


The Chemistry of Phenylimidotechnetium(V) Complexes with Isocyanides: Steric and Electronic Factors

 ,
,
Abstract
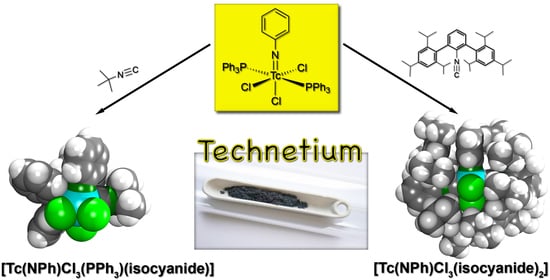
1. Introduction
2. Results and Discussion
2.1. The Ligands
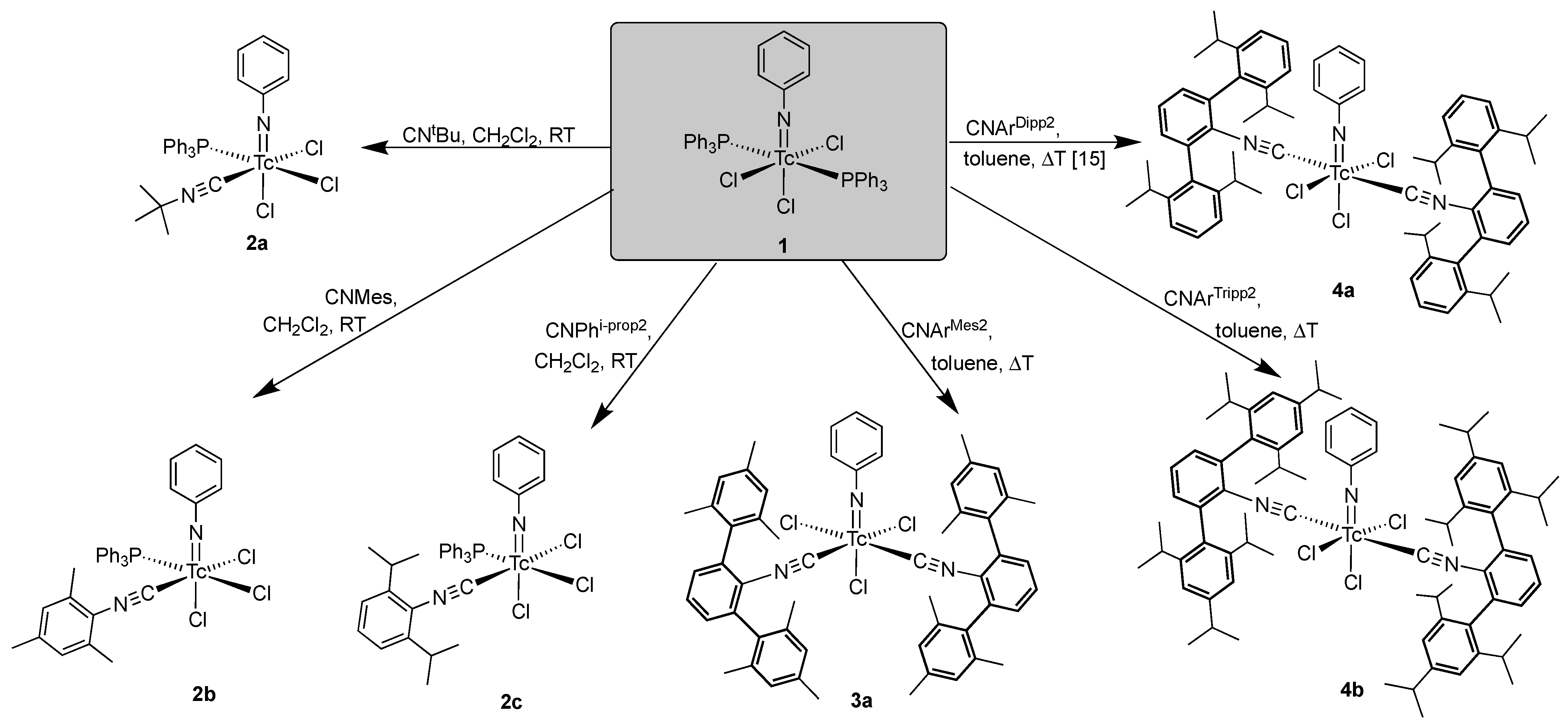
2.2. Reactions of [Tc(NPh)Cl3(PPh3)2] with Alkyl and (Alkyl-Substituted) Aryl Isocyanides
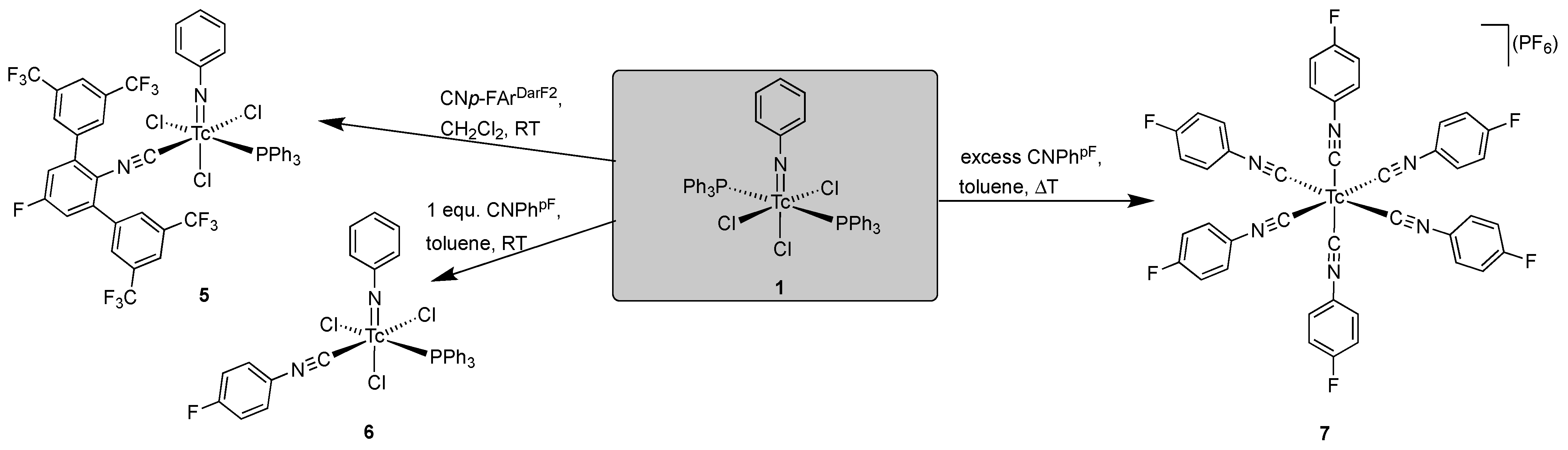
2.3. Reactions of [Tc(NPh)Cl3(PPh3)2] with Fluorine-Substituted Aryl Isocyanides
3. Materials and Methods
3.1. Radiation Precautions
3.2. Syntheses
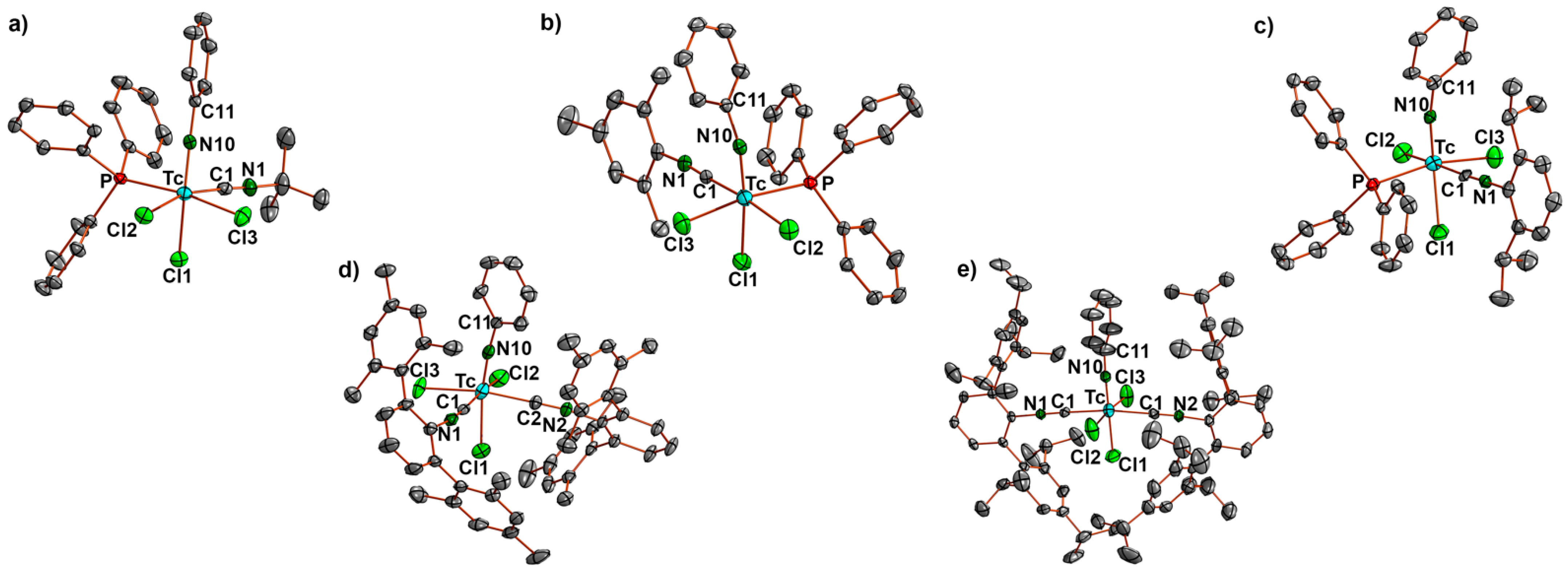
- [Tc(NPh)Cl3(PPh3)(CNtBu)] (2a): Green needles. Yield: 15 mg, 47%. IR (cm−1): 3057 (w), 2984 (w), 2918 (w), 2207 (s, νC≡N), 1437 (s), 1191 (m), 1092 (m), 749 (m), 695 (s), 525 (s). 