
Computational Bioprospecting Guggulsterone against ADP Ribose Phosphatase of SARS-CoV-2
 , , , , and
, , , , and
Abstract
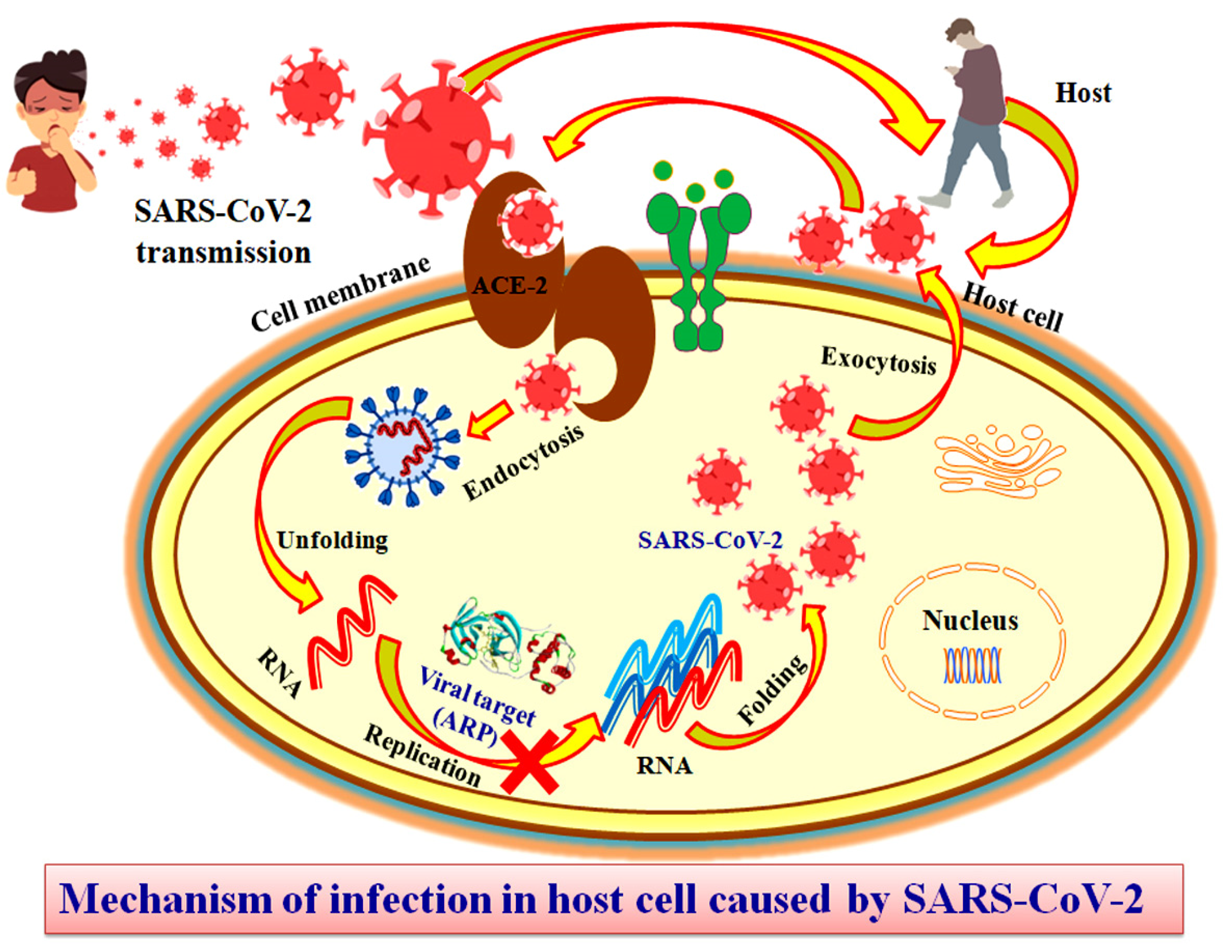
1. Introduction
2. Experimental
2.1. Selection and Preparation of Target Protein
2.2. Binding Site Identification
2.3. Molecular Docking
2.4. Validation of Docking Methodology
2.5. In-Silico Screening
2.6. Analysis of Docking Results
2.7. Molecular Dynamic Simulation
3. Results
3.1. Selection and Preparation of Target Protein
3.2. Binding Site Identification
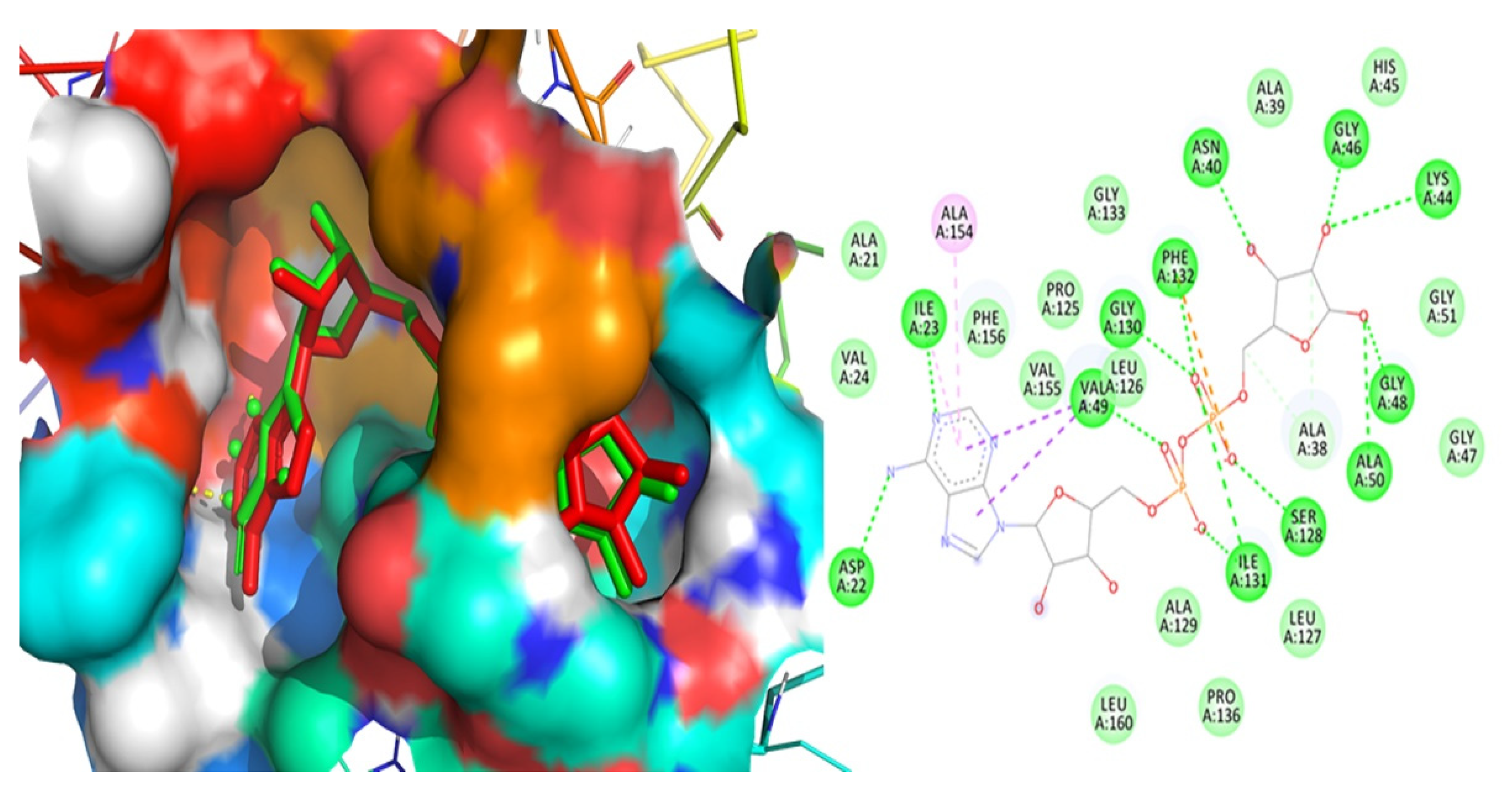
3.3. Validation of Docking Methodology
3.4. Virtual Screening
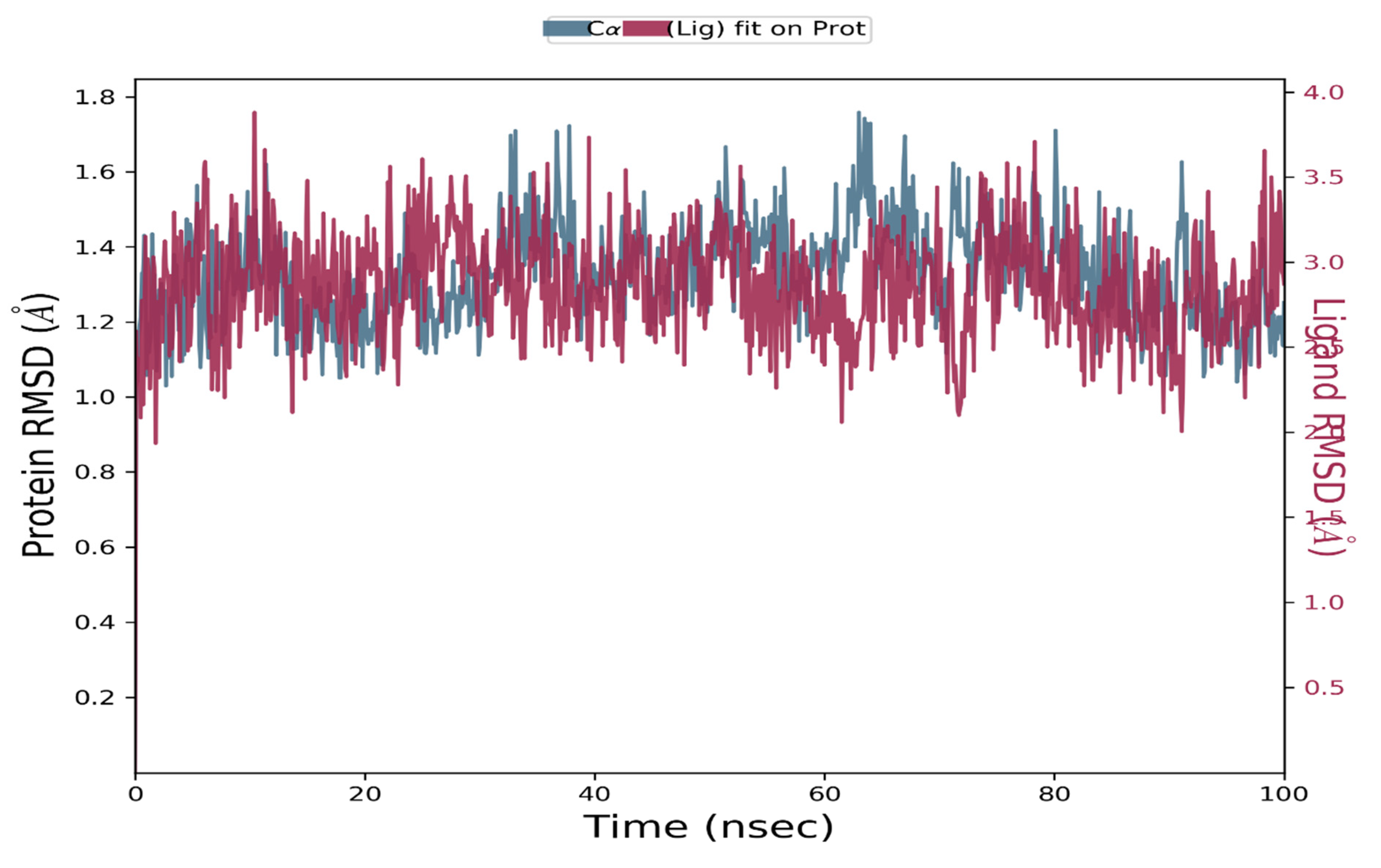
3.5. Molecular Dynamic Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elfiky, A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020, 248, 117477. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020, 253, 117592. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.U.; Froeyen, M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharm. Anal. 2020, 10, 320–328. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Zhang, T.; Xu, J.; Shang, S. The mechanism and treatment of gastrointestinal symptoms in patients with COVID-19. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G245–G252. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Pandey, S.K.; Kumar, S.; Kumar, A.; Rai, D.; Munjal, K.; Rani, M.; Narayan, S. Change in bone mineral density in premenopausal women with rheumatoid arthritis managed with or without prednisolone. Int. J. Health Clin. Res. 2020, 3, 201–210. [Google Scholar]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Sohraby, F.; Bagheri, M.; Aryapour, H. Performing an In Silico Repurposing of Existing Drugs by Combining Virtual Screening and Molecular Dynamics Simulation. Methods Mol. Biol. 2019, 1903, 23–43. [Google Scholar] [PubMed]
- De Clercq, E. Antiviral agents: Characteristic activity spectrum depending on the molecular target with which they interact. Adv. Virus Res. 1993, 42, 1–55. [Google Scholar] [PubMed]
- Kausar, S.; Said Khan, F.; Ishaq Mujeeb Ur Rehman, M.; Akram, M.; Riaz, M.; Rasool, G.; Hamid Khan, A.; Saleem, I.; Shamim, S.; Malik, A. A review: Mechanism of action of antiviral drugs. Int. J. Immunopathol. Pharm. 2021, 35, 20587384211002621. [Google Scholar] [CrossRef] [PubMed]
