21757. [Google Scholar] [CrossRef]
- Barnett, M.J.; Solow-Cordero, D.E.; Long, S.R. A high-throughput system to identify inhibitors of Candidatus Liberibacter asiaticus transcription regulators. Proc. Natl. Acad. Sci. USA 2019, 116, 18009–18014. [Google Scholar] [CrossRef]
- Pagliai, F.A.; Gonzalez, C.F.; Lorca, G.L. Identification of a ligand binding pocket in LdtR from Liberibacter asiaticus. Front. Microbiol. 2015, 6, 1314. [Google Scholar] [CrossRef]
- Gardner, C.L.; da Silva, D.R.; Pagliai, F.A.; Pan, L.; Padgett-Pagliai, K.A.; Blaustein, R.A.; Merli, M.L.; Zhang, D.; Pereira, C.; Teplitski, M. Assessment of unconventional antimicrobial compounds for the control of ‘Candidatus Liberibacter asiaticus’, the causative agent of citrus greening disease. Sci. Rep. 2020, 10, 5395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Han, Q.; Mao, X.; Liu, J.; Wang, W.; Li, D.; Zhou, F.; Ke, Y.; Xu, L.; Hu, L. Discovery of novel SecA inhibitors against “Candidatus Liberibacter asiaticus” through virtual screening and biological evaluation. Chem. Biol. Drug Des. 2021, 98, 395–404. [Google Scholar] [CrossRef]
- Gardner, C.L.; Pagliai, F.A.; Pan, L.; Bojilova, L.; Torino, M.I.; Lorca, G.L.; Gonzalez, C.F. Drug repurposing: Tolfenamic acid inactivates PrbP, a transcriptional accessory protein in Liberibacter asiaticus. Front. Microbiol. 2016, 7, 1630. [Google Scholar] [CrossRef] [PubMed]
- Wulff, N.A.; Zhang, S.; Setubal, J.C.; Almeida, N.F.; Martins, E.C.; Harakava, R.; Kumar, D.; Rangel, L.T.; Foissac, X.; Bové, J.M. The complete genome sequence of ‘Candidatus Liberibacter americanus’, associated with citrus huanglongbing. Mol. Plant-Microbe Interact. 2014, 27, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Blacutt, A.; Ginnan, N.; Dang, T.; Bodaghi, S.; Vidalakis, G.; Ruegger, P.; Peacock, B.; Viravathana, P.; Vieira, F.C.; Drozd, C. An in vitro pipeline for screening and selection of citrus-associated microbiota with potential anti-“Candidatus Liberibacter asiaticus” properties. Appl. Environ. Microbiol. 2020, 86, e02883-19. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Pontius, J.; Richelle, J.; Wodak, S. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Release, S. 4: Glide; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Wizard, P.P. Epik Version 2.8; Schrödinger LLC: New York, NY, USA, 2014. [Google Scholar]
- Release, S. 1: Maestro; Schrödinger LLC: New York, NY, USA, 2017. [Google Scholar]
- Release, S. 4: Desmond Molecular Dynamics System; DE Shaw Research: New York, NY, USA, 2017. [Google Scholar]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate—DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Template | Chain | GMQE | QSQE | Seq Identity | Seq Similarity | Membrane Protein | Refs. |
|---|---|---|---|---|---|---|---|
| 5iuy | A | 0.71 | 0.51 | 0.30 | 0.35 | OprN | [40] |
| 4mt0 | A | 0.71 | 0.47 | 0.26 | 0.33 | MtrE | [44] |
| 3pik | A | 0.71 | 0.43 | 0.24 | 0.32 | CusC | [45] |
| No. | Peptide | Anti-Gram-Negative | Definition a | APD3 ID | Length | Hydrophobic Residue% | Net Charge |
|---|---|---|---|---|---|---|---|
| 1 | LSPNLLKSLL | No | Temporin H (linear) | AP00859 | 10 | 50 | 2 |
| 2 | LRQSQFVGSR | Yes | Urechistachykinin I (linear) | AP01480 | 10 | 30 | 3 |
| 3 | AAGMGFFGAR | Yes | Urechistachykinin II (linear) | AP01481 | 10 | 60 | 2 |
| 4 | KTKKKLLKKT | Yes | Colistin A (cyclic) | AP02204 | 10 | 20 | 6 |