1H NMR (CD2Cl2, ppm): δ = 7.82 (mc, 6H, o-H (PPh3)), 7.65 (t, J = 7.46 Hz, 1H, p-H (arom. NPh)), 7.46 (mc, 3H, p-H (PPh3)), 7.39 (mc, 6H, m-H (PPh3)), 7.30 (d, J = 7.84 Hz, 2H, o-H (arom. NPh)), 7.17 (t, J = 7.75 Hz, 2H, m-H (arom. NPh)), 1.38 (s, 9H, (CH3)3).
- [Tc(NPh)Cl3(PPh3)(CNMes)] (2b): Green needles. Yield: 10 mg, 28%. IR (cm−1): 3057 (w), 2920 (w), 2187 (s, νC≡N), 1480 (w), 1435 (m), 1310 (w), 1092(m), 747 (m), 693 (s), 523 (s). 1H NMR (CD2Cl2, ppm): 7.87 (mc, 6H, o-H(PPh3)), 7.66 (t, J = 7.47 Hz, 1H, p-H (arom. NPh)), 7.41–7.31 (m, 11H, m-/p-H(PPh3), o-H (arom. NPh)), 7.18 (t, J = 7.77 Hz, 2H, m-H (arom. NPh)), 6.87 (s, 2H, m-H (arom. CNMes)), 2.32 (s, 3H, p-CH3 (CNMes)), 1.94 (s, 6H, o-CH3 (CNMes)).
