
Proton Affinity in the Chemistry of Beta-Octamolybdate: HPLC-ICP-AES, NMR and Structural Studies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
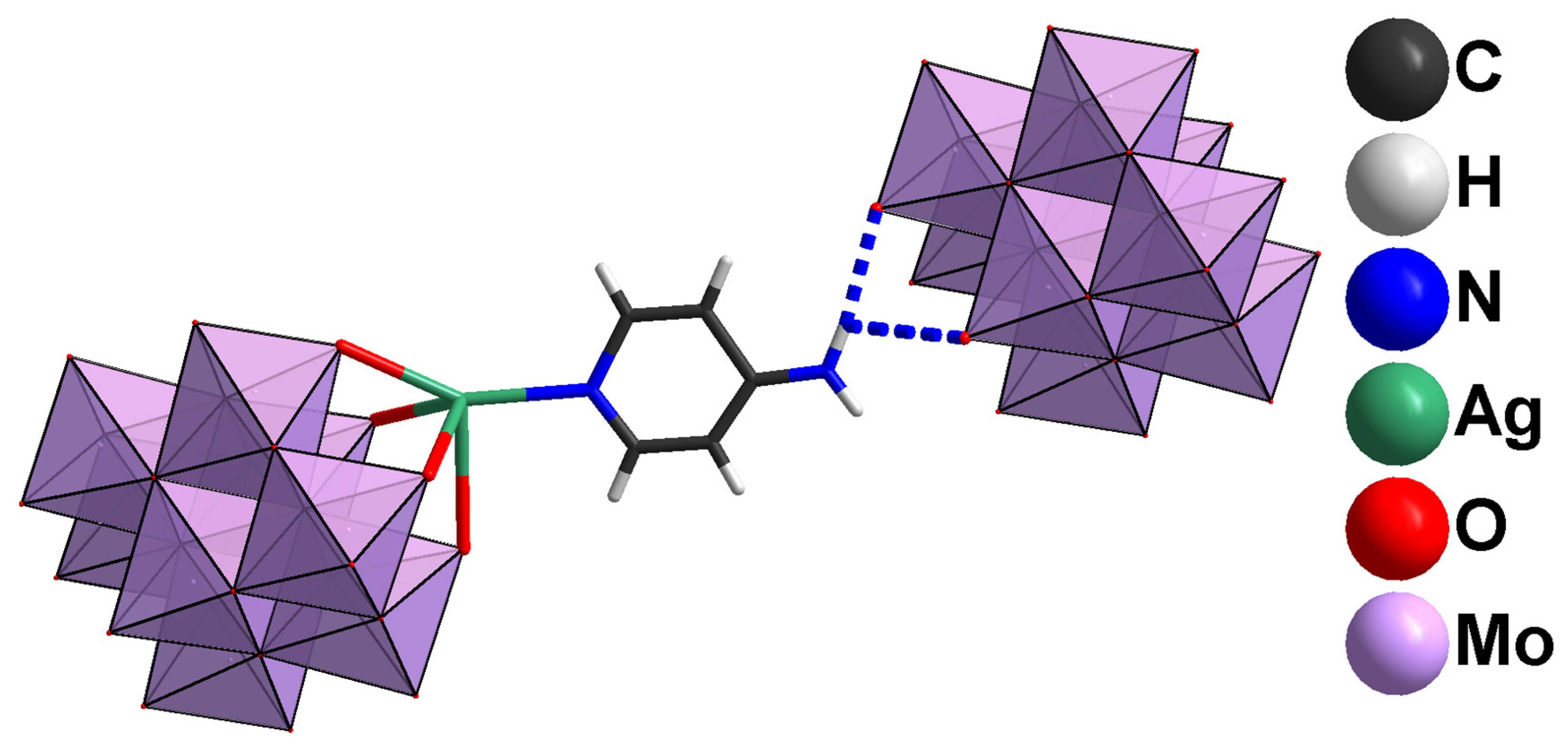
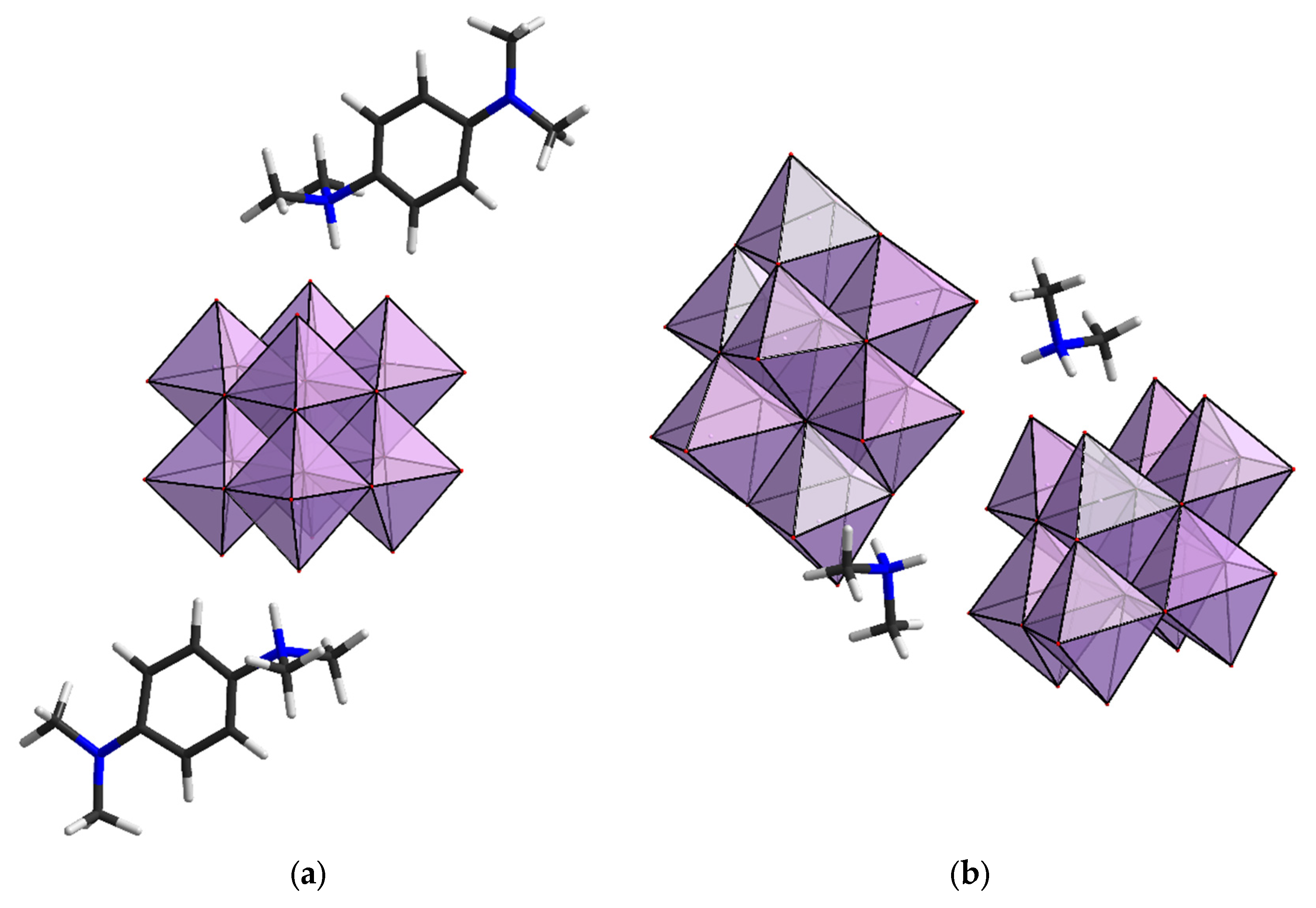
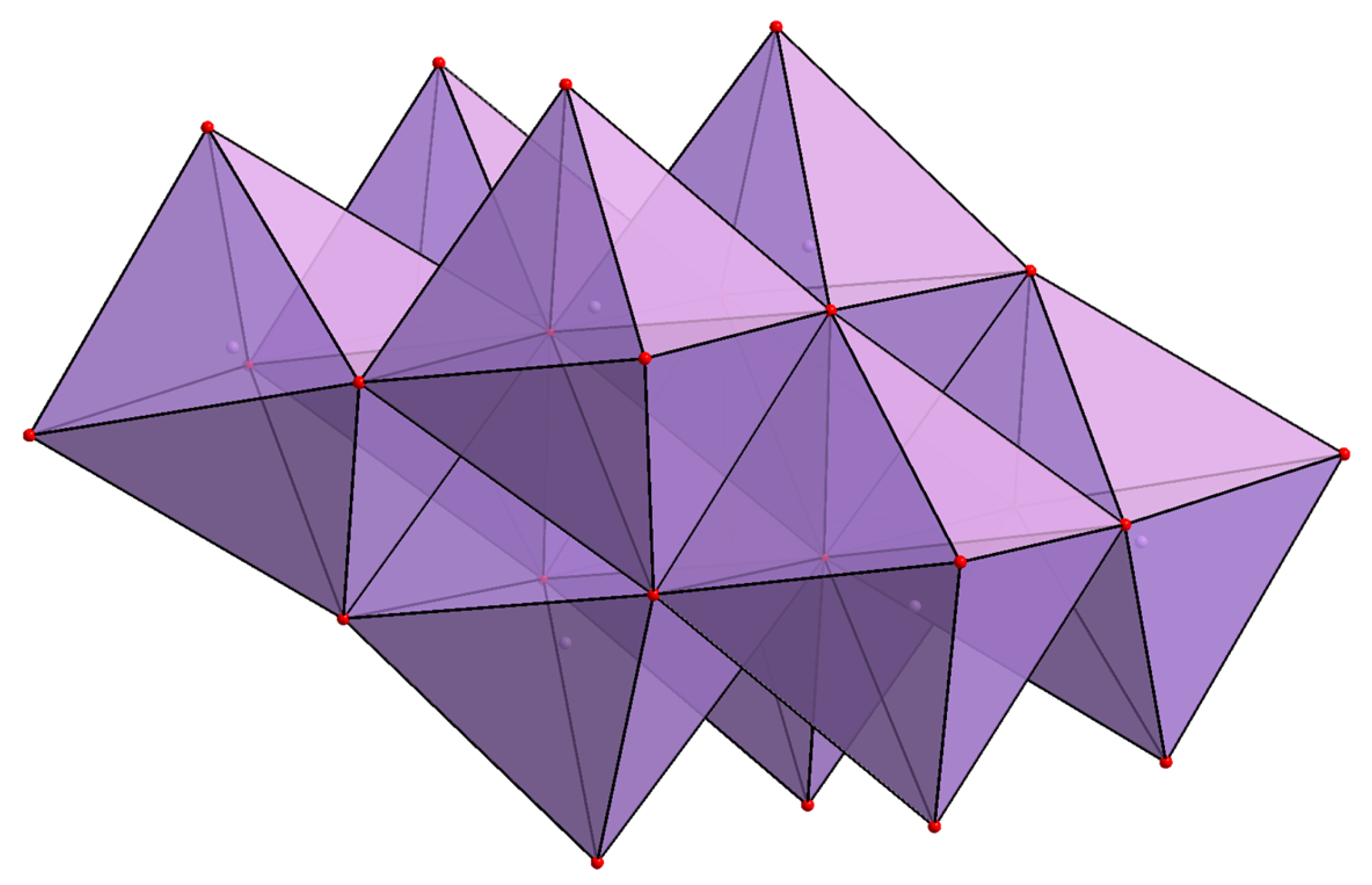
2.1. Structural Analysis
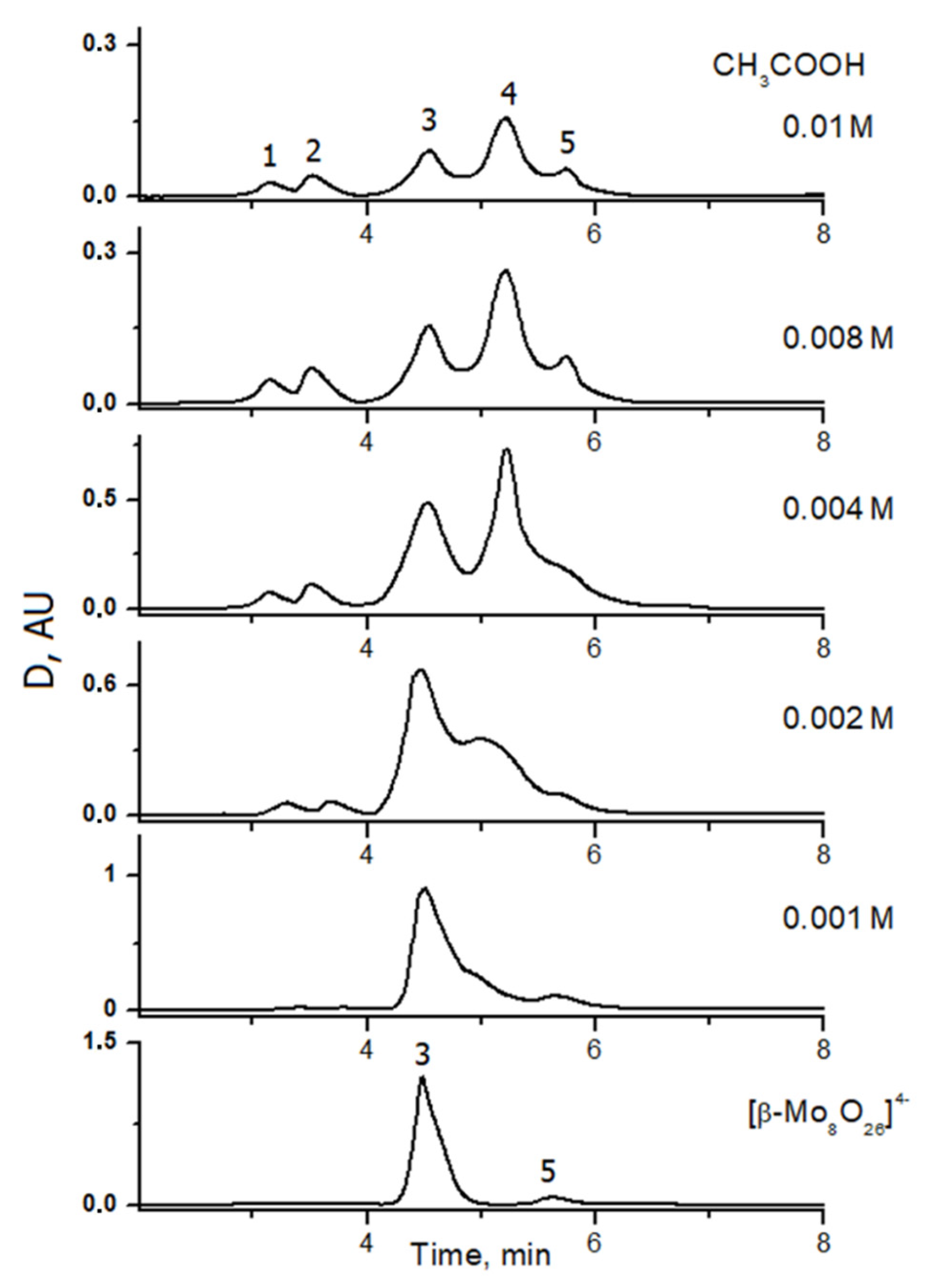
2.2. Reactivity of [β-Mo8O26]4−
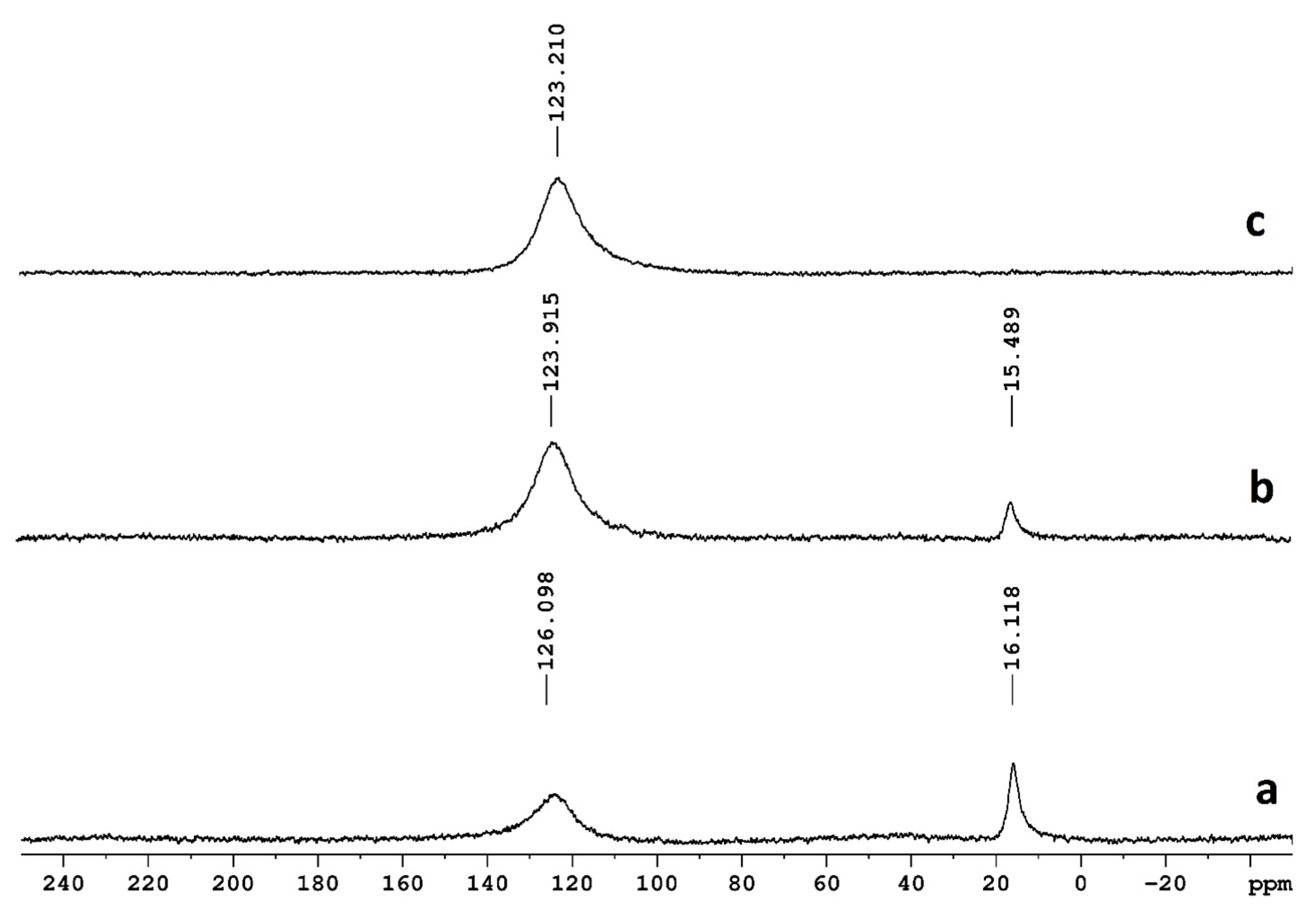
2.3. NMR
3. Materials and Methods
3.1. Physical Methods
3.2. NMR
3.3. X-ray Diffraction on Single Crystals
3.4. XRPD
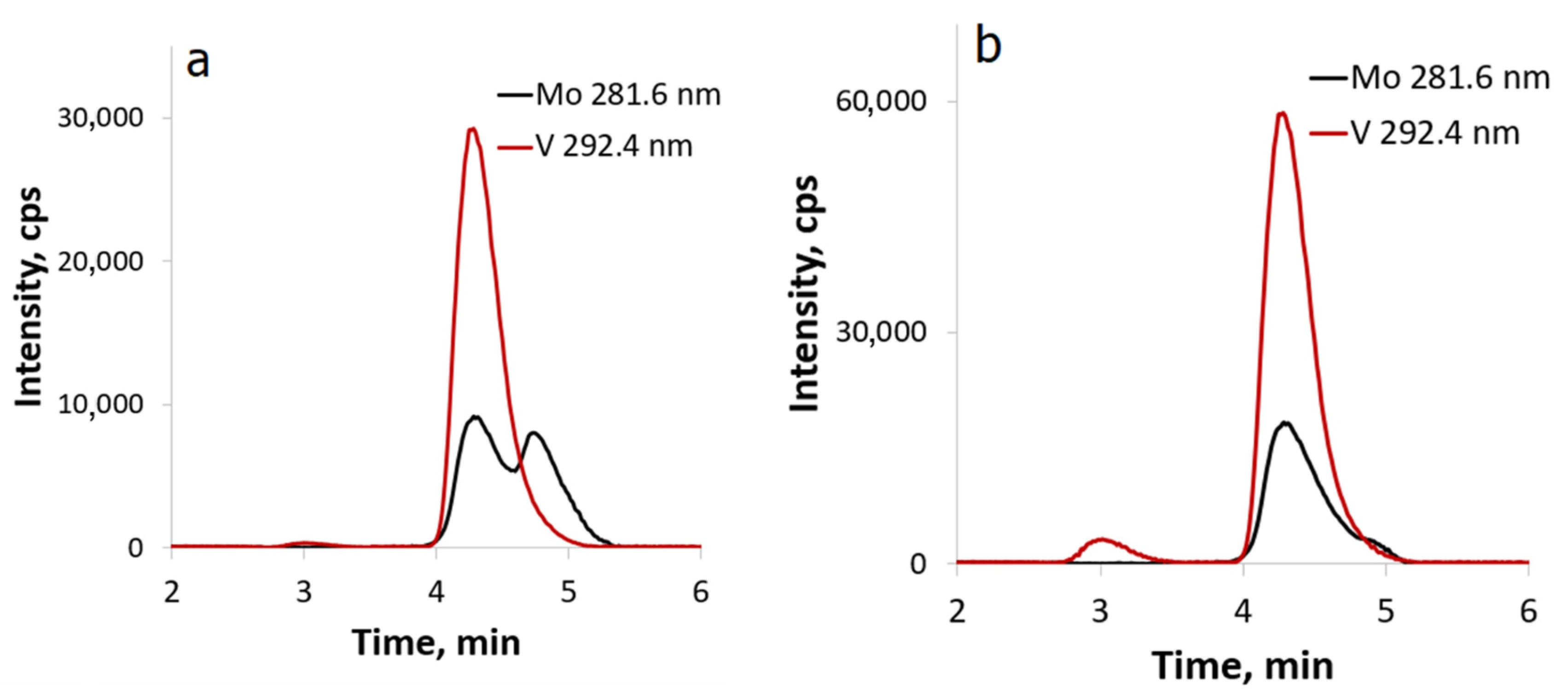
3.5. HPLC-ICP-AES and HPLC
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ishikita, H.; Saito, K. Proton transfer reactions and hydrogen-bond networks in protein environments. J. R. Soc. Interface 2014, 11, 20130518. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.M. Proton-Coupled Electron Transfer: A Reaction Chemist’s View. Annu. Rev. Phys. Chem. 2004, 55, 363–390. [Google Scholar] [CrossRef] [PubMed]
- Hammes-Schiffer, S.; Stuchebrukhov, A.A. Theory of Coupled Electron and Proton Transfer Reactions. Chem. Rev. 2010, 110, 6939–6960. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chang, M.C.Y.; Damrauer, N.H.; Nocera, D.G. Proton-coupled electron transfer: A unifying mechanism for biological charge transport, amino acid radical initiation and propagation, and bond making/breaking reactions of water and oxygen. Biochim. Biophys. Acta—Bioenerg. 2004, 1655, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Tyburski, R.; Liu, T.; Glover, S.D.; Hammarström, L. Proton-Coupled Electron Transfer Guidelines, Fair and Square. J. Am. Chem. Soc. 2021, 143, 560–576. [Google Scholar] [CrossRef]
- Darcy, J.W.; Koronkiewicz, B.; Parada, G.A.; Mayer, J.M. A Continuum of Proton-Coupled Electron Transfer Reactivity. Acc. Chem. Res. 2018, 51, 2391–2399. [Google Scholar] [CrossRef]
- Agarwal, R.G.; Coste, S.C.; Groff, B.D.; Heuer, A.M.; Noh, H.; Parada, G.A.; Wise, C.F.; Nichols, E.M.; Warren, J.J.; Mayer, J.M. Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications. Chem. Rev. 2022, 122, 1–49. [Google Scholar] [CrossRef]