- Mujwar, S. Computational repurposing of tamibarotene against triple mutant variant of SARS-CoV-2. Comput. Biol. Med. 2021, 136, 104748. [Google Scholar] [CrossRef] [PubMed]
- Darlenski, R.; Tsankov, N. COVID-19 pandemic and the skin: What should dermatologists know? Clin. Dermatol. 2020, 38, 785–787. [Google Scholar] [CrossRef]
- Kumar, A.; Mansoori, N.; Kavitamunjal; Dahiya, G.; Dwivedi, V.; Choudhary, M. Comparative efficacy study of a polyherbal formulation with other available drugs in propiobacterium acnes induced rat model. Eur. J. Biomed. 2019, 6, 362–374. [Google Scholar]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Marciniak, B.; Drozda, R.; Kontek, R. Targeting carbonic anhydrase IX and XII isoforms with small molecule inhibitors and monoclonal antibodies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1278–1298. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Mujwar, S. Repurposing metocurine as main protease inhibitor to develop novel antiviral therapy for COVID-19. J. Struct. Chem. 2020, 31, 2487–2499. [Google Scholar] [CrossRef] [PubMed]
- Mondal, R.; Lahiri, D.; Deb, S.; Bandyopadhyay, D.; Shome, G.; Sarkar, S.; Paria, S.R.; Thakurta, T.G.; Singla, P.; Biswas, S.C. COVID-19: Are we dealing with a multisystem vasculopathy in disguise of a viral infection? J. Thromb. Thrombolysis 2020, 50, 567–579. [Google Scholar] [CrossRef]
- Michalska, K.; Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Stols, L.; Endres, M.; Joachimiak, A. Crystal structures of SARS-CoV-2 ADP-ribose phosphatase (ADRP): From the apo form to ligand complexes. IUCrJ 2020, 7, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Saikatendu, K.S.; Joseph, J.S.; Subramanian, V.; Clayton, T.; Griffith, M.; Moy, K.; Velasquez, J.; Neuman, B.W.; Buchmeier, M.J.; Stevens, R.C.; et al. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1’’-phosphate dephosphorylation by a conserved domain of nsP3. Structure 2005, 13, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Mujwar, S.; Sun, L.; Fidan, O. In silico evaluation of food-derived carotenoids against SARS-CoV-2 drug targets: Crocin is a promising dietary supplement candidate for COVID-19. J. Food Biochem. 2022, 46, e14219. [Google Scholar] [CrossRef] [PubMed]