- [Tc(NPh)Cl3(PPh3)(CNPhi-prop2)] (2c): Green-yellow, dichroic needles. Yield: 26 mg, 70%. IR (cm−1): 3055 (w), 2695 (m), 2922 (w), 2183 (s, νC≡N), 1572 (m), 1477 (m), 1433 (s), 980 (w), 804 (w), 746 (m), 692 (m), 525 (m). 1H NMR (CD2Cl2, ppm): 7.89 (mc, 6H, o-H(PPh3)), 7.69 (t, J = 8.0 Hz, 1H, p-H (arom. NPh)), 7.43–7.29 (m, 12H, m-/p-H (PPh3), H(CNPhi-prop2)), 7.20 (t, J = 8.0 Hz, 2H, m-H (arom. NPh)), 7.14 (d, J = 7.7 Hz, 2H, o-H (arom. NPh)), 2.68 (h, J = 6.8 Hz, i-prop CH), 0.97 (d, J = 7.0 Hz, i-prop CH3), 0.89 (d, J = 7.0 Hz, i-prop CH3).
- Subsequently, cis-[Tc(NPh)Cl3(CNArMes2)2] (3a): [Tc(NPh)Cl3(PPh3)] (1) (82 mg, 0.1 mmol) was suspended in toluene (5 mL). CNArMes2 (68 mg, 0.2 mmol) was added, and the reaction mixture was heated under reflux for one hour. It became dark green and homogenous upon heating. The resultant solution was slowly evaporated at 5 °C. After one day, the first crop of a few yellow-green needles (compound 3a) suitable for X-ray diffraction were obtained and analyzed by IR spectroscopy. Upon further evaporation of the solvent, more of the aforementioned needles was obtained along with other green crystals of different shapes, which were not suitable for X-ray diffraction. They were filtered off and washed with small amounts of n-pentane and studied by 1H NMR spectroscopy. Three sets of resonances were observed in the methyl region, suggesting the presence of at least three isomers which could be cis/trans-[Tc(NPh)Cl3(CNArMes2)2] or cis/trans-[Tc(NPh)Cl3(PPh3)(CNArMes2)]. 3a: Yellow-green needles. IR (cm−1): 3058 (w), 2919 (m), 2851 (w), 2178 (s, νC≡N), 1572 (m), 1433 (m), 1308 (w), 1094 (w), 845 (w), 749 (w), 695 (m), 521 (m). The isomeric mixture was of [Tc(NPh)Cl3(CNArMes2)2] and [Tc(NPh)Cl3(PPh3)(CNArMes2)]: IR (cm−1): 3058 (w), 2919 (m), 2851 (w), 2178 (s, νC≡N), 1572 (m), 1433 (m), 1308 (w), 1094 (w), 845 (w), 749 (w), 695 (m), 521 (m). 1H NMR (CD2Cl2, ppm): 7.78–6.58 (m, aryl), 2.20 (s, CH3), 2.11 (s, CH3), 2.05 (s, CH3), 2.02 (s, CH3), 1.96 (s, CH3), 1.94 (s, CH3).
- trans-[Tc(NPh)Cl3(CNArTripp2)2] (4b): [Tc(NPh)Cl3(PPh3)] (1) (41 mg, 0.05 mmol) was suspended in toluene (5 mL). CNArTripp2 (51 mg, 0.1 mmol) was added, and the reaction mixture was heated under reflux for one hour. It became slightly red and homogenous upon heating. The resultant solution was slowly evaporated at 5 °C. After five days, green-yellow crystals suitable for X-ray diffraction were obtained. They were filtered off and washed with small amounts of cold MeOH and n-pentane, and then dried under a reduced pressure. Yield: 39 mg, 72%. IR (cm−1): 3057 (w), 2957 (s), 2866 (s), 2184 (s, νC≡N), 1607 (m), 1576 (m), 1570 (m), 1461 (s), 1439 (s), 1362 (s), 1316 (m), 1191 (m), 1121 (m), 1071 (w), 943 (w), 874 (s), 807 (m), 722 (m), 697 (s), 543 (s). 1H NMR (CD2Cl2, ppm): 7.73 (t, J = 7.33 Hz, 1H, p-H (arom. NPh)), 7.50 (t, J = 6.8 Hz, 2H, CNArTripp2), 7.41 (d, J = 7.46 Hz, 2H, o-H (arom. NPh)), 7.26 (d, J = 7.46 Hz, 4H, CNArTripp2), 7.14 (t, J = 6.68 Hz, 2H, m-H (arom. NPh)), 6.92 (s, 8H, CNArTripp2), 2.83 (h, J = 6.1 Hz, 4H, (i-prop CH)), 2.38 (h, J = 6.3 Hz, 8H, (i-prop CH)), 1.32 (d, J = 6.52 Hz, 24H, (i-prop CH3)), 1.01 (d, J = 6.0 Hz, 24H, (i-prop CH3)), 0.96 (d, J = 6.0 Hz, 24H, (i-prop CH3)).