- Warren, J.J.; Tronic, T.A.; Mayer, J.M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar] [CrossRef]
- Siewert, I. Proton-Coupled Electron Transfer Reactions Catalysed by 3 d Metal Complexes. Chem.—A Eur. J. 2015, 21, 15078–15091. [Google Scholar] [CrossRef]
- Cukier, R.I. Proton-Coupled Electron Transfer Reactions: Evaluation of Rate Constants. J. Phys. Chem. 1996, 100, 15428–15443. [Google Scholar] [CrossRef]
- Huynh, M.T.; Mora, S.J.; Villalba, M.; Tejeda-Ferrari, M.E.; Liddell, P.A.; Cherry, B.R.; Teillout, A.-L.; Machan, C.W.; Kubiak, C.P.; Gust, D.; et al. Concerted One-Electron Two-Proton Transfer Processes in Models Inspired by the Tyr-His Couple of Photosystem II. ACS Cent. Sci. 2017, 3, 372–380. [Google Scholar] [CrossRef]
- Odella, E.; Mora, S.J.; Wadsworth, B.L.; Huynh, M.T.; Goings, J.J.; Liddell, P.A.; Groy, T.L.; Gervaldo, M.; Sereno, L.E.; Gust, D.; et al. Controlling Proton-Coupled Electron Transfer in Bioinspired Artificial Photosynthetic Relays. J. Am. Chem. Soc. 2018, 140, 15450–15460. [Google Scholar] [CrossRef]
- Odella, E.; Wadsworth, B.L.; Mora, S.J.; Goings, J.J.; Huynh, M.T.; Gust, D.; Moore, T.A.; Moore, G.F.; Hammes-Schiffer, S.; Moore, A.L. Proton-Coupled Electron Transfer Drives Long-Range Proton Translocation in Bioinspired Systems. J. Am. Chem. Soc. 2019, 141, 14057–14061. [Google Scholar] [CrossRef]
- Odella, E.; Mora, S.J.; Wadsworth, B.L.; Goings, J.J.; Gervaldo, M.A.; Sereno, L.E.; Groy, T.L.; Gust, D.; Moore, T.A.; Moore, G.F.; et al. Proton-coupled electron transfer across benzimidazole bridges in bioinspired proton wires. Chem. Sci. 2020, 11, 3820–3828. [Google Scholar] [CrossRef]
- Goldsmith, Z.K.; Lam, Y.C.; Soudackov, A.V.; Hammes-Schiffer, S. Proton Discharge on a Gold Electrode from Triethylammonium in Acetonitrile: Theoretical Modeling of Potential-Dependent Kinetic Isotope Effects. J. Am. Chem. Soc. 2019, 141, 1084–1090. [Google Scholar] [CrossRef]
- Warburton, R.E.; Hutchison, P.; Jackson, M.N.; Pegis, M.L.; Surendranath, Y.; Hammes-Schiffer, S. Interfacial Field-Driven Proton-Coupled Electron Transfer at Graphite-Conjugated Organic Acids. J. Am. Chem. Soc. 2020, 142, 20855–20864. [Google Scholar] [CrossRef]
