


Photocatalytic Oxidation of Carbon Monoxide Using Synergy of Redox-Separated Photocatalyst and Ozone


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
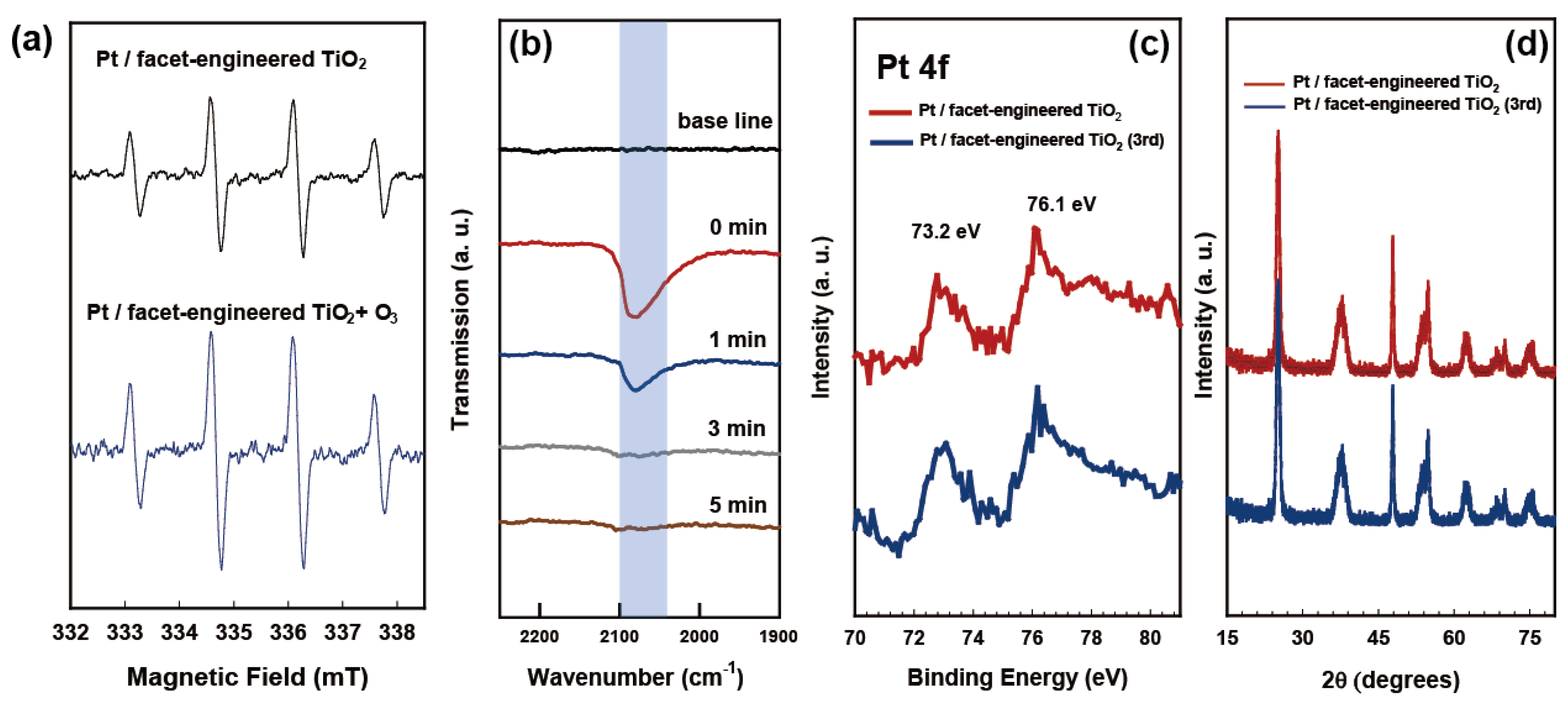{kind=link}
{kind=link}
Abstract
1. Introduction
2. Result and Discussion
2.1. Structural Properties of Redox-Separated Photocatalysts
2.2. Photocatalytic Oxidation of CO
2.3. Charge Transfer Properties of Redox-Separated Photocatalyst
2.4. Ozone-Assisted Photocatalytic Oxidation of CO
3. Experimental Section
3.1. Chemicals
3.2. Preparation of Redox-Separated Photocatalysts
3.3. CO Oxidation Measurements
3.4. In Situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy (In Situ DRIFTS)
3.5. Material Characterizations
3.6. Photoelectrochemical Measurements
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rose, J.J.; Wang, L.; Xu, Q.; McTiernan, C.F.; Shiva, S.; Tejero, J.; Gladwin, M.T. Carbon monoxide poisoning: Pathogenesis, management, and future directions of therapy. Am. J. Respir. Crit. Care Med. 2017, 195, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Raub, J.A.; Mathieu-Nolf, M.; Hampson, N.B.; Thom, S.R. Carbon monoxide poisoning—A public health perspective. Toxicology 2000, 145, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Spengler, J.D.; Sexton, K. Indoor air pollution: A public health perspective. Science 1983, 221, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, L.; Ren, J.; Li, R. CO adsorption on activated carbon-supported Cu-based adsorbent prepared by a facile route. Sep. Purif. Technol. 2010, 76, 89–93. [Google Scholar] [CrossRef]
- Yin, Y.; Wen, Z.; Shi, L.; Zhang, Z.; Yang, Z.; Xu, C.; Sun, H.; Wang, S.; Yuan, A. Cuprous/vanadium sites on MIL-101 for selective CO adsorption from gas mixtures with superior stability. ACS Sustain. Chem. Eng. 2019, 7, 11284–11292. [Google Scholar] [CrossRef]
- Heiz, U.; Sanchez, A.; Abbet, S.; Schneider, W.-D. Catalytic oxidation of carbon monoxide on monodispersed platinum clusters: Each atom counts. J. Am. Chem. Soc. 1999, 121, 3214–3217. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Z.; Liu, C. Perspective on CO oxidation over Pd-based catalysts. Catal. Sci. Technol. 2015, 5, 69–81. [Google Scholar] [CrossRef]
- Gatla, S.; Aubert, D.; Agostini, G.; Mathon, O.; Pascarelli, S.; Lunkenbein, T.; Willinger, M.G.; Kaper, H. Room-Temperature CO Oxidation Catalyst: Low-Temperature Metal–Support Interaction between Platinum Nanoparticles and Nanosized Ceria. ACS Catal. 2016, 6, 6151–6155. [Google Scholar] [CrossRef]
- Deng, X.-Q.; Zhu, B.; Li, X.-S.; Liu, J.-L.; Zhu, X.; Zhu, A.-M. Visible-light photocatalytic oxidation of CO over plasmonic Au/TiO2: Unusual features of oxygen plasma activation. Appl. Catal. B: Environ. 2016, 188, 48–55. [Google Scholar] [CrossRef]