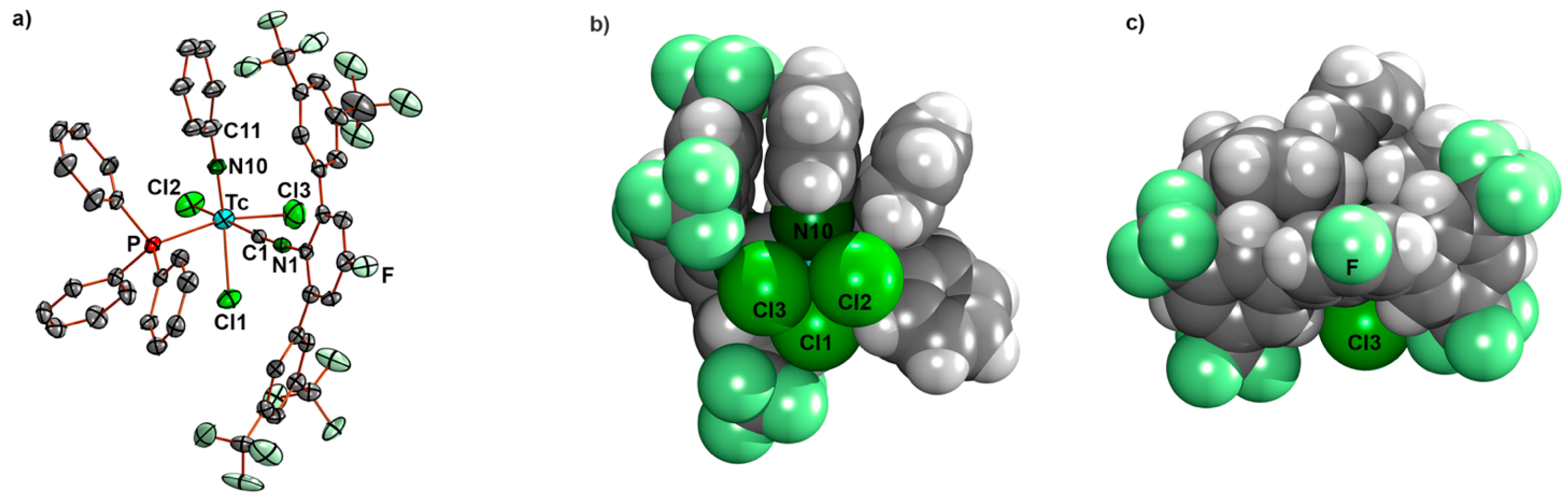
- [Tc(NPh)Cl3(PPh3)(CNp-FArDarF2)] (5): [Tc(NPh)Cl3(PPh3)2] (1) (41 mg, 0.05 mmol) was dissolved in CH2Cl2 (5 mL). CNp-FArDarF2 (30 mg, 0.055 mmol) was added, and the dark green solution was stirred for 20 min at room temperature. A pale green solid was precipitated by the addition of an excess of n-hexane (approximately 30 mL). The immediately formed precipitate was washed with pentane and a small amount of diethyl ether, redissolved in CH2Cl2 (1 mL), and overlayered with n-hexane. Pale green columns were formed together with brown oil. The crystals were separated and washed with pentane alongside a small amount of diethyl ether, and the crystallization procedure was repeated in the described way. The resultant single crystals were suitable for X-ray diffraction. Pale green needles. Yield: 22 mg, 40%. IR (cm−1): 3057 (w), 2176 (vs, νC≡N), 1482 (w), 1435 (m), 1364 (m), 1279 (s), 1179 (s), 1135 (s), 1092 (m), 905 (w), 743 (m), 701 (m), 693 (m) 520 (m). 1H NMR (CD2Cl2, ppm): 7.90 (s, 4H, CNp-FArDarF2), 7.61 (s, 2H, CNp-FArDarF2), 7.55 (t, J = 7.1 Hz, 1H, p-H (arom. NPh)), 7.46 (t, J = 8.4 Hz, 6H, o-H (PPh3)), 7.31 (t, J = 6.9 Hz, 3H, p-H (PPh3)), 7.25 (d, J = 7.7 Hz, 2H, m-H (CNp-FArDarF2)), 7.16 (t, J = 6.0 Hz, 6H, m-H (PPh3)), 6.89 (t, J = 7.2 Hz, 2H, m-H (arom. NPh)), 6.71 (d, J = 6.9 Hz, 2H, o-H (arom. NPh)). 19F NMR (CD2Cl2, ppm): −65.0 (s, 12F, m,m’-CF3 (CNp-FArDarF2)), −107.9 (s, 1F, p-F (CNp-FArDarF2)).
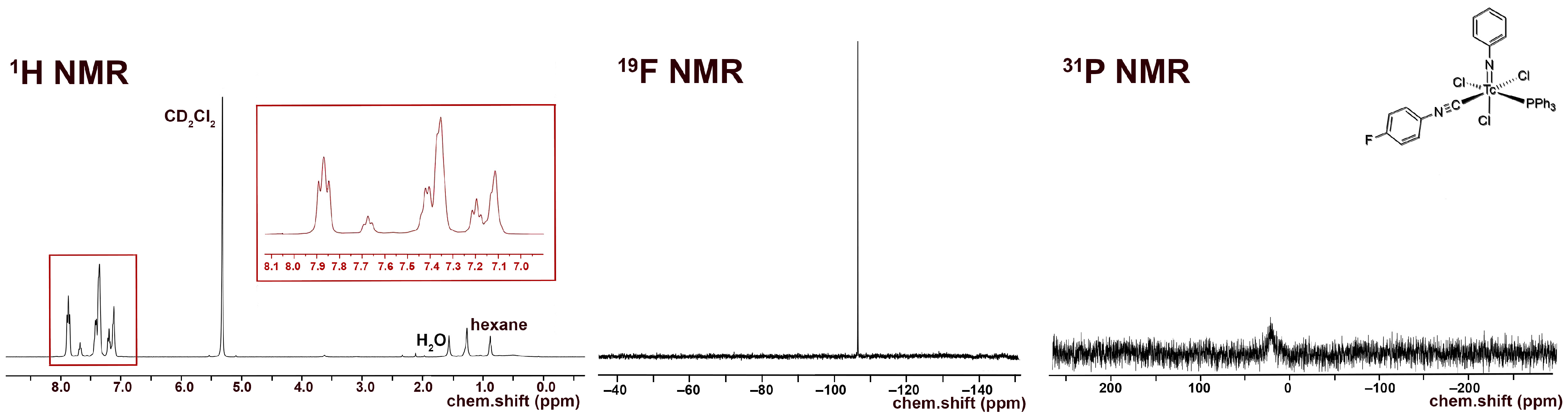
- [Tc(NPh)Cl3(PPh3)(CNPhF)] (6): 100 µL of a solution prepared from CNPhpF (160 µL) and toluene (940 µL was added to a suspension of [Tc(NPh)Cl3(PPh3)2] (1) (41 mg, 0.05 mmol) in CH2Cl2 (5 mL). The mixture was gently heated and held at a temperature of 30 °C until the reaction mixture became homogenous (approximately 2 min). Then, n-hexane (20 mL) was immediately added, which resulted in the formation of a pale green precipitate. The obtained solid (pure compound 6 with some incorporated n-hexane) was filtered off, washed with diethyl ether alongside n-hexane, and then dried under reduced pressure. Complex 6 is stable as a solid but decomposes at room temperature in solvents such as dichloromethane or acetone. Yield: 20 mg, 57%. IR (cm−1): 3422 (br), 3058 (w), 2923 (w), 2186 (vs, νC≡N), 1570 (w), 1499 (s), 1435 (m), 1239 (w), 1092 (m), 990 (w), 841 (m), 749 (m), 697 (s), 521 (s). 1H NMR (CD2Cl2, ppm): 7.80 (mc, 6H, o-H(PPh3)), 7.68 (t, J = 8.0 Hz, 1H, p-H (arom. NPh)), 7.46–7.31 (m, 11H), 7.20 (t, J = 8.0 Hz, 2H, m-H (arom.)), 7.17–7.07 (m, 4H), 1.27 (CH2, 0.2 n-hexane), 0.88 (CH3, 0.2 n-hexane). 19F NMR (CD2Cl2, ppm): −106.5.