- Mujwar, S.; Deshmukh, R.; Harwansh, R.K.; Gupta, J.K.; Gour, A. Drug Repurposing Approach for Developing Novel Therapy Against Mupirocin-Resistant Staphylococcus aureus. ASSAY Drug Dev. Technol. 2019, 17, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Mujwar, S.; Harwansh, R.K. In Silico Bioprospecting of Taraxerol as a Main Protease Inhibitor of SARS-CoV-2 to Develop Therapy against COVID-19. Struct. Chem. 2022, 33, 1517–1528. [Google Scholar] [CrossRef]
- Mujwar, S.; Kumar, V. Computational Drug Repurposing Approach to Identify Potential Fatty Acid-Binding Protein-4 Inhibitors to Develop Novel Antiobesity Therapy. ASSAY Drug Dev. Technol. 2020, 18, 318–327. [Google Scholar] [CrossRef]
- Mujwar, S.; Tripathi, A. Repurposing Benzbromarone as Antifolate to Develop Novel Antifungal Therapy for Candida Albicans. J. Mol. Model. 2021, 28, 193. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Mujwar, S.; Pardasani, K.R. Prediction of riboswitch as a potential drug target and design of its optimal inhibitors for Mycobacterium tuberculosis. Int. J. Comput. Biol. Drug Des. 2015, 8, 326–347. [Google Scholar] [CrossRef]
- Agrawal, N.; Upadhyay, P.; Mujwar, S.; Mishra, P. Analgesic, anti-inflammatory activity and docking study of 2-(substituted phenyl)-3-(naphthalen1-yl)thiazolidin-4-ones. J. Indian Chem. Soc. 2020, 97, 39–46. [Google Scholar]
- Kaur, A.; Mujwar, S.; Adlakha, N. In-silico analysis of riboswitch of Nocardia farcinica for design of its inhibitors and pharmacophores. Int. J. Comput. Biol. Drug Des. 2016, 9, 261–276. [Google Scholar] [CrossRef]
- Kaushal, S.K.; Brijendra, S.; Mujwar, S.; Prakash, B.S. Molecular Docking based analysis to elucidate the DNA Topoisomerase IIbeta as the potential target for the Ganoderic acid, A natural therapeutic agent in cancer therapy. Curr. Comput. Aided Drug Des. 2019, 16, 176–189. [Google Scholar]
- Minaz, N.; Razdan, R.; Hammock, B.D.; Mujwar, S.; Goswami, S.K. Impact of diabetes on male sexual function in streptozotocin-induced diabetic rats: Protective role of soluble epoxide hydrolase inhibitor. Biomed. Pharmacother. 2019, 115, 108897. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Mishra, I.M.R.; Mujwar, S.; Chandra, P.; Sachan, N. A retrospect on antimicrobial potential of thiazole scaffold. J. Heterocycl. Chem. 2020, 57, 2304–2329. [Google Scholar] [CrossRef]
