
Transformation of Amides to Thioamides Using an Efficient and Novel Thiating Reagent

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
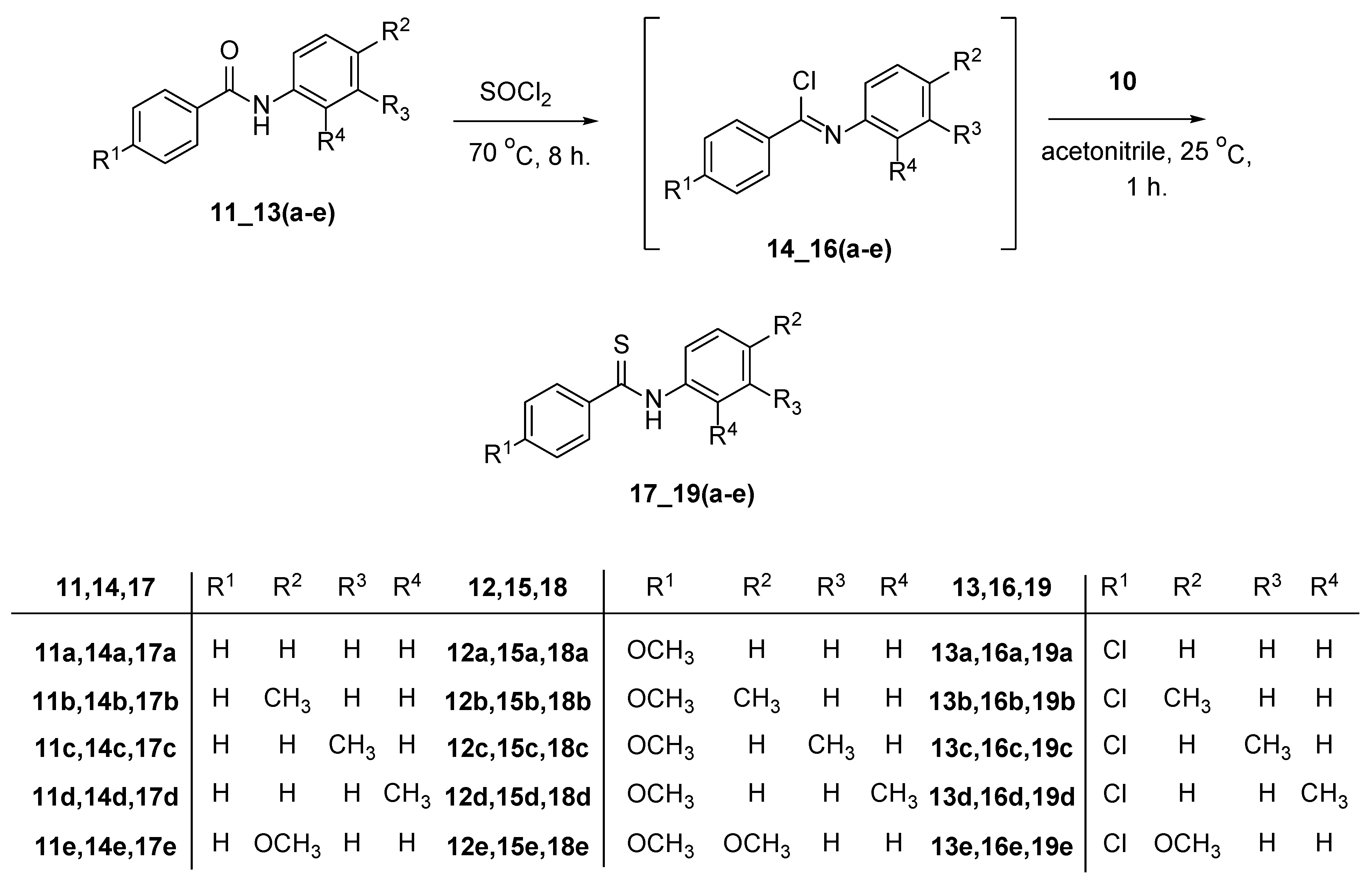
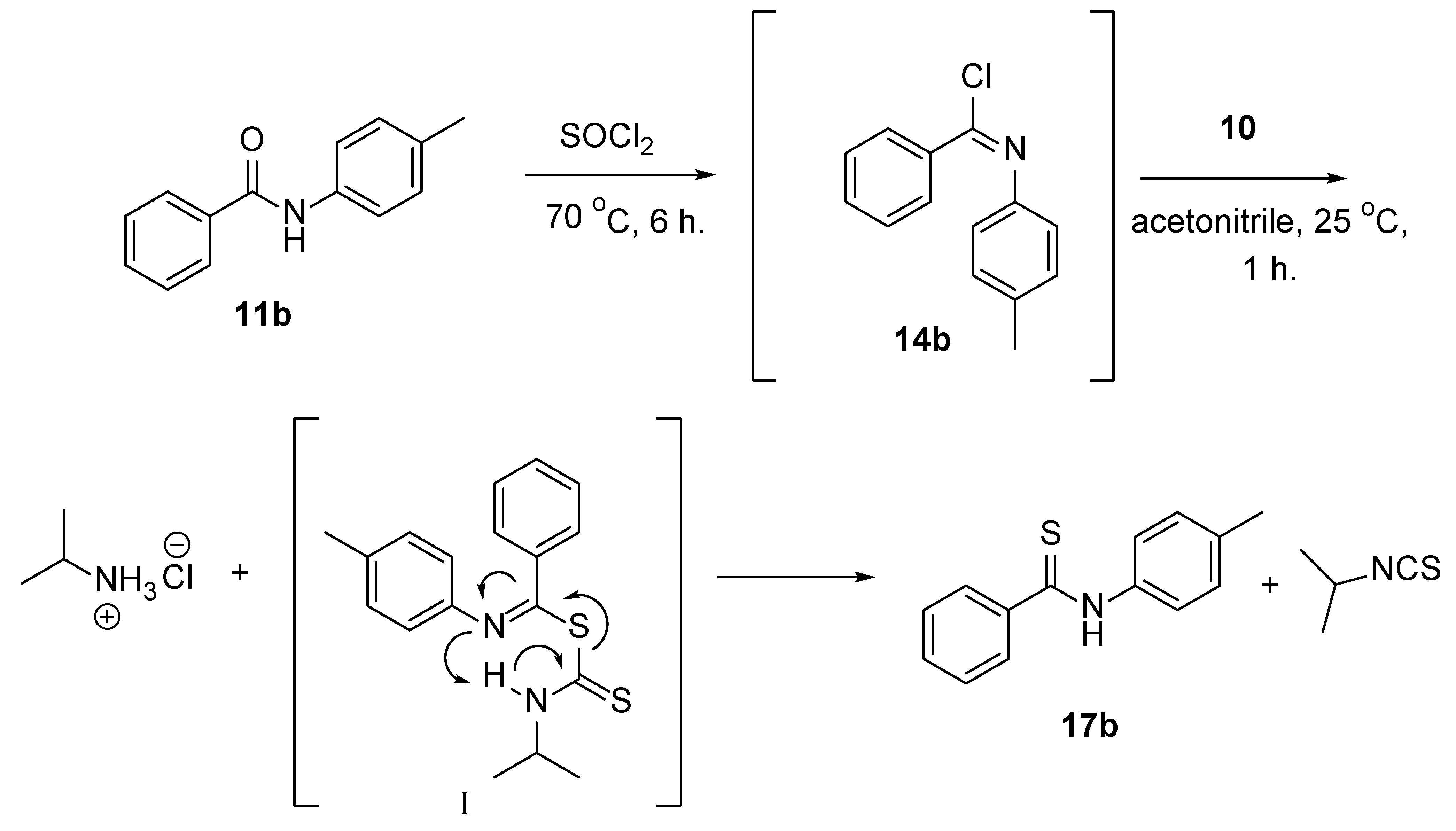
2. Discussion
3. Conclusions
4. Experimental
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Mahanta, N.; Szantai-Kis, D.M.; Petersson, E.J.; Mitchell, D.A. Biosynthesis and Chemical Applications of Thioamides. ACS Chem. Biol. 2019, 14, 142. [Google Scholar] [CrossRef] [PubMed]
- Jagodzin´, T.S. Ski. Thioamides as Useful Synthons in the Synthesis of Heterocycles. Chem. Rev. 2003, 103, 197–228. [Google Scholar] [CrossRef] [PubMed]
- Murai, T.; Aso, H.; Tatematsu, Y.; Itoh, Y.; Niwa, H.; Kato, S. Reaction and Characterization of Thioamide Dianions Derived from N-Benzyl Thioamides. J. Org. Chem. 2003, 68, 8514–8519. [Google Scholar] [CrossRef] [PubMed]
- Hurd, R.N.; DeLaMater, G. The Preparation and Chemical Properties of Thionamides. Chem. Rev. 1961, 61, 45–86. [Google Scholar] [CrossRef]
- Pathak, U.; Pandey, L.K.; Mathur, S.; Suryanarayana, M.V.S. Ketoximes to N-substituted thioamides viaPSCl3 mediated Beckmann rearrangement. Chem. Commun. 2009, 36, 5409. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, Y.; Xie, L.G. Dehydrative Beckmann rearrangement and the following cascade reactions. Chin. Chem. Lett. 2022, 33, 2407. [Google Scholar] [CrossRef]
- Yadav, A.K.; Srivastava, V.P.; Yadav, L.D.S. O,O-Diethyl dithiophosphoric acid mediated direct synthesis of thioamides from aldehydes and ketones. Tetrahedron Lett. 2012, 53, 7113. [Google Scholar] [CrossRef]
- Pathare, S.P.; Chaudhari, P.S.; Akamanchi, K.G. Sulfated tungstate: An efficient catalyst for synthesis of thioamides via Kindler reaction. Appl. Catal. A Gen. 2012, 425, 125. [Google Scholar] [CrossRef]