- [Tc(CNPhpF)6](PF6) (7): [Tc(NPh)Cl3(PPh3)2] (1) (41 mg, 0.05 mmol) was suspended in toluene (5 mL). CNPhpF (45.9 μL, 0.5 mmol) was added, and the solution was heated under reflux for one hour. The reaction mixture became homogeneous upon heating and changed its color to pale yellow within the first ten minutes. Then, a solid started to precipitate. The reaction mixture was cooled to room temperature and filtered. The obtained solid was washed with a small amount of toluene and redissolved in MeOH. NH4(PF6) (0.5 g) was dissolved in a water/MeOH mixture (5 mL, 1:1) which was added. A colorless solid precipitated, which was filtered off and washed sequentially with water, MeOH, and Et2O. Yield: 12 mg, 26%. IR (cm−1): 2918 (w), 2087 (s, νC≡N), 1501 (m), 1235 (m), 1154 (m), 836 (m), 558 (w). 1H NMR (CD2Cl2, ppm): 7.46 (mc, 12H, o-H (CNPhpF)), 7.16 (t, J = 8.5 Hz, 12H, m-H (CNPhpF)). 19F NMR (CD2Cl2, ppm): −73.4 (d, 1J (19F-31P) = 670 Hz, 6F, PF6), −109.2 (s, 6F, (CNPhpF)). 99Tc NMR (CD2Cl2, ppm): −1886 (s, ν1/2 = 42 Hz).
3.3. X-ray Crystallography
3.4. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kronauge, J.F.K.; Mindiola, D.J. The value of Stable Metal-Carbon Bonds in Nuclear Medicine and the Cardiolite Story. Organometallics 2016, 35, 3432–3435. [Google Scholar] [CrossRef]
- Abrams, M.J.; Davison, A.; Jones, A.G.; Costello, C.; Pang, H. Synthesis and Characterization of Hexakis(alkyl isocyanide) and Hexakis(aryl isocyanide) Complexes of Technetium(I). Inorg. Chem. 1983, 22, 2798–2800. [Google Scholar] [CrossRef]
- Abrams, M.J.; Davison, A.; Faggiani, R.; Jones, A.G.; Lock, C.J.L. Chemistry and Structure of Hexakis(thiourea-S)technetium(III) Trichloride Tetrahydrate, [Tc(SC(NH2)2)6]Cl3⋅4H2O. Inorg. Chem. 1984, 23, 3284–3288. [Google Scholar] [CrossRef]
- Technetium-99m Radiopharmaceuticals: Manufacture of Kits; IAEA Technical Reports Series No. 466; International Atomic Energy Agency: Vienna, Austria, 2008; pp. 126–129.
- Herman, L.W.; Sharma, V.; Kronauge, J.F.; Barbaric, E.; Herman, L.; Piwnica-Worms, D. Novel Hexakis(areneisonitrile)technetium(I) Complexes as Radioligands Targeted to the Multidrug Resistance P-Glycoprotein. J. Med. Chem. 1995, 38, 2955–2963. [Google Scholar] [CrossRef] [PubMed]
- Piwnica-Worms, D.; Kronauge, J.F.; Holman, B.L.; Davison, A.; Jones, A.G. Comparative Myocardial Uptake Characteristics of Hexakis (Alkylisonitrile) Technetium(I) Complexes. Investig. Radiol. 1989, 24, 25–29. [Google Scholar] [CrossRef]
- Kronauge, J.F.; Davison, A.; Roseberrry, A.M.; Costello, C.E.; Maleknia, S.; Jones, A.G. Synthesis, and Identification of the Monocation Tc(CPI)6+ in Tc(CNC(CH3)2COOCH3)6Cl and Its Hydrolysis Products. Inorg. Chem. 1991, 30, 4265–4271. [Google Scholar] [CrossRef]
- World Nuclear Association. Radioisotopes in Medicine. Available online: https://www.world-nuclear.org/information-library/non-power-nuclear-applications/radioisotopes-research/radioisotopes-in-medicine.aspx (accessed on 21 August 2022).
