
New Nucleic Base-Tethered Trithiolato-Bridged Dinuclear Ruthenium(II)-Arene Compounds: Synthesis and Antiparasitic Activity

 ,
,  ,
,  and
and
Abstract

1. Introduction
2. Results
2.1. Synthesis
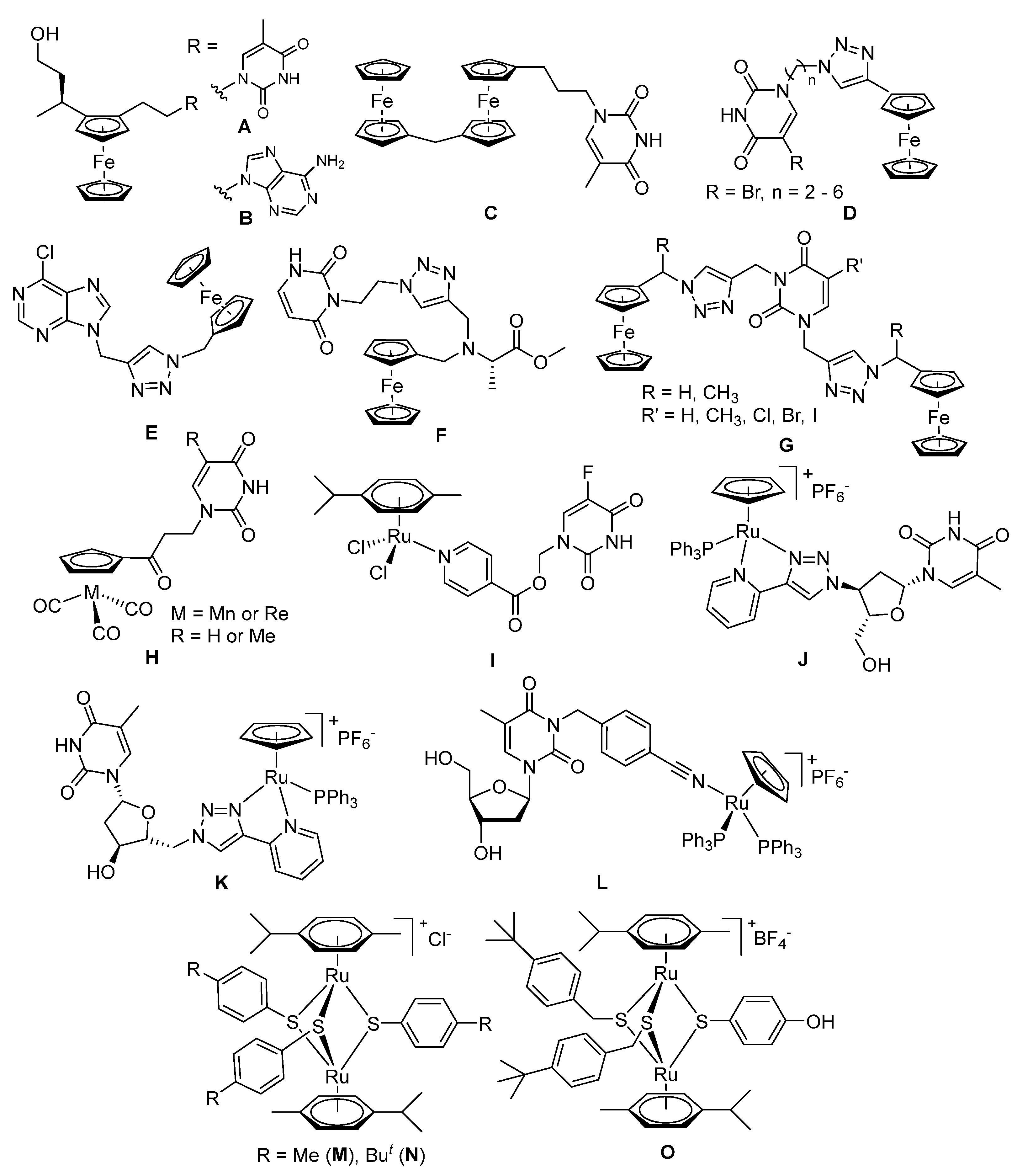