- Okamoto, K.; Yamamoto, T.; Kanbara, T. Efficient Synthesis of Thiobenzanilides by Willgerodt-Kindler Reaction with Base Catalysts. SYNLETT 2007, 17, 2687. [Google Scholar]
- Yin, Z.; Zheng, B.; Ai, F. Nano n-propylsulfonated magnetic γ-Fe2O3 as an efficient and reusable catalyst for the synthesis of thioamides. Phosphorus Sulfur Silicon 2013, 188, 1412. [Google Scholar] [CrossRef]
- Xu, H.; Deng, H.; Li, Z.; Xiang, H.; Zhou, X. Synthesis of Thioamides by Catalyst-Free Three-Component Reactions in Water. Eur. J. Org. Chem. 2013, 31, 7054–7057. [Google Scholar] [CrossRef]
- Do, N.T.; Tran, K.M.; Phan, H.T.; To, T.A.; Nguyen, T.T.; Phan, N.T.S. Functionalization of activated methylene C–H bonds with nitroarenes and sulfur for the synthesis of thioamides. Org. Biomol. Chem. 2019, 40, 8987. [Google Scholar] [CrossRef]
- Doszczak, L.; Rachon, J. Synthesis of S-thioacyl dithiophosphates, efficient and chemoselective thioacylating agents J. Chem. Soc. Perkin Trans. 2002, 10, 1271. [Google Scholar] [CrossRef]
- Doszczak, L.; Rachon, J. New, efficient and chemoselective method of thioacylation, starting from carboxylic acidsPresented in part at 13th ICOS, Warsaw 2000, Poland, pp. 139. Chem. Commun. 2000, 21, 2093. [Google Scholar] [CrossRef]
- Varma, R.S.; Kumar, D. Microwave-accelerated solvent-free synthesis of thioketones, thiolactones, thioamides, thionoesters, and thioflavonoids. Org. Lett. 1999, 1, 697–700. [Google Scholar] [CrossRef]
- Cho, D.; Ahn, J.; De Castro, K.A.; Ahn, H.; Rheeb, H. P4S10/dimethicone tandem: Efficient reagent for thionation of various aromatic amides and esters. Tetrahedron 2010, 66, 5583–5585. [Google Scholar] [CrossRef]
- Fathalla, W.; Ali, I.A.I.; Pazdera, P. A novel method for heterocyclic amide–thioamide transformations. Beilstein J. Org. Chem. 2017, 13, 174–181. [Google Scholar] [CrossRef]