- Patil, P.; Ahmadian-Moghaddam, M.; Dömling, A. Isocyanide 2.0. Green Chem. 2020, 22, 6902–6911. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Mokhtarzadeh, C.C.; Ripatti, D.S.; Havrylyuk, I.; Kamezawa, R.; Moore, C.E.; Rheingold, A.L.; Figueroa, J.S. Comparative Measure of the Electronic Influence of Highly Substituted Aryl Isocyanides. Inorg. Chem. 2015, 54, 2936–2944. [Google Scholar] [CrossRef]
- Claude, G.; Genz, J.; Weh, D.; Roca Jungfer, M.; Hagenbach, A.; Gembicky, M.; Figueroa, J.S.; Abram, U. Mixed-isocyanide Complexes of Technetium under Steric and Electronic Control. Inorg. Chem. 2022, 61, 16163–16176. [Google Scholar] [CrossRef]
- Hahn, F.E.; Imhof, L.; Lügger, T. Synthesis of trichlorooxo-bis(2,6-dimethylphenylisocyanide) rhenium(V) and crystal structure of μ-oxo-bis[dichlorooxo-bis(2,6-dimethylphenylisocyanide) dirhenium(V)]. Inorg. Chim. Acta 1998, 269, 347–349. [Google Scholar] [CrossRef]
- Bryan, J.; Stenkamp, R.E.; Tulip, T.H.; Mayer, J.M. Oxygen atom transfer among rhenium, sulfur, and phosphorus. Characterization and reactivity of Re(O)Cl3(Me2S)(OPPh3) and Re(O)Cl3(CNCMe3)2. Inorg. Chem. 1987, 26, 2283–2288. [Google Scholar] [CrossRef]
- Figueroa, J.S.; Abram, U. Oxidorhenium(V) and Rhenium(III) Complexes with m-Terphenyl Isocyanides. Z. Anorg. Allg. Chem. 2020, 646, 909–914. [Google Scholar] [CrossRef]
- Claude, G.; Salsi, F.; Hagenbach, A.; Gembicky, M.; Neville, M.; Chan, C.; Figueroa, J.S.; Abram, U. Structural and Redox Variations in Technetium Complexes Supported by m-Terphenyl Isocyanides. Organometallics 2020, 39, 2287–2294. [Google Scholar] [CrossRef]
- Nicholson, T.; Davison, A.; Jones, A.G. The synthesis of a technetium(V) phenylimido complex from pertechnetate. The single crystal X-ray structure of [TcCl3(NPh)(PPh3)2]-CH2Cl2. Inorg. Chim. Acta 1991, 187, 51–57. [Google Scholar] [CrossRef]
- Chatt, J.; Garforth, J.D.; Johnson, N.P.; Rowe, G.A. Nitrido- and Arylimido-complexes of Rhenium. J. Chem. Soc. 1964, 1012–1020. [Google Scholar] [CrossRef]
- Nicholson, T.; Storm, S.L.; Davis, W.M.; Davison, A.; Jones, A.G. The synthesis and characterization of [TcCl3(NPh)(Ph2PCH2CH2PPh2)] and [TcCl3(NPh)(PPh3)2]. The single crystal X-ray structure of [TcCl3(NPh)(Ph2PCH2CH2PPh2)]. Inorg. Chim. Acta 1992, 196, 27–34. [Google Scholar] [CrossRef]
- Rochon, F.F.; Melanson, R.; Kong, P.-C. Synthesis and Crystal Structures of (Phosphine)technetium(V) Complexes with Phenylimido and Phenyldiazenido Ligands Using the Precursor PhNHNHCOCH3. Inorg. Chem. 1995, 34, 2273–2277. [Google Scholar] [CrossRef]
- Nicholson, T.; Davison, A.; Zubieta, J.A.; Chen, C.; Jones, A.G. The synthesis and characterization of a cationic technetium(V) phenylimido complex. The X-ray crystal structure of [TcCl2(NPh)(PMe2Ph)3](BPh4). Inorg. Chim. Acta 1995, 230, 205–208. [Google Scholar] [CrossRef]
- Kuhn, B.; Abram, U. Phenylimido Complexes of Technetium and Rhenium with Maleonitriledithiolate. Z. Anorg. Allg. Chem. 2011, 637, 242–245. [Google Scholar] [CrossRef]
- Porchia, M.; Tisato, F.; Refosco, F.; Bolzati, C.; Cavazza-Ceccato, M.; Dolmella, A. New Approach to the Chemistry of Technetium(V) and Rhenium(V) Phenylimido Complexes: Novel [M(NPh)PNP]3+ Metal Fragments (M = Tc, Re; PNP = Aminodiphosphine) Suitable for the Synthesis of Stable Mixed-Ligand Compounds. Inorg. Chem. 2005, 44, 4766–4776. [Google Scholar] [CrossRef]
- Kuznetsov, M.L. Theoretical studies of transition metal complexes with nitriles and isocyanides. Russ. Chem. Rev. 2002, 71, 265–282. [Google Scholar] [CrossRef]
- Csonka, I.P.; Szepes, L.; Modelli, A. Donor–acceptor properties of isonitriles studied by photoelectron spectroscopy and electron transmission spectroscopy. J. Mass Spectrom. 2004, 39, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Saillard, J.Y.; Le Beuze, A.; Simonneaux, G.; Le Maux, P.; Jaouen, G. Variation of R in the isocyanide series RNC: As an example of the concept of controlled modifications of the properties of ligands for organometallic synthesis. J. Mol. Struct. (Theochem) 1981, 86, 149–154. [Google Scholar] [CrossRef]
- Guy, M.P.; Guy, J.T., Jr.; Bennett, D.W. A theoretical comparison of the electronic structures of alkyl and aryl isocyanide ligands. J. Mol. Struct. (Theochem) 1985, 122, 95–99. [Google Scholar] [CrossRef]
- Hieber, W.; Lux, F.; Herget, C. Über Kohlenoxidverbindungen des Technetiums. Z. Naturforsch. 1965, 20b, 1159–1165. [Google Scholar] [CrossRef]