- Lam, Y.-C.; Soudackov, A.V.; Hammes-Schiffer, S. Theory of Electrochemical Proton-Coupled Electron Transfer in Diabatic Vibronic Representation: Application to Proton Discharge on Metal Electrodes in Alkaline Solution. J. Phys. Chem. C 2020, 124, 27309–27322. [Google Scholar] [CrossRef]
- Sarkar, S.; Maitra, A.; Lake, W.R.; Warburton, R.E.; Hammes-Schiffer, S.; Dawlaty, J.M. Mechanistic Insights about Electrochemical Proton-Coupled Electron Transfer Derived from a Vibrational Probe. J. Am. Chem. Soc. 2021, 143, 8381–8390. [Google Scholar] [CrossRef]
- Yadav, S.; Sharma, A.K.; Kumar, P. Nanoscale Self-Assembly for Therapeutic Delivery. Front. Bioeng. Biotechnol. 2020, 8, 127. [Google Scholar] [CrossRef]
- Tan, M.; Tian, P.; Zhang, Q.; Zhu, G.; Liu, Y.; Cheng, M.; Shi, F. Self-sorting in macroscopic supramolecular self-assembly via additive effects of capillary and magnetic forces. Nat. Commun. 2022, 13, 5201. [Google Scholar] [CrossRef]
- Li, K.; Hu, J.-M.; Qin, W.-M.; Guo, J.; Cai, Y.-P. Precise heteroatom doping determines aqueous solubility and self-assembly behaviors for polycyclic aromatic skeletons. Commun. Chem. 2022, 5, 104. [Google Scholar] [CrossRef]
- Cui, Z.; Jin, G.-X. Construction of a molecular prime link by interlocking two trefoil knots. Nat. Synth. 2022, 1, 635–640. [Google Scholar] [CrossRef]
- Woods, J.F.; Gallego, L.; Pfister, P.; Maaloum, M.; Vargas Jentzsch, A.; Rickhaus, M. Shape-assisted self-assembly. Nat. Commun. 2022, 13, 3681. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Qiu, Y.; Zhang, L.; Liu, W.-G.; Mao, H.; Chen, H.; Feng, Y.; Cai, K.; Shen, D.; Song, B.; et al. Electron-catalysed molecular recognition. Nature 2022, 603, 265–270. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Boncheva, M. Beyond molecules: Self-assembly of mesoscopic and macroscopic components. Proc. Natl. Acad. Sci. USA 2002, 99, 4769–4774. [Google Scholar] [CrossRef]
- Gartner, F.M.; Graf, I.R.; Frey, E. The time complexity of self-assembly. Proc. Natl. Acad. Sci. USA 2022, 119, e2116373119. [Google Scholar] [CrossRef]