- Mujwar, S.; Shah, K.; Gupta, J.K.; Gour, A. Docking based screening of curcumin derivatives: A novel approach in the inhibition of tubercular DHFR. Int. J. Comput. Biol. Drug Des. 2021, 14, 297–314. [Google Scholar] [CrossRef]
- Gupta, N.; Qayum, A.; Singh, S.; Mujwar, S.; Sangwan, P.L. Isolation, Anticancer Evaluation, Molecular Docking, Drug likeness and ADMET Studies of Secondary Metabolites from Psoralea corylifolia seeds. ChemistrySelect 2022, 7, e202202115. [Google Scholar] [CrossRef]
- Mujwar, S.; Pardasani, K.R. Prediction of Riboswitch as a potential drug target for infectious diseases: An Insilico case study of anthrax. J. Med. Imaging Health Inform. 2015, 5, 7–16. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A. Using autodock for ligand–receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8.14. 1–8.14. 40. [Google Scholar] [CrossRef]
- Mujwar, S. Computational bioprospecting of andrographolide derivatives as potent cyclooxygenase-2 inhibitors. Biomed. Biotechnol. Res. J. 2021, 5, 446. [Google Scholar] [CrossRef]
- Pradhan, P.; Soni, N.K.; Chaudhary, L.; Mujwar, S.; Pardasani, K.R. In-silico prediction of riboswitches and design of their potent inhibitors for H1N1, H2N2 and H3N2 strains of influenza virus. Biosci. Biotechnol. Res. Asia 2015, 12, 2173–2186. [Google Scholar] [CrossRef]
- Shah, K.; Mujwar, S.; Gupta, J.K.; Shrivastava, S.K.; Mishra, P. Molecular Docking and In Silico Cogitation Validate Mefenamic Acid Prodrugs as Human Cyclooxygenase-2 Inhibitor. ASSAY Drug Dev. Technol. 2019, 17, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Mujwar, S.; Krishna, G.; Gupta, J.K. Computational Design and Biological Depiction of Novel Naproxen Derivative. ASSAY Drug Dev. Technol. 2020, 18, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Mujwar, S.; Szymanowska, A.; Marciniak, B.; Bukowski, K.; Mojzych, M.; Kontek, R. Preparation of Novel Pyrazolo [4, 3-e] tetrazolo [1, 5-b][1, 2, 4] triazine Sulfonamides and Their Experimental and Computational Biological Studies. Int. J. Mol. Sci. 2022, 23, 5892. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Raghvendra, S.U.; Kalra, T.; Munjal, K. Applications of metabolomics-a systematic study of the unique chemical fingerprints: An overview. Int. J. Pharm. Sci. Rev. Res. 2010, 3, 83–86. [Google Scholar]