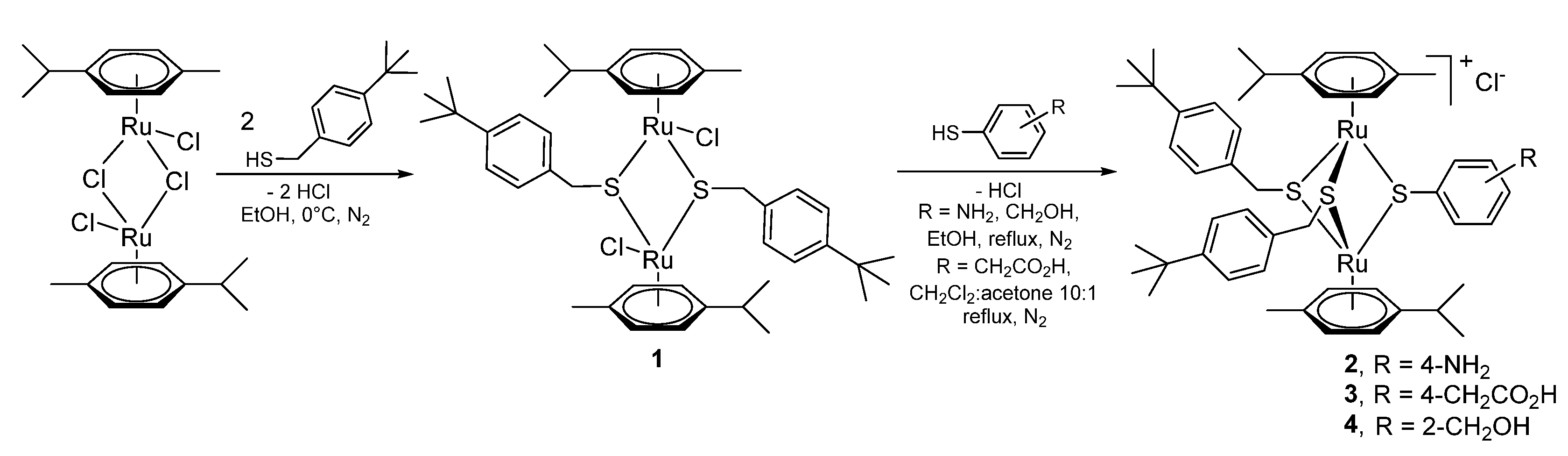
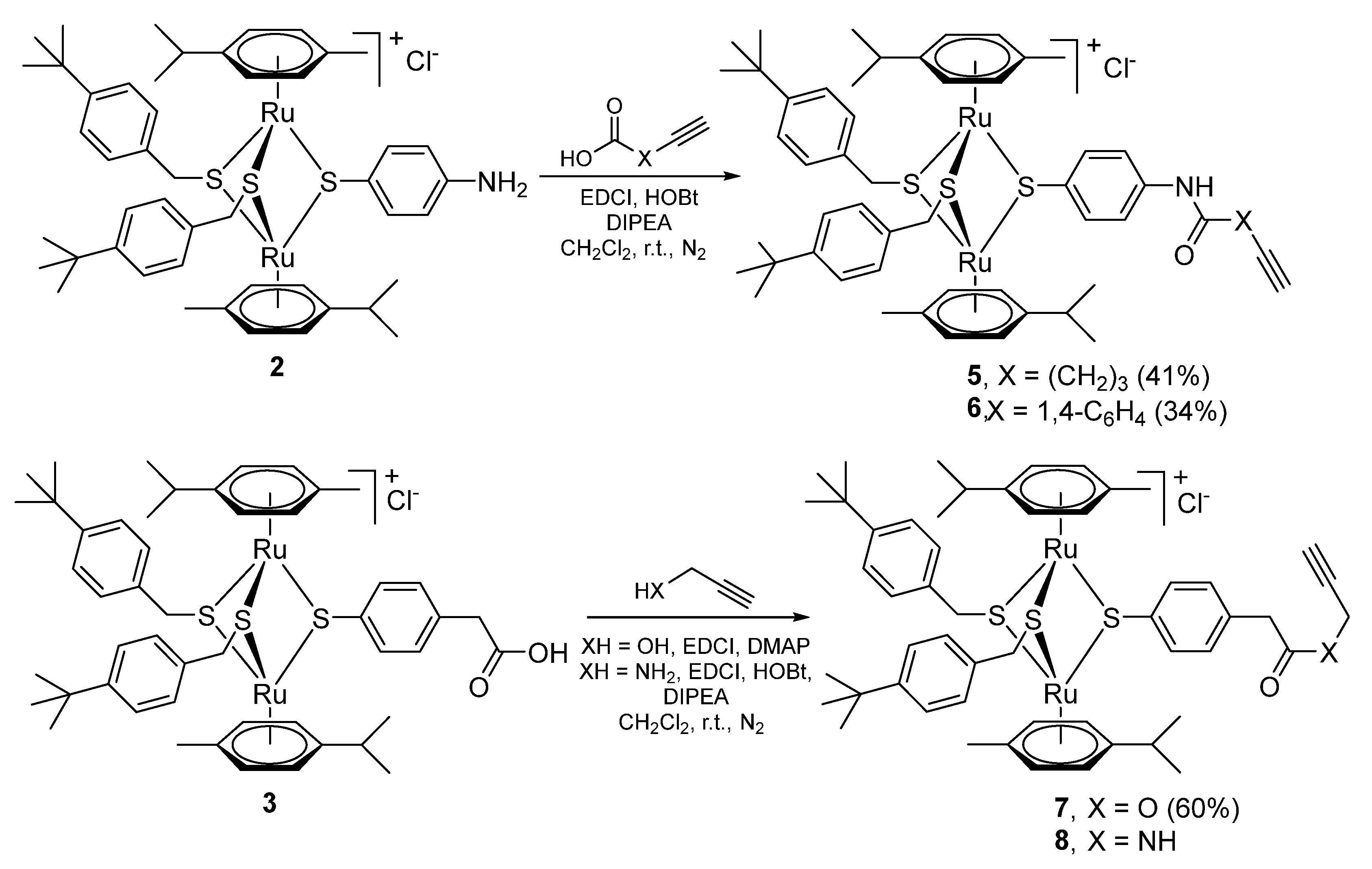
2.1.1. Synthesis of the Trithiolato-Bridged Dinuclear