- Deamer, D.; Singaram, S.; Rajamani, S.; Kompanichenko, V.; Guggenheim, S. Self-assembly processes in the prebiotic environment. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1809–1818. [Google Scholar] [CrossRef]
- Wang, Z.; Gupta, R.K.; Luo, G.; Sun, D. Recent Progress in Inorganic Anions Templated Silver Nanoclusters: Synthesis, Structures and Properties. Chem. Rec. 2020, 20, 389–402. [Google Scholar] [CrossRef]
- Gao, G.-G.; Cheng, P.-S.; Mak, T.C.W. Acid-Induced Surface Functionalization of Polyoxometalate by Enclosure in a Polyhedral Silver−Alkynyl Cage. J. Am. Chem. Soc. 2009, 131, 18257–18259. [Google Scholar] [CrossRef]
- Shi, J.-Y.; Gupta, R.K.; Deng, Y.-K.; Sun, D.; Wang, Z. Recent Advances in the Asymmetrical Templation Effect of Polyoxometalate in Silver Clusters. Polyoxometalates 2022, 1, 9140010. [Google Scholar] [CrossRef]
- Fabre, B.; Falaise, C.; Cadot, E. Polyoxometalates-Functionalized Electrodes for (Photo)Electrocatalytic Applications: Recent Advances and Prospects. ACS Catal. 2022, 12, 12055–12091. [Google Scholar] [CrossRef]
- Wu, Y.; Bi, L. Research Progress on Catalytic Water Splitting Based on Polyoxometalate/Semiconductor Composites. Catalysts 2021, 11, 524. [Google Scholar] [CrossRef]
- Kuznetsov, A.E.; Geletii, Y.V.; Hill, C.L.; Morokuma, K.; Musaev, D.G. Dioxygen and Water Activation Processes on Multi-Ru-Substituted Polyoxometalates: Comparison with the “Blue-Dimer” Water Oxidation Catalyst. J. Am. Chem. Soc. 2009, 131, 6844–6854. [Google Scholar] [CrossRef]
- Lauinger, S.M.; Piercy, B.D.; Li, W.; Yin, Q.; Collins-Wildman, D.L.; Glass, E.N.; Losego, M.D.; Wang, D.; Geletii, Y.V.; Hill, C.L. Stabilization of Polyoxometalate Water Oxidation Catalysts on Hematite by Atomic Layer Deposition. ACS Appl. Mater. Interfaces 2017, 9, 35048–35056. [Google Scholar] [CrossRef]
- Anwar, N.; Sartorel, A.; Yaqub, M.; Wearen, K.; Laffir, F.; Armstrong, G.; Dickinson, C.; Bonchio, M.; McCormac, T. Surface Immobilization of a Tetra-Ruthenium Substituted Polyoxometalate Water Oxidation Catalyst Through the Employment of Conducting Polypyrrole and the Layer-by-Layer (LBL) Technique. ACS Appl. Mater. Interfaces 2014, 6, 8022–8031. [Google Scholar] [CrossRef]