- Gupta, N.; Qayum, A.; Singh, S.; Mujwar, S.; Sangwan, P.L. Isolation, Cytotoxicity Evaluation, Docking, ADMET and Drug Likeness Studies of Secondary Metabolites from the Stem Bark of Anthocephalus cadamba (Roxb.). ChemistrySelect 2022, 7, e202202950. [Google Scholar] [CrossRef]
- Agrawal, N.; Mujwar, S.; Goyal, A.; Gupta, J.K. Phytoestrogens as Potential Antiandrogenic Agents Against Prostate Cancer: An In Silico Analysis. Lett. Drug Des. Discov. 2022, 19, 69–78. [Google Scholar] [CrossRef]
- Rani, I.; Kalsi, A.; Kaur, G.; Sharma, P.; Gupta, S.; Gautam, R.K.; Chopra, H.; Bibi, S.; Ahmad, S.U.; Singh, I.; et al. Surgery, Modern drug discovery applications for the identification of novel candidates for COVID-19 infections. Ann. Med. Surg. 2022, 80, 104125. [Google Scholar] [CrossRef] [PubMed]
- Shinu, P.; Sharma, M.; Gupta, G.L.; Mujwar, S.; Kandeel, M.; Kumar, M.; Nair, A.B.; Goyal, M.; Singh, P.; Attimarad, M.; et al. Computational Design, Synthesis, and Pharmacological Evaluation of Naproxen-Guaiacol Chimera for Gastro-Sparing Anti-Inflammatory Response by Selective COX2 Inhibition. Molecules 2022, 27, 6905. [Google Scholar] [CrossRef] [PubMed]
- Low, Z.Y.; Farouk, I.A.; Lal, S.K. Drug Repositioning: New Approaches and Future Prospects for Life-Debilitating Diseases and the COVID-19 Pandemic Outbreak. Viruses 2020, 12, 1058. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Kumari, L.; Badwaik, H.R.; Shanmugarajan, D. Computational Approaches for Drug Repositioning and Repurposing to Combat SARS-CoV-2 Infection. In Computational Approaches for Novel Therapeutic and Diagnostic Designing to Mitigate SARS-CoV-2 Infection; Academic Press: Cambridge, MA, USA, 2022; pp. 247–265. [Google Scholar] [CrossRef]
- Malik, J.; Munjal, K.; Deshmukh, R. Attenuating effect of standardized lyophilized Cinnamomum zeylanicum bark extract against streptozotocin-induced experimental dementia of Alzheimer’s type. J. Basic Clin. Physiol. Pharmacol. 2015, 26, 275–285. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar]
- Mukherjee, P.K.; Bahadur, S.; Harwansh, R.K.; Biswas, S.; Banerjee, S. Paradigm shift in natural product research: Traditional medicine inspired approaches. Phytochem. Rev. 2017, 16, 803–826. [Google Scholar] [CrossRef]