- Kolobov, N.; Svintsitskiy, D.; Kozlova, E.; Selishchev, D.; Kozlov, D. UV-LED photocatalytic oxidation of carbon monoxide over TiO2 supported with noble metal nanoparticles. Chem. Eng. J. 2017, 314, 600–611. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, S.; Chen, F. Pt–Ru Bimetal Alloy Loaded TiO2 Photocatalyst and Its Enhanced Photocatalytic Performance for CO Oxidation. J. Phys. Chem. C 2016, 120, 9732–9739. [Google Scholar]
- Hwang, S.; Lee, M.C.; Choi, W. Highly enhanced photocatalytic oxidation of CO on titania deposited with Pt nanoparticles: Kinetics and mechanism. Appl. Catal. B Environ. 2003, 46, 49–63. [Google Scholar] [CrossRef]
- Weon, S.; He, F.; Choi, W. Status and challenges in photocatalytic nanotechnology for cleaning air polluted with volatile organic compounds: Visible light utilization and catalyst deactivation. Environ. Sci. Nano 2019, 6, 3185–3214. [Google Scholar] [CrossRef]
- Weon, S.; Choi, E.; Kim, H.; Kim, J.Y.; Park, H.-J.; Kim, S.-m.; Kim, W.; Choi, W. Active {001} Facet Exposed TiO2 Nanotubes Photocatalyst Filter for Volatile Organic Compounds Removal: From Material Development to Commercial Indoor Air Cleaner Application. Environ. Sci. Technol. 2018, 52, 9330–9340. [Google Scholar]
- Chu, C.; Zhu, Q.; Pan, Z.; Gupta, S.; Huang, D.; Du, Y.; Weon, S.; Wu, Y.; Muhich, C.; Stavitski, E.; et al. Spatially separating redox centers on 2D carbon nitride with cobalt single atom for photocatalytic H2O2 production. Proc. Natl. Acad. Sci. USA 2020, 117, 6376–6382. [Google Scholar] [CrossRef]
- Ong, W.-J.; Tan, L.-L.; Chai, S.-P.; Yong, S.-T.; Mohamed, A.R. Highly reactive {001} facets of TiO2-based composites: Synthesis, formation mechanism and characterization. Nanoscale 2014, 6, 1946–2008. [Google Scholar] [CrossRef]
- Xiong, Z.; Lei, Z.; Li, Y.; Dong, L.; Zhao, Y.; Zhang, J. A review on modification of facet-engineered TiO2 for photocatalytic CO2 reduction. J. Photochem. Photobiol. C Photochem. Rev. 2018, 36, 24–47. [Google Scholar]
- Ono, L.K.; Yuan, B.; Heinrich, H.; Cuenya, B.R. Formation and Thermal Stability of Platinum Oxides on Size-Selected Platinum Nanoparticles: Support Effects. J. Phys. Chem. C 2010, 114, 22119–22133. [Google Scholar] [CrossRef]
- Svintsitskiy, D.A.; Kibis, L.S.; Stadnichenko, A.I.; Koscheev, S.V.; Zaikovskii, V.I.; Boronin, A.I. Highly Oxidized Platinum Nanoparticles Prepared through Radio-Frequency Sputtering: Thermal Stability and Reaction Probability towards CO. ChemPhysChem 2015, 16, 3318–3324. [Google Scholar]
- Weon, S.; Kim, J.; Choi, W. Dual-components modified TiO2 with Pt and fluoride as deactivation-resistant photocatalyst for the degradation of volatile organic compound. Appl. Catal. B Environ. 2018, 220, 1–8. [Google Scholar] [CrossRef]
- Gambardella, P.; Šljivančanin, Ž.; Hammer, B.; Blanc, M.; Kuhnke, K.; Kern, K. Oxygen dissociation at Pt steps. Phys. Rev. Lett. 2001, 87, 056103. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K.; Zhang, S.; Zhang, M.; Yang, J.; Jin, Z. Effect of photocatalytic activity of CO oxidation on Pt/TiO2 by strong interaction between Pt and TiO2 under oxidizing atmosphere. J. Mol. Catal. A Chem. 2006, 258, 83–88. [Google Scholar] [CrossRef]