- Azmani, K.; Besora, M.; Soriano-López, J.; Landolsi, M.; Teillout, A.-L.; de Oliveira, P.; Mbomekallé, I.-M.; Poblet, J.M.; Galán-Mascarós, J.-R. Understanding polyoxometalates as water oxidation catalysts through iron vs. cobalt reactivity. Chem. Sci. 2021, 12, 8755–8766. [Google Scholar] [CrossRef]
- Soriano-López, J.; Musaev, D.G.; Hill, C.L.; Galán-Mascarós, J.R.; Carbó, J.J.; Poblet, J.M. Tetracobalt-polyoxometalate catalysts for water oxidation: Key mechanistic details. J. Catal. 2017, 350, 56–63. [Google Scholar] [CrossRef]
- Fertig, A.A.; Brennessel, W.W.; McKone, J.R.; Matson, E.M. Concerted Multiproton–Multielectron Transfer for the Reduction of O 2 to H 2 O with a Polyoxovanadate Cluster. J. Am. Chem. Soc. 2021, 143, 15756–15768. [Google Scholar] [CrossRef]
- Wang, X.-L.; Zhang, Y.; Chen, Y.-Z.; Wang, Y.; Wang, X. Two polymolybdate-directed Zn(ii) complexes tuned by a new bis-pyridine-bis-amide ligand with a diphenylketone spacer for efficient ampere sensing and dye adsorption. CrystEngComm 2022, 24, 5289–5296. [Google Scholar] [CrossRef]
- Huang, X.; Cui, Y.; Liu, G.; Wang, H.; Ren, J.; Zhang, Y.; Shen, G.; Lv, L.; Wang, H.-W.; Chen, Y.-F. Imidazole-Dependent Assembly of Copper Polymolybdate Frameworks for One-Pot Sulfide Oxidation and C–H Activation. Energy Fuels 2022, 36, 1665–1675. [Google Scholar] [CrossRef]
- Liu, J.; Huang, M.; Hua, Z.; Dong, Y.; Feng, Z.; Sun, T.; Chen, C. Polyoxometalate-Based Metal Organic Frameworks: Recent Advances and Challenges. ChemistrySelect 2022, 7, e202200546. [Google Scholar] [CrossRef]
- Talib, S.H.; Yu, X.; Lu, Z.; Ahmad, K.; Yang, T.; Xiao, H.; Li, J. A polyoxometalate cluster-based single-atom catalyst for NH3 synthesis via an enzymatic mechanism. J. Mater. Chem. A 2022, 10, 6165–6177. [Google Scholar] [CrossRef]
- Paul, A.; Das Adhikary, S.; Kapurwan, S.; Konar, S. En route to artificial photosynthesis: The role of polyoxometalate based photocatalysts. J. Mater. Chem. A 2022, 10, 13152–13169. [Google Scholar] [CrossRef]