- Huang, F.; Li, Y.; Leung, E.L.-H.; Liu, X.; Liu, K.; Wang, Q.; Lan, Y.; Li, X.; Yu, H.; Cui, L.; et al. A review of therapeutic agents and Chinese herbal medicines against SARS-COV-2 (COVID-19). Pharmacol. Res. 2020, 158, 104929. [Google Scholar] [CrossRef] [PubMed]
- Fernández, S.; Wasowski, C.; Paladini, A.C.; Marder, M. Sedative and sleep-enhancing properties of linarin, a flavonoid-isolated from Valeriana officinalis. Pharmacol. Biochem. Behav. 2004, 77, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Munjal, K.; Ahmad, S.; Gupta, A.; Haye, A.; Amin, S.; Mir, S.R. Polyphenol-Enriched Fraction and the Compounds Isolated from Garcinia Indica Fruits Ameliorate Obesity through Suppression of Digestive Enzymes and Oxidative Stress. Pharmacogn. Mag. 2020, 16, 236–245. [Google Scholar]
- Singh, S.; Sharma, S.; Ali, M. Fatty acid esters from roots of amaranthus hybridus linn. Asian J. Chem. 2011, 23, 232–234. [Google Scholar]
- Meltzer, D.O.; Best, T.J.; Zhang, H.; Vokes, T.; Arora, V.; Solway, J. Association of Vitamin D Status and Other Clinical Characteristics With COVID-19 Test Results. JAMA Netw. Open 2020, 3, e2019722. [Google Scholar] [CrossRef]
- Boulkrane, M.S.; Ilina, V.; Melchakov, R.; Fedotova, J.; Drago, F.; Gozzo, L.; Das, U.N.; Abd El-Aty, A.M.; Baranenko, D. COVID-19 Disease and Vitamin D: A Mini-Review. Front. Pharmacol. 2020, 11, 604579. [Google Scholar] [CrossRef] [PubMed]
- Munjal, K.; Sharma, S.; Sharma, S.; Kumar, D.; Choudhary, A.; Berwal, R.; Kumar, A. Comparison of serum 25-hydroxyvitamin D levels after a single oral dose of vitamin D3 formulations in mild vitamin D3 deficiency. J. Pharmacol. Pharmacother. 2021, 12, 163–167. [Google Scholar]
- Deng, R. Therapeutic effects of guggul and its constituent guggulsterone: Cardiovascular benefits. Cardiovasc. Drug Rev. 2007, 25, 375–390. [Google Scholar] [CrossRef]
- Brobst, D.E.; Ding, X.; Creech, K.L.; Goodwin, B.; Kelley, B.; Staudinger, J.L. Guggulsterone activates multiple nuclear receptors and induces CYP3A gene expression through the pregnane X receptor. J. Pharmacol. Exp. Ther. 2004, 310, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef]
- Fidan, O.; Mujwar, S.; Kciuk, M. Discovery of adapalene and dihydrotachysterol as antiviral agents for the Omicron variant of SARS-CoV-2 through computational drug repurposing. Mol. Divers. 2022, 1–13. [Google Scholar] [CrossRef]