Ruthenium(II)-Arene Intermediates 2–9
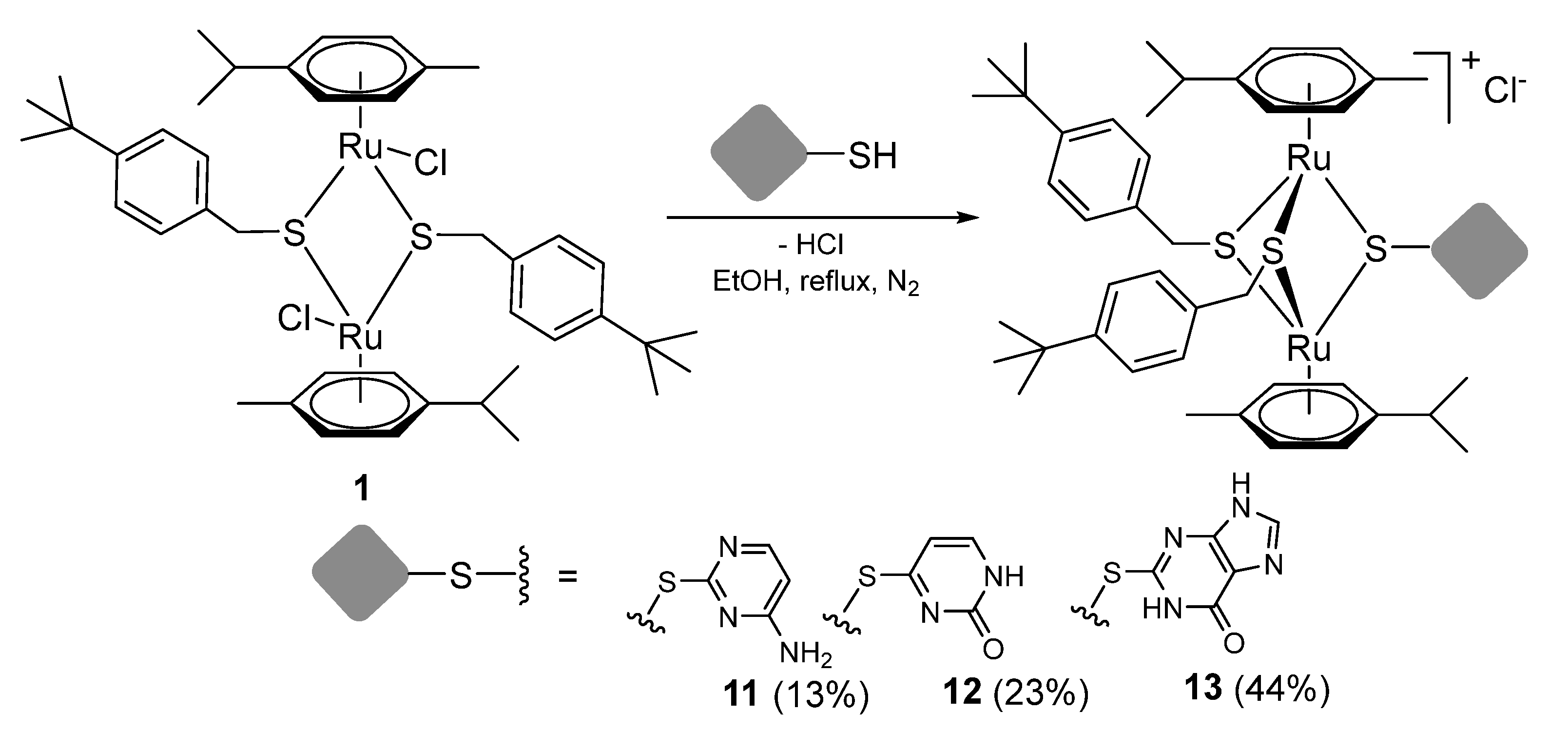
2.1.2. Synthesis of Compounds 11–13 (Family 1)