- Liu, S.; Cui, C.; Dai, Y.; Liu, G.; Qiao, S.; Tao, Y.; Zhang, Y.; Shen, G.; Li, Z.; Huang, X. Two silver–containing polyoxometalate–based inorganic–organic hybrids as heterogeneous bifunctional catalysts for construction of C–C bonds and decontamination of sulfur mustard simulant. J. Solid State Chem. 2022, 316, 123547. [Google Scholar] [CrossRef]
- Silva, D.F.; Viana, A.M.; Santos-Vieira, I.; Balula, S.S.; Cunha-Silva, L. Ionic Liquid-Based Polyoxometalate Incorporated at ZIF-8: A Sustainable Catalyst to Combine Desulfurization and Denitrogenation Processes. Molecules 2022, 27, 1711. [Google Scholar] [CrossRef]
- Veríssimo, M.I.S.; Evtuguin, D.V.; Gomes, M.T.S.R. Polyoxometalate Functionalized Sensors: A Review. Front. Chem. 2022, 10, 840657. [Google Scholar] [CrossRef]
- Ren, W.; Li, B.; Li, S.; Li, Y.; Gao, Z.; Chen, X.; Zang, H. Synthesis and Proton Conductivity of Two Molybdate Polymers Based on [Mo8O26]4− Anions. ChemistrySelect 2022, 7, e202201337. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Shaikh, S.N.; Zubieta, J. Derivatized polyoxomolybdates. Synthesis and characterization of oxomolybdate clusters containing coordinatively bound diazenido units. Crystal and molecular structure of the octanuclear oxomolybdate (NHEt3)2(n-Bu4N)2[Mo8O20(NNPh)6] and comparison to the structures of the parent oxomolybdate .alpha.-(n-Bu4N)4[Mo8O26] and the tetranuclear (diazenido)oxomolybdates (n-Bu4N)2[Mo4O10(OMe)2(NNPh)2] and (n-Bu4N)2[Mo4O8(OMe)2(NNC6H4NO2)4]. Inorg. Chem. 1987, 26, 4079–4089. [Google Scholar]
- Cindrić, M.; Veksli, Z.; Kamenar, B. Polyoxomolybdates and polyoxomolybdovanadates—from structure to functions: Recent results. Croat. Chem. Acta 2009, 82, 345–362. [Google Scholar]
- Chupina, A.V.; Shayapov, V.; Novikov, A.S.; Volchek, V.V.; Benassi, E.; Abramov, P.A.; Sokolov, M.N. [{AgL}2Mo8O26]n− complexes: A combined experimental and theoretical study. Dalton Trans. 2020, 49, 1522–1530. [Google Scholar] [CrossRef]
- Abramov, P.A.; Komarov, V.Y.; Pischur, D.A.; Sulyaeva, V.S.; Benassi, E.; Sokolov, M.N. Solvatomorphs of (Bu4N)2[{Ag(N2-py)}2Mo8O26]: Structure, colouration and phase transition. CrystEngComm 2021, 23, 8527–8537. [Google Scholar] [CrossRef]