- Lorenz, B.; Findeisen, M.; Olk, B.; Schmidt, K. Technetium(I) Complexes Tc(CO)3BrL2 (L = Phosphine, Pyridine, Isocyanide). Z. Anorg. Allg. Chem. 1988, 566, 160–168. [Google Scholar] [CrossRef]
- Alberto, R.; Schibli, R.; Egli, A.; Hermann, W.A.; Artus, G.; Abram, U.; Kaden, T.A. Metal carbonyl syntheses. XXII. Low-pressure carbonylation of [MOCl4]− and [MO4]−. The technetium(I) and rhenium(l) complexes [NEt4]2[MCl3(CO)3]. J. Organomet. Chem. 1995, 492, 217–224. [Google Scholar] [CrossRef]
- Alberto, R.; Schibli, R.; Angst, D.; Schubiger, P.A.; Abram, U.; Abram, S.; Kaden, T.A. Application of technetium and rhenium carbonyl chemistry to nuclear medicine. Preparation of [NEt4]2[TcCl3(CO)3] from [NBu4][TcO4] and structure of [NEt4][Tc2(µ-Cl)3(CO)6]; structures of the model complexes [NEt4][Re2(µ-OEt)2(µ-OAc)(CO)6] and [ReBr({-CH2S(CH2)2Cl}2)(CO)3]. Transit. Met. Chem. 1997, 22, 597–601. [Google Scholar]
- Ditri, T.; Fox, B.; Moore, C.; Rheingold, A.; Figueroa, J. Effective Control of Ligation and Geometric Isomerism: Direct Comparison of Steric Properties Associated with Bis-mesityl and Bis-diisopropylphenyl m-Terphenyl Isocyanides. Inorg. Chem. 2009, 48, 8362–8375. [Google Scholar] [CrossRef]
- Stewart, M.A.; Moore, C.E.; Ditri, T.B.; Labios, L.A.; Rheingold, A.L.; Figueroa, J.S. Electrophilic functionalization of well-behaved manganese monoanions supported by m-terphenyl isocyanides. Chem. Commun. 2011, 47, 406–408. [Google Scholar] [CrossRef]
- Ditri, T.B.; Carpenter, A.E.; Ripatti, D.S.; Moore, C.E.; Rheingold, A.L.; Figueroa, J.S. Chloro- and Trifluoromethyl-Substituted Flanking-Ring m-Terphenyl Isocyanides: η6-Arene Binding to Zero-Valent Molybdenum Centers and Comparison to Alkyl-Substituted Derivatives. Inorg. Chem. 2013, 52, 13216–13229. [Google Scholar] [CrossRef] [PubMed]
- Salsi, S.; Neville, M.; Drance, M.; Hagenbach, A.; Chan, C.C.; Figueroa, J.S.; Abram, U. A closed-shell monomeric rhenium(1-) anion provided my m-terphenyl isocyanide ligation. Chem. Commun. 2020, 56, 7009–7012. [Google Scholar] [CrossRef] [PubMed]
- Salsi, F.; Neville, M.; Drance, M.; Hagenbach, A.; Figueroa, J.S.; Abram, U. {MI(CO)X(CNp-FArDArF2)4} (DArF = 3,5-(CF3)2C6H3; M = Re, Tc; X = Br, Cl) Complexes: Convenient Platforms for the Synthesis of Low-valent Rhenium and Technetium Compounds. Organometallics 2021, 40, 1336–1343. [Google Scholar] [CrossRef]
- Tasi, G.; Pálinkó, I. Using Molecular Electrostatic Potential Maps for Similarity Studies. Top. Curr. Chem. 1995, 174, 46–71. [Google Scholar]
- Kikuchi, O.; Yamaguchi, K.; Morihashi, K.; Yokoyama, Y.; Nakayama, M. Molecular Electrostatic Potential Map Analysis of Metal Cation Interaction with Nucleophilic Molecules. Bull. Chem. Soc. Jpn. 1993, 66, 2412–2414. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Chan, C.; Rheingold, A.L.; Figueroa, J.S. A Well-Defined Isocyano Analogue of HCo(CO)4. 1: Synthesis, Decomposition, and Catalytic 1,1-Hydrogenation of Isocyanodes. Organometallics 2016, 35, 2319–2326. [Google Scholar] [CrossRef]
- Scholtysik, C.; Roca Jungfer, M.; Hagenbach, A.; Abram, U. Reactions of [ReOCl3(PPh3)2] with 4-Fluoroaniline. Z. Anorg. Allg. Chem. 2018, 644, 1451–1455. [Google Scholar] [CrossRef]
- Scholtysik, C.; Njiki Noufele, C.; Hagenbach, A.; Abram, U. Complexes of Technetium(V) and Rhenium(V) with β-Diketonates. Inorg. Chem. 2019, 58, 5241–5252. [Google Scholar] [CrossRef]
- Wendlandt, D.; Bauche, J.; Luc, P. Hyperfine structure in Tc I: Experiment and theory. J. Phys. B At. Mol. Phys. 1977, 10, 1989–2002. [Google Scholar] [CrossRef]
- Mikhalev, V.A. 99mTc NMR Spectroscopy. Radiochemistry 2005, 47, 319–333. [Google Scholar] [CrossRef]
- Abram, U.; Lorenz, L.; Kaden, L.; Scheller, D. Nitrido Complexes of Technetium with Tertiary Phosphines and Arsines. Polyhedron 1988, 7, 285–289. [Google Scholar] [CrossRef]
- O’Connell, L.A.; Pearlstein, R.M.; Davison, A.; Thornback, J.R.; Kronauge, J.F.; Jones, A.G. Technetium-99 NMR spectroscopy: Chemical shift trends and long range coupling effects. Inorg. Chim. Acta 1989, 161, 39–43. [Google Scholar] [CrossRef]
- Ackermann, J.; Abdulkader, A.; Scholtysik, C.; Roca Jungfer, M.; Hagenbach, A.; Abram, U. [TcI(NO)X(Cp)(PPh3)] Complexes (X− = I−, I3−, SCN−, CF3SO3−, or CF3COO−) and Their Reactions. Organometallics 2019, 38, 4471–4478. [Google Scholar] [CrossRef]
- Roca Jungfer, M.; Elsholz, L.; Abram, U. Technetium Hydrides Revisited: Syntheses, Structures, and Reactions of [TcH3(PPh3)4] and [TcH(CO)3(PPh3)2]. Organometallics 2021, 40, 3095–3112. [Google Scholar] [CrossRef]