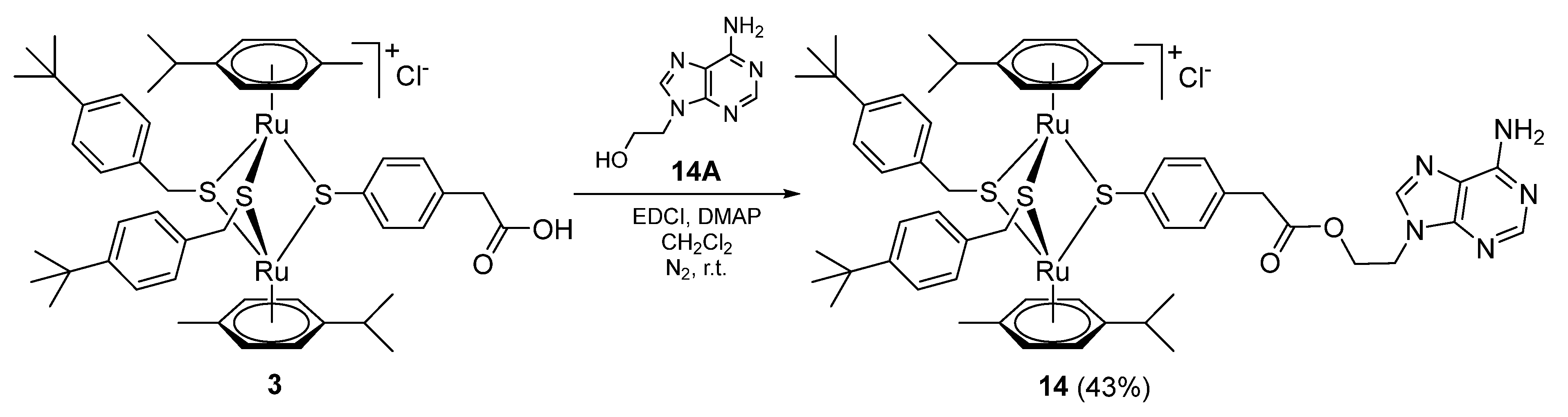
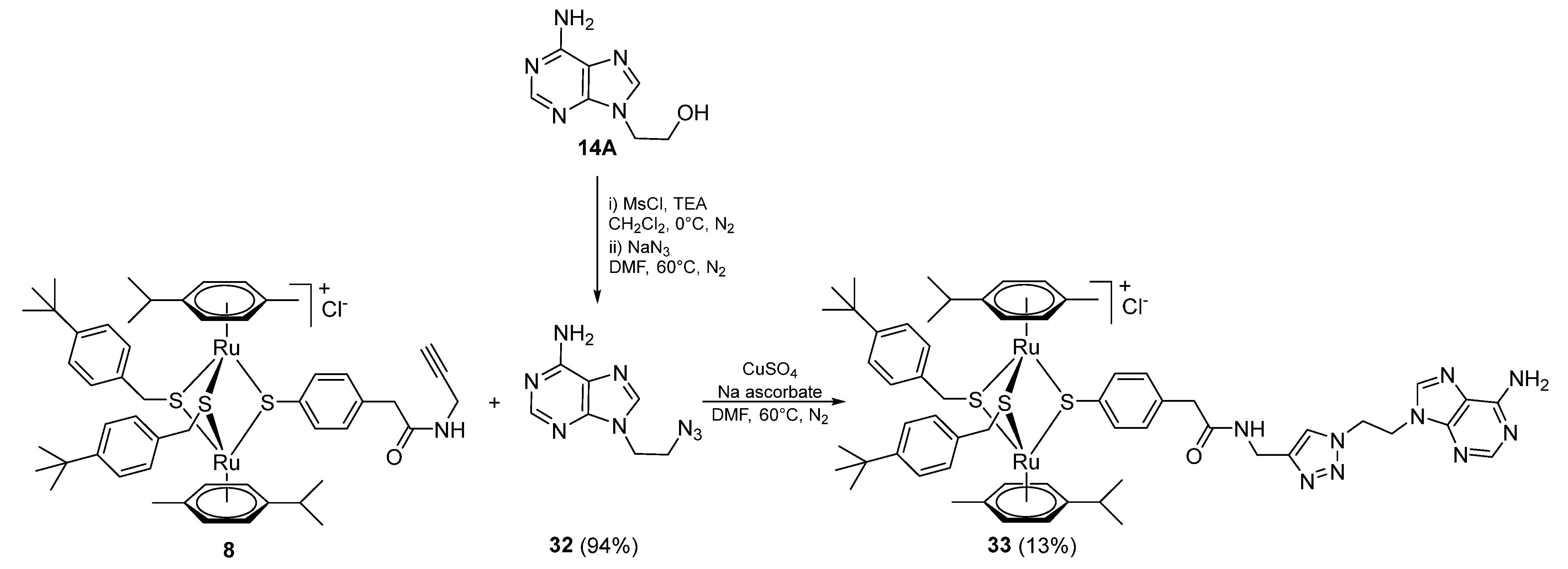
2.1.3. Synthesis of Compound 14 (Family 2)