- Komlyagina, V.I.; Romashev, N.F.; Kokovkin, V.V.; Gushchin, A.L.; Benassi, E.; Sokolov, M.N.; Abramov, P.A. Trapping of Ag+ into a Perfect Six-Coordinated Environment: Structural Analysis, Quantum Chemical Calculations and Electrochemistry. Molecules 2022, 27, 6961. [Google Scholar] [CrossRef]
- Karoui, H.; Ritchie, C. Microwave-assisted synthesis of organically functionalized hexa-molybdovanadates. New J. Chem. 2018, 42, 25–28. [Google Scholar] [CrossRef]
- Shuvaeva, O.V.; Zhdanov, A.A.; Romanova, T.E.; Abramov, P.A.; Sokolov, M.N. Hyphenated techniques in speciation analysis of polyoxometalates: Identification of individual [PMo12−xVxO40]−3−x (x = 1–3) in the reaction mixtures by high performance liquid chromatography and atomic emission spectrometry with inductively coupled. Dalton Trans. 2017, 46, 3541–3546. [Google Scholar] [CrossRef]
- Mukhacheva, A.A.; Volcheck, V.V.; Sheven, D.G.; Yanshole, V.V.; Kompankov, N.B.; Haouas, M.; Abramov, P.A.; Sokolov, M.N. Coordination capacity of Keggin anions as polytopic ligands: Case study of [VNb12O40]15−. Dalton Trans. 2021, 50, 7078–7084. [Google Scholar] [CrossRef]
- Mukhacheva, A.A.; Shmakova, A.A.; Volchek, V.V.; Romanova, T.E.; Benassi, E.; Gushchin, A.L.; Yanshole, V.; Sheven, D.G.; Kompankov, N.B.; Abramov, P.A.; et al. Reactions of [Ru(NO)Cl5]2− with pseudotrilacunary {XW9O33}9− (X = As III, Sb III ) anions. Dalton Trans. 2019, 48, 15989–15999. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Volchek, V.V.; Yanshole, V.V.; Fedorenko, A.D.; Kompankov, N.B.; Kokovkin, V.V.; Gushchin, A.L.; Abramov, P.A.; Sokolov, M.N. Coordination of Pt(IV) by {P8W48} Macrocyclic Inorganic Cavitand: Structural, Solution, and Electrochemical Studies. Inorg. Chem. 2022, 61, 14560–14567. [Google Scholar] [CrossRef]
- Chupina, A.V.; Mukhacheva, A.A.; Abramov, P.A.; Sokolov, M.N. Complexation and Isomerization of [β-Mo8O26]4− in the Presence of Ag+ and DMF. J. Struct. Chem. 2020, 61, 299–308. [Google Scholar] [CrossRef]
- Pantyukhina, V.S.; Volchek, V.V.; Komarov, V.Y.; Korolkov, I.V.; Kokovkin, V.V.; Kompankov, N.B.; Abramov, P.A.; Sokolov, M.N. Tubular polyoxoanion [(SeMo6O21)2(C2O4)3]10− and its transformations. New J. Chem. 2021, 45, 6745–6752. [Google Scholar] [CrossRef]