- Weschler, C.J. Ozone in indoor environments: Concentration and chemistry. Indoor Air 2000, 10, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cao, J.; Fu, W.; Zhang, J.; Qian, G.; Yang, J.; Chen, D.; Zhou, X.; Yuan, W.; Duan, X. Molecular-Level Insights into the Notorious CO Poisoning of Platinum Catalyst. Angew. Chem. Int. Ed. 2022, 61. [Google Scholar] [CrossRef]
- Ding, K.; Gulec, A.; Johnson, A.M.; Schweitzer, N.M.; Stucky, G.D.; Marks, L.D.; Stair, P.C. Identification of active sites in CO oxidation and water-gas shift over supported Pt catalysts. Science 2015, 350, 189–192. [Google Scholar] [CrossRef]
- Weon, S.; Suh, M.-J.; Chu, C.; Huang, D.; Stavitski, E.; Kim, J.-H. Site-Selective Loading of Single-Atom Pt on TiO2 for Photocatalytic Oxidation and Reductive Hydrodefluorination. ACS EST Eng. 2021, 1, 512–522. [Google Scholar] [CrossRef]
- Weon, S.; Choi, W. TiO2 Nanotubes with Open Channels as Deactivation-Resistant Photocatalyst for the Degradation of Volatile Organic Compounds. Environ. Sci. Technol. 2016, 50, 2556–2563. [Google Scholar] [CrossRef]
- Lee, M.G.; Yang, J.W.; Kwon, H.R.; Jang, H.W. Crystal facet and phase engineering for advanced water splitting. CrystEngComm 2022, 24, 5838–5864. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, R.; Li, Z.; Li, A.; Wang, S.; Liang, Z.; Liao, S.; Li, C. The dependence of photocatalytic activity on the selective and nonselective deposition of noble metal cocatalysts on the facets of rutile TiO2. J. Catal. 2016, 337, 36–44. [Google Scholar] [CrossRef]
- Liu, G.; Yang, H.G.; Pan, J.; Yang, Y.Q.; Lu, G.Q.; Cheng, H.-M. Titanium Dioxide Crystals with Tailored Facets. Chem. Rev. 2014, 114, 9559–9612. [Google Scholar] [CrossRef]
- Lardhi, S.; Cavallo, L.; Harb, M. Significant Impact of Exposed Facets on the BiVO4 Material Performance for Photocatalytic Water Splitting Reactions. J. Phys. Chem. Lett. 2020, 11, 5497–5503. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Du, M.; Ma, Y.; Zhu, Q.; Xing, M.; Zhang, J. Integration of redox cocatalysts for artificial photosynthesis. Energy Environ. Sci. 2021, 14, 5260–5288. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weon, S. Photocatalytic Oxidation of Carbon Monoxide Using Synergy of Redox-Separated Photocatalyst and Ozone. Molecules 2022, 27, 8482. https://doi.org/10.3390/molecules27238482
Weon S. Photocatalytic Oxidation of Carbon Monoxide Using Synergy of Redox-Separated Photocatalyst and Ozone. Molecules. 2022; 27(23):8482. https://doi.org/10.3390/molecules27238482
Chicago/Turabian StyleWeon, Seunghyun. 2022. "Photocatalytic Oxidation of Carbon Monoxide Using Synergy of Redox-Separated Photocatalyst and Ozone" Molecules 27, no. 23: 8482. https://doi.org/10.3390/molecules27238482
APA StyleWeon, S. (2022). Photocatalytic Oxidation of Carbon Monoxide Using Synergy of Redox-Separated Photocatalyst and Ozone. Molecules, 27(23), 8482. https://doi.org/10.3390/molecules27238482