- Coppens, P. The Evaluation of Absorption and Extinction in Single-Crystal Structure Analysis. Crystallographic Computing; Muksgaard: Copenhagen, Denmark, 1979. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Putz, H.; Brandenburg, K. (Eds.) DIAMOND, Crystal and Molecular Structure Visualization Crystal Impact; Version 4.6.5; GbR: Bonn, Germany, 2021. [Google Scholar]
- Bennett, L.; Melchers, B.; Proppe, B. Curta: A General-Purpose High-Performance Computer at ZEDAT, Freie Universität Berlin. Internal Report. Available online: https://refubium.fu-berlin.de/handle/fub188/26993 (accessed on 21 August 2022). [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, C.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 17. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Sundermann, A. Correlation consistent valence basis sets for use with the Stuttgart-Dresden-Bonn relativistic effective core potentials: The atoms Ga-Kr and In-Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Spitznagel, G.W.; Clark, T.; von Rague Schleyer, P.; Hehre, W.J. An evaluation of the performance of diffuse function-augmented basis sets for second row elements, Na-Cl. J. Comput. Chem. 1987, 8, 1109–1116. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isocyanide | Exposed VdW Surface, ES (Å2) | Extrema for Potential Energies at VdW Surface, EP (kcal/mol) | Average Potential Energies at VdW Surface, AP (kcal/mol) | SADAP | ||
|---|---|---|---|---|---|---|
| ESpos | ESneg | EPmin | EPmax | APoverall | ||
| CNArTripp2 | 0.00 | 22.23 | −38.01 | −9.31 | −26.65 | −3.33 |
| CNArDipp2 | 0.00 | 22.13 | −37.64 | −8.87 | −26.04 | −3.28 1 |
| CNPhi-prop2 | 0.00 | 25.99 | −35.47 | −5.16 | −21.20 | −2.38 |
| CNArMes2 | 0.00 | 30.10 | −39.20 | −7.01 | −25.11 | −2.37 |
| CNMes | 0.00 | 28.89 | −36.79 | −6.83 | −21.68 | −2.26 |
| CNtBu | 0.00 | 31.43 | −39.63 | −5.86 | −22.00 | −2.15 1 |
| CNnBu | 0.00 | 31.58 | −38.51 | −5.52 | −21.36 | −2.07 1 |
| MIBI | 0.02 | 29.07 | −35.96 | 2.30 | −18.65 | −1.80 |
| CNH | 0.00 | 31.12 | −31.48 | −4.62 | −16.42 | −1.69 1 |
| CNPhp-F | 1.67 | 29.53 | −32.02 | 7.05 | −14.10 | −1.25 1 |
| CNp-FArDArF2 | 20.50 | 6.74 | −11.49 | 69.07 | 16.88 | 2.73 1 |
| Tc-N10 | Tc-Cl1 | Tc-Cl2 | Tc-Cl3 | Tc-C1 | Tc-C2 | C1-N1 | C2-N2 | Tc-N10-C11 | N10-Tc-Cl1 | |
|---|---|---|---|---|---|---|---|---|---|---|
| 2a | 1.711(2) | 2.4131(6) | 2.3909(6) | 2.4154(6) | 2.057(2) | 1.133(3) | 163.5(2) | 160.78(6) | ||
| 2b | 1.705(2) | 2.3972(7) | 2.4033(6) | 2.4388(6) | 2.037(3) | 1.152(3) | 164.5(2) | 165.24(7) | ||
| 2c | 1.725(6) | 2.413(2) | 2.421(2) | 2.409(2) | 2.030(8) | 1.18(1) | 167.4(6) | 165.2(2) | ||
| 3a | 1.699(3) | 2.400(1) | 2.391(1) | 2.3877(8) | 2.056(4) | 2.034(3) | 1.147(4) | 1.160(4) | 171.7(3) | 166.0(1) |
| 4b * | 1.714(5) 1.692(5) | 2.347(2) 2.361(2) | 2.393(2) 2399(2) | 2.395(2) 2.417(2) | 2.076(5) 2.095(5) | 2.095(5) 2.095(5) | 1.150(6) 1.138(6) | 1.139(6) 1.135(6) | 177.7(2) 178.9(2) | 176.9(6) 178.9(2) |
| 2a (CNtBu) | 2b (CNMes) | 2c (CNPhi-prop2) | 3a (CNArMes2) | 4a * (CNArDipp2) | 4b (CNArTripp2) | 5 (CNp-FArDarF2) | 6 (CNPhpF) | 7 (CNPhpF) | |
|---|---|---|---|---|---|---|---|---|---|
| 2207 | 2187 | 2183 | 2177 | 2187 | 2184 | 2176 | 2186 | 2087 | |
| Ligand | 2135 | 2114 | 2113 | 2120 | 2124 | 2114 | 2119 | 2129 | 2129 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claude, G.; Zeh, L.; Roca Jungfer, M.; Hagenbach, A.; Figueroa, J.S.; Abram, U. The Chemistry of Phenylimidotechnetium(V) Complexes with Isocyanides: Steric and Electronic Factors. Molecules 2022, 27, 8546. https://doi.org/10.3390/molecules27238546
Claude G, Zeh L, Roca Jungfer M, Hagenbach A, Figueroa JS, Abram U. The Chemistry of Phenylimidotechnetium(V) Complexes with Isocyanides: Steric and Electronic Factors. Molecules. 2022; 27(23):8546. https://doi.org/10.3390/molecules27238546
Chicago/Turabian StyleClaude, Guilhem, Laura Zeh, Maximilian Roca Jungfer, Adelheid Hagenbach, Joshua S. Figueroa, and Ulrich Abram. 2022. "The Chemistry of Phenylimidotechnetium(V) Complexes with Isocyanides: Steric and Electronic Factors" Molecules 27, no. 23: 8546. https://doi.org/10.3390/molecules27238546
APA StyleClaude, G., Zeh, L., Roca Jungfer, M., Hagenbach, A., Figueroa, J. S., & Abram, U. (2022). The Chemistry of Phenylimidotechnetium(V) Complexes with Isocyanides: Steric and Electronic Factors. Molecules, 27(23), 8546. https://doi.org/10.3390/molecules27238546