| 5 | FLPLIGRVLSGIL | Yes | Temporin A (linear) | AP00094 | 13 | 61 | 2 |
| 6 | FWQKMSFA | Yes | Plantaricin JLA-9 (linear) | AP02677 | 8 | 62 | 1 |
| 7 | WWWLRKIW | Yes | TetraF2W-RK (linear) | AP02856 | 8 | 75 | 3 |
| 8 | WNWSKSF | Yes | Darobactin (bicyclic) | AP03168 | 7 | 42 | 1 |
| Definition | Docking Score | Glide Model | H-Bond | π-π Stack | π-Cation |
|---|---|---|---|---|---|
| Temporin H | −8.971 | −120.362 | G: ASP314; H: ASN315, SER316; I: ASN315, SER316 | ||
| Urechistachykinin I | −8.610 | −88.383 | G: ASP314, ASN315; H: ASP314, ASN315, SER316; I: SER316 | ||
| Urechistachykinin II | −9.332 | −146.077 | G: LYS113, HIE313; H: ASN315, SER316, PHE317; I: SER316; | I: PHE317 | |
| Colistin A | −7.325 | −45.045 | G: ASP314, SER316, PHE317, TYR320; H: ARG107, ASP314; I: SER316 | G: TYR320 | |
| Temporin A | −2.787 | 2.406 | G: ASP314, ASN315 | ||
| Plantaricin JLA-9 | −9.002 | −109.183 | G: HIE313, ASP314; H: ASP314, ASN315, SER316; I: SER316 | I: TYR320 | H: ASP314 |
| TetraF2W-RK | −8.700 | −122.832 | G: ASP314, ASN315, ASN321; I: SER316 | G: PHE317 | |
| Darobactin | −9.605 | −144.059 | G: ASP314, ASN315; H: ASN315, SER316; I: ASN312 | H: ASP314, PHE317 | |
| MRL-494 | −7.678 | −66.449 | G: ASP314; H: ASP314, ASN315; |
| Compound Name | ΔGbind Overall (kcal/mol) | ΔG Coulomb Energy (kcal/mol) | ΔG Covalent Binding Energy (kcal/mol) | ΔG Lipophilic. Energy (kcal/mol) | ΔG Van der Waals Energy (kcal/mol) | ΔG Generalized Born Electrostatic Solvation Energy (kcal/mol) | ΔG Ligand Strain (kcal/mol) |
|---|---|---|---|---|---|---|---|
| Darobactin | −42.09 ± 11.20 | −7.78 ± 40.00 | 1.49 ± 4.29 | −8.10 ± 4.60 | −48.49 ± 7.58 | 23.70 ± 35.44 | 17.69 ± 9.98 |
| Plantaricin JLA-9 | −51.84 ± 10.90 | −8.24 ± 14.95 | 1.34 ± 3.48 | −13.09 ± 3.39 | −46.83 ± 10.72 | 18.40 ± 12.91 | 10.30 ± 6.76 |
| Urechistachykinin II | −46.15 ± 12.09 | −19.16 ± 24.41 | −1.27 ± 4.50 | −13.88 ± 2.79 | −48.25 ± 8.78 | 39.78 ± 20.54 | 29.48 ± 5.98 |
| MRL-494 | −63.55 ± 8.51 | −39.08 ± 26.15 | 5.64 ± 2.20 | −12.55 ± 1.97 | −43.96 ± 4.64 | 34.24 ± 25.02 | 7.35 ± 2.88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Mulgaonkar, N.; Mallawarachchi, S.; Ramasamy, M.; Padilla, C.S.; Irigoyen, S.; Coaker, G.; Mandadi, K.K.; Fernando, S. Evaluation of Candidatus Liberibacter Asiaticus Efflux Pump Inhibition by Antimicrobial Peptides. Molecules 2022, 27, 8729. https://doi.org/10.3390/molecules27248729
Wang H, Mulgaonkar N, Mallawarachchi S, Ramasamy M, Padilla CS, Irigoyen S, Coaker G, Mandadi KK, Fernando S. Evaluation of Candidatus Liberibacter Asiaticus Efflux Pump Inhibition by Antimicrobial Peptides. Molecules. 2022; 27(24):8729. https://doi.org/10.3390/molecules27248729
Chicago/Turabian StyleWang, Haoqi, Nirmitee Mulgaonkar, Samavath Mallawarachchi, Manikandan Ramasamy, Carmen S. Padilla, Sonia Irigoyen, Gitta Coaker, Kranthi K. Mandadi, and Sandun Fernando. 2022. "Evaluation of Candidatus Liberibacter Asiaticus Efflux Pump Inhibition by Antimicrobial Peptides" Molecules 27, no. 24: 8729. https://doi.org/10.3390/molecules27248729
APA StyleWang, H., Mulgaonkar, N., Mallawarachchi, S., Ramasamy, M., Padilla, C. S., Irigoyen, S., Coaker, G., Mandadi, K. K., & Fernando, S. (2022). Evaluation of Candidatus Liberibacter Asiaticus Efflux Pump Inhibition by Antimicrobial Peptides. Molecules, 27(24), 8729. https://doi.org/10.3390/molecules27248729