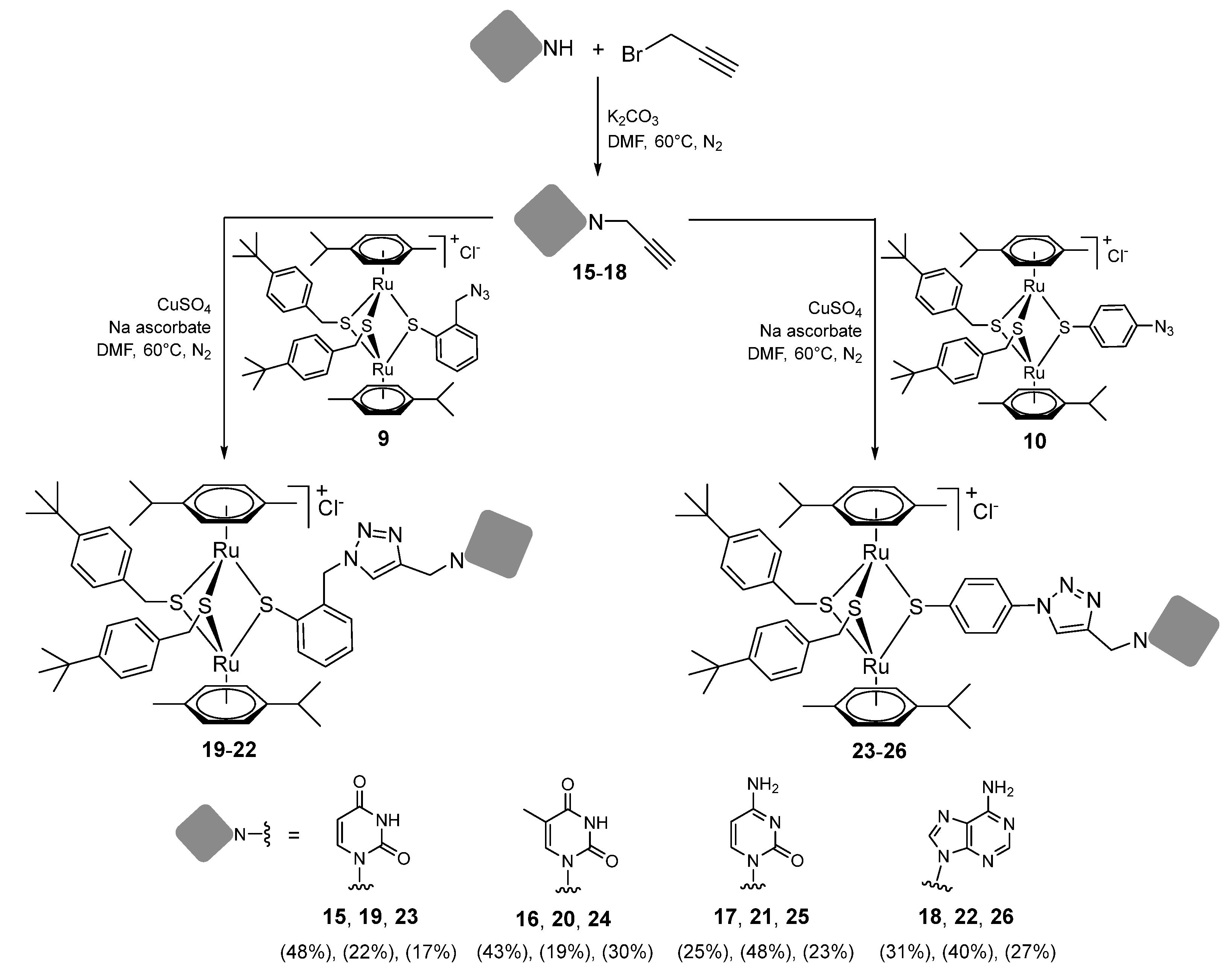
2.1.4. Synthesis of Compounds 15–26 (Family 3)
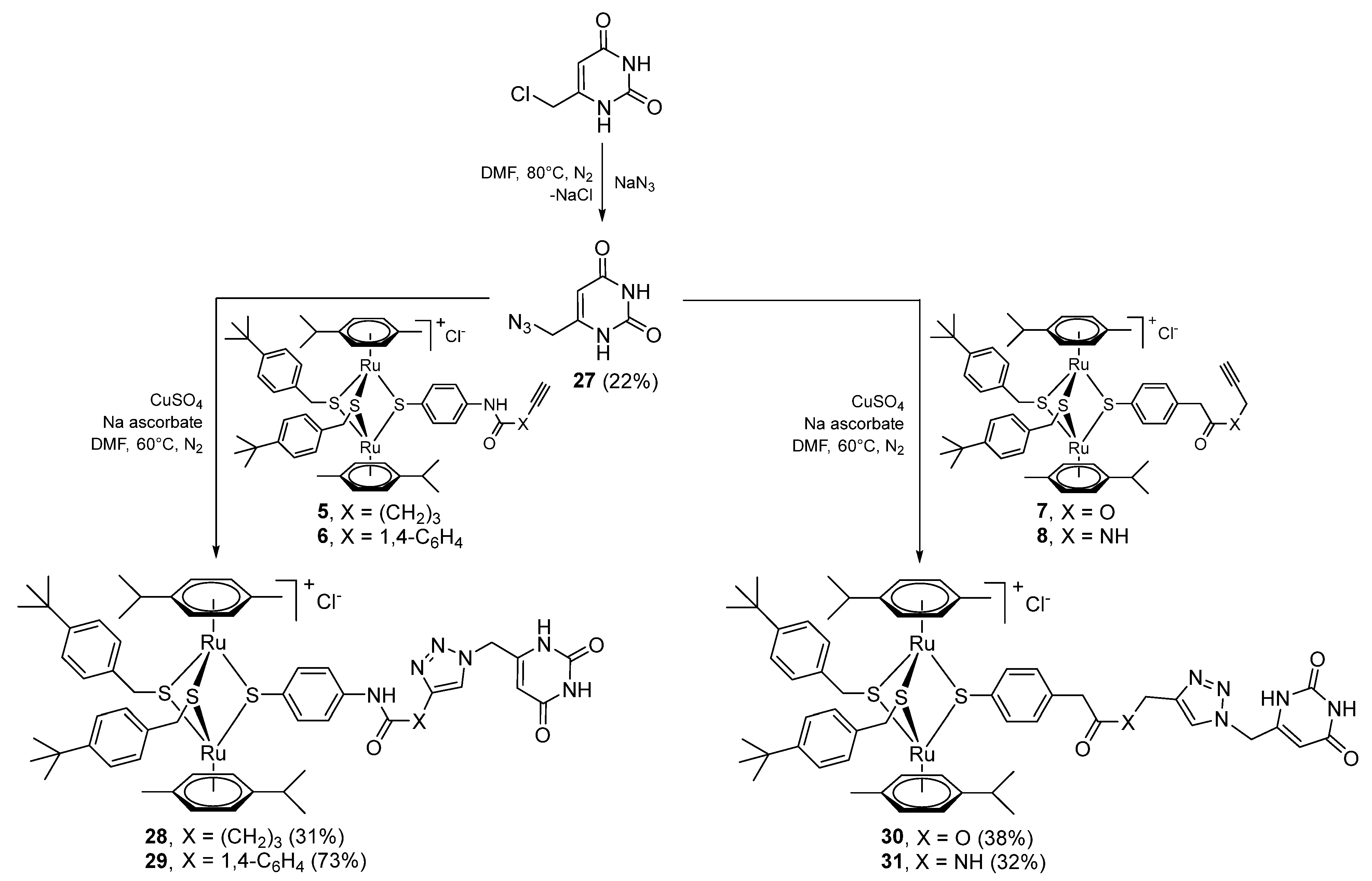