- Pandit, S.; Mukherjee, P.K.; Mukherjee, K.; Gajbhiye, R.; Venkatesh, M.; Ponnusankar, S.; Bhadra, S. Cytochrome P450 inhibitory potential of selected Indian spices—Possible food drug interaction. Food Res. Int. 2012, 45, 69–74. [Google Scholar] [CrossRef]
- Enmozhi, S.K.; Raja, K.; Sebastine, I.; Joseph, J. Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. J. Biomol. Struct. Dyn. 2021, 39, 3092–3098. [Google Scholar] [CrossRef]
- Tallei, T.E.; Tumilaar, S.G.; Niode, N.J.; Fatimawali; Kepel, B.J.; Idroes, R.; Effendi, Y.; Sakib, S.A.; Emran, T.B. Potential of Plant Bioactive Compounds as SARS-CoV-2 Main Protease (Mpro) and Spike (S) Glycoprotein Inhibitors: A Molecular Docking Study. Scientifica 2020, 2020, 6307457. [Google Scholar] [CrossRef] [PubMed]
- Farmanpour-Kalalagh, K.; Beyraghdar Kashkooli, A.; Babaei, A.; Rezaei, A.; van der Krol, A.R. Artemisinins in Combating Viral Infections Like SARS-CoV-2, Inflammation and Cancers and Options to Meet Increased Global Demand. Front. Plant Sci. 2022, 13, 780257. [Google Scholar] [CrossRef]
- Chen, W.C.; Wei, C.K.; Hossen, M.; Hsu, Y.C.; Lee, J.C. (E)-Guggulsterone Inhibits Dengue Virus Replication by Upregulating Antiviral Interferon Responses through the Induction of Heme Oxygenase-1 Expression. Viruses 2021, 13, 712. [Google Scholar] [CrossRef]
- Bouslama, L.; Kouidhi, B.; Alqurashi, Y.M.; Chaieb, K.; Papetti, A. Virucidal Effect of Guggulsterone Isolated from Commiphora gileadensis. Planta Med. 2019, 85, 1225–1232. [Google Scholar] [CrossRef]
- Preethi, L.; Ganamurali, N.; Dhanasekaran, D.; Sabarathinam, S. Therapeutic use of Guggulsterone in COVID-19 induced obesity (COVIBESITY) and significant role in immunomodulatory effect. Obes. Med. 2021, 24, 100346. [Google Scholar] [CrossRef]
- Mishra, A.; Pathak, Y.; Choudhir, G.; Kumar, A.; Mishra, S.K.; Tripathi, V. Anticancer natural compounds as potential inhibitors of novel coronavirus (COVID19) main protease: An in-silico study. Cancer Res. 2021, 81, 712. [Google Scholar] [CrossRef]
- Scholtes, C.; André, P.; Trépo, C.; Cornu, C.; Remontet, L.; Ecochard, R.; Bejan-Angoulvant, T.; Gueyffier, F. Farnesoid X receptor targeting for hepatitis C: Study protocol for a proof-of-concept trial. Therapie 2012, 67, 423–427. [Google Scholar] [CrossRef]