- Megahed, M.; Fathalla, W.; El-sheikh, A. Synthesis and Antimicrobial Activity of Methyl 2-(2-(2-Arylquinazolin-4-yl)oxy) Acetylamino Alkanoates. J. Heterocycl. Chem. 2018, 55, 2799. [Google Scholar] [CrossRef]
- El Rayes, S.M.; Abou-Elmagd, A.; Gomaa, M.S.; Ali, I.A.I.; Fathalla, W.; Pottoo, F.; Khan, F. Convenient Synthesis and Anticancer Activity of Methyl 2-[3-(3-Phenyl-quinoxalin-2-ylsulfanyl)propanamido]alkanoates and N-Alkyl 3-((3-Phenyl-quinoxalin-2-yl)sulfanyl)propanamides. ACS Omega 2019, 4, 18555–18566. [Google Scholar] [CrossRef]
- Soliman, M.H.A.; Ali, I.A.I.; El-Sakka, S.S.A.; Mohamed, O.E.A. Novel quinoline derivatives as antitumor agents against HepG2 cells: Synthesis, characterization, in silico, in vitro and docking studies J. Mol. Struct. 2022, 1254, 132325. [Google Scholar] [CrossRef]
- Fathalla, W.; Pazdera, P. Recent Progress in the Synthesis of 2,3-Dihydrobenzofurans. Org. Prep. Proced. Int. 2018, 50, 385. [Google Scholar] [CrossRef]
- Fathalla, W.; Pazdera, P.; Khalifa, M.E.; Ali, I.A.I.; El Rayes, S.M. Novel domino synthesis of 2-(2,3.4-substituted phenyl) quinazolin-4-amine. J. Heterocycl. Chem. 2022, 59, 933. [Google Scholar] [CrossRef]
- Fathalla, W.; Ali, I.A.I.; Marek, J.; Pazdera, P. Syntheses and reactivity of alkyl 1-[N-(2-chloro-5-nitro-phenyl)-benzimidoyl] thioureas. J. Sulfur Chem. 2012, 33, 49–63. [Google Scholar] [CrossRef]
- Nahakpam, L.; Chipem, F.A.S.; Chingakham, B.S.; Laitonjam, W.S. Decomposition of benzoylthioureas into benzamides and thiobenzamides under solvent-free conditions using iodine–alumina as the catalyst and its mechanistic study by density functional theory. New J. Chem. 2015, 39, 2240. [Google Scholar] [CrossRef]
- Downer-Riley, N.K.; Jackson, Y.A. Conversion of thiobenzamides to benzothiazoles via intramolecular cyclization of the aryl radical cation. Tetrahedron 2008, 64, 7741. [Google Scholar] [CrossRef]