- Shmakova, A.A.; Akhmetova, M.M.; Volchek, V.V.; Romanova, T.E.; Korolkov, I.; Sheven, D.G.; Adonin, S.A.; Abramov, P.A.; Sokolov, M.N. A HPLC-ICP-AES technique for the screening of [XW11NbO40]n− aqueous solutions. New J. Chem. 2018, 42, 7940–7948. [Google Scholar] [CrossRef]
- Korpar-Čolig, B.; Cindrić, M.; Matković-Čalogović, D.; Vrdoljak, V.; Kamenar, B. Synthesis and characterization of some new acetato complexes of molybdenum(IV), (V) and (VI). Polyhedron 2002, 21, 147–153. [Google Scholar] [CrossRef]
- Wang, S.; Mo, S.; Liu, Z.-G. Synthesis and characterization of a new octamolybdate (NH4)4[Mo8O24(C3H2O2)2]·4H2O. Russ. J. Inorg. Chem. 2012, 57, 430–433. [Google Scholar] [CrossRef]
- Brito, J.A.; Teruel, H.; Massou, S.; Gómez, M. 95Mo NMR: A useful tool for structural studies in solution. Magn. Reson. Chem. 2009, 47, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Fedotov, M.A.; Maksimovskaya, R.I. NMR structural aspects of the chemistry of V, Mo, W polyoxometalates. J. Struct. Chem. 2006, 47, 952–978. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volchek, V.V.; Kompankov, N.B.; Sokolov, M.N.; Abramov, P.A. Proton Affinity in the Chemistry of Beta-Octamolybdate: HPLC-ICP-AES, NMR and Structural Studies. Molecules 2022, 27, 8368. https://doi.org/10.3390/molecules27238368
Volchek VV, Kompankov NB, Sokolov MN, Abramov PA. Proton Affinity in the Chemistry of Beta-Octamolybdate: HPLC-ICP-AES, NMR and Structural Studies. Molecules. 2022; 27(23):8368. https://doi.org/10.3390/molecules27238368
Chicago/Turabian StyleVolchek, Victoria V., Nikolay B. Kompankov, Maxim N. Sokolov, and Pavel A. Abramov. 2022. "Proton Affinity in the Chemistry of Beta-Octamolybdate: HPLC-ICP-AES, NMR and Structural Studies" Molecules 27, no. 23: 8368. https://doi.org/10.3390/molecules27238368
APA StyleVolchek, V. V., Kompankov, N. B., Sokolov, M. N., & Abramov, P. A. (2022). Proton Affinity in the Chemistry of Beta-Octamolybdate: HPLC-ICP-AES, NMR and Structural Studies. Molecules, 27(23), 8368. https://doi.org/10.3390/molecules27238368