- Szapary, P.O.; Wolfe, M.L.; Bloedon, L.T.; Cucchiara, A.J.; DerMarderosian, A.H.; Cirigliano, M.D.; Rader, D.J. Guggulipid for the treatment of hypercholesterolemia: A randomized controlled trial. JAMA 2003, 290, 765–772. [Google Scholar] [CrossRef]
- Nohr, L.A.; Rasmussen, L.B.; Straand, J. Resin from the mukul myrrh tree, guggul, can it be used for treating hypercholesterolemia? A randomized, controlled study. Complement. Ther. Med. 2009, 17, 16–22. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Macromolecule | x-Axis | y-Axis | z-Axis | Spacing (Ả) | x-Center | y-Center | z-Center |
|---|---|---|---|---|---|---|---|
| 6w02 | 40 | 40 | 40 | 0.452 | 3.501 | −5.927 | −22.574 |
| Macromolecule | Binding Residues | RMSD | Binding Energy (kcal/mol) |
|---|---|---|---|
| 6w02 | Asp22, Ile23, Val49, Ala154, Gly130, Phe132, Asn40, Gly46, Lys44, Gly48, Ala50, Ser128, and Ile131 | 1.32 | −9.59 |
| S. No. | Name | Structure | Binding Energy (kcal/mol) |
|---|---|---|---|
| 1. | Guggulsterone |  | −10.24 |
| 2. | Mahanimbine |  | −10.16 |
| 3. | Linarin |  | −9.93 |
| 4 | Withanolide |  | −9.79 |
| 5. | Andrographolide |  | −9.55 |
| 6. | Artemisinin |  | −9.44 |
| 7. | Mahanine |  | −9.39 |
| 8. | Silybin |  | −9.13 |
| 9. | Quercetin |  | −9.03 |
| 10. | Luteolin |  | −9.02 |
| 11. | ZINC82673 |  | −8.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kciuk, M.; Mujwar, S.; Rani, I.; Munjal, K.; Gielecińska, A.; Kontek, R.; Shah, K. Computational Bioprospecting Guggulsterone against ADP Ribose Phosphatase of SARS-CoV-2. Molecules 2022, 27, 8287. https://doi.org/10.3390/molecules27238287
Kciuk M, Mujwar S, Rani I, Munjal K, Gielecińska A, Kontek R, Shah K. Computational Bioprospecting Guggulsterone against ADP Ribose Phosphatase of SARS-CoV-2. Molecules. 2022; 27(23):8287. https://doi.org/10.3390/molecules27238287
Chicago/Turabian StyleKciuk, Mateusz, Somdutt Mujwar, Isha Rani, Kavita Munjal, Adrianna Gielecińska, Renata Kontek, and Kamal Shah. 2022. "Computational Bioprospecting Guggulsterone against ADP Ribose Phosphatase of SARS-CoV-2" Molecules 27, no. 23: 8287. https://doi.org/10.3390/molecules27238287
APA StyleKciuk, M., Mujwar, S., Rani, I., Munjal, K., Gielecińska, A., Kontek, R., & Shah, K. (2022). Computational Bioprospecting Guggulsterone against ADP Ribose Phosphatase of SARS-CoV-2. Molecules, 27(23), 8287. https://doi.org/10.3390/molecules27238287