- Varun, B.V.; Sood, A.; Prabhu, K.R. A metal-free and a solvent-free synthesis of thio-amides and amides: An efficient Friedel–Crafts arylation of isothiocyanates and isocyanates. RSC Adv. 2014, 4, 60798. [Google Scholar] [CrossRef]
- Stevens, F.G.; McCall, C.J.; Lelievald, P.; Alexander, P.; Richter, A.; Davies, D.E. Structural Studies on Bioactive Compounds. 23. Synthesis of Polyhydroxylated 2-Phenylbenzothiazoles and a Comparison of their Cytotoxicities and Pharmacological Properties with Genistein and Quercetin. J. Med. Chem. 1994, 37, 1689–1695. [Google Scholar] [CrossRef]
- Kobayashi, K.; Fujiwara, D.; Tanmatsu, M. One-Pot Synthesis of 2-Substituted 1H-Isoindole-1,3(2H)-dithiones from Secondary Benzothioamides and Isothiocyanates. Heterocycles 2018, 96, 902–909. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomaa, M.S.; El Enany, G.; Fathalla, W.; Ali, I.A.I.; El Rayes, S.M. Transformation of Amides to Thioamides Using an Efficient and Novel Thiating Reagent. Molecules 2022, 27, 8275. https://doi.org/10.3390/molecules27238275
Gomaa MS, El Enany G, Fathalla W, Ali IAI, El Rayes SM. Transformation of Amides to Thioamides Using an Efficient and Novel Thiating Reagent. Molecules. 2022; 27(23):8275. https://doi.org/10.3390/molecules27238275
Chicago/Turabian StyleGomaa, Mohamed S., Gaber El Enany, Walid Fathalla, Ibrahim A. I. Ali, and Samir. M. El Rayes. 2022. "Transformation of Amides to Thioamides Using an Efficient and Novel Thiating Reagent" Molecules 27, no. 23: 8275. https://doi.org/10.3390/molecules27238275
APA StyleGomaa, M. S., El Enany, G., Fathalla, W., Ali, I. A. I., & El Rayes, S. M. (2022). Transformation of Amides to Thioamides Using an Efficient and Novel Thiating Reagent. Molecules, 27(23), 8275. https://doi.org/10.3390/molecules27238275