2.1.5. Synthesis of Compounds 27–33 (Family 4)
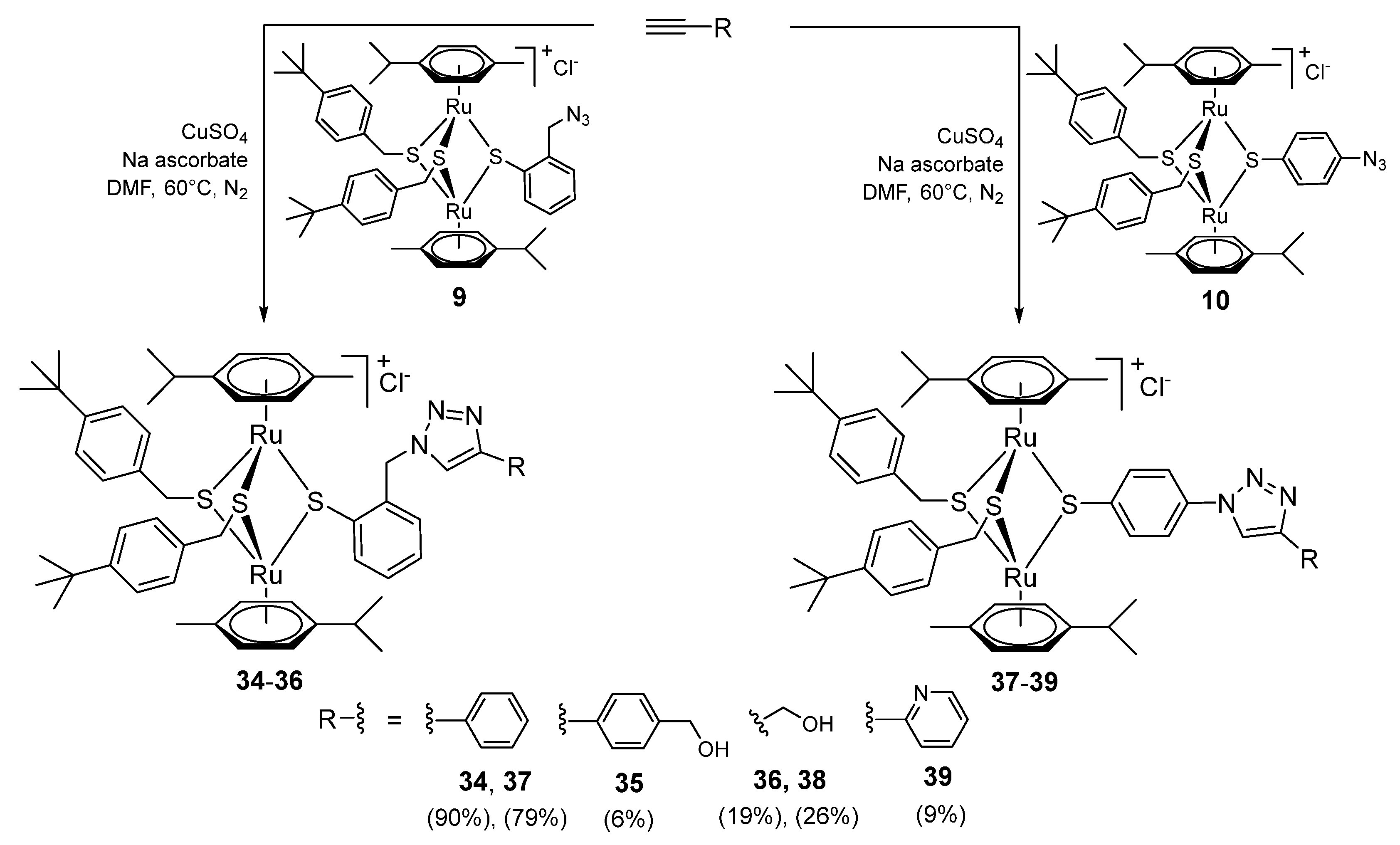
2.1.6. Synthesis of Compounds 34–39 (Family 5)
2.1.7. Stability of the Compounds
2.2. Assessment of the In Vitro Activity against T. gondii β-gal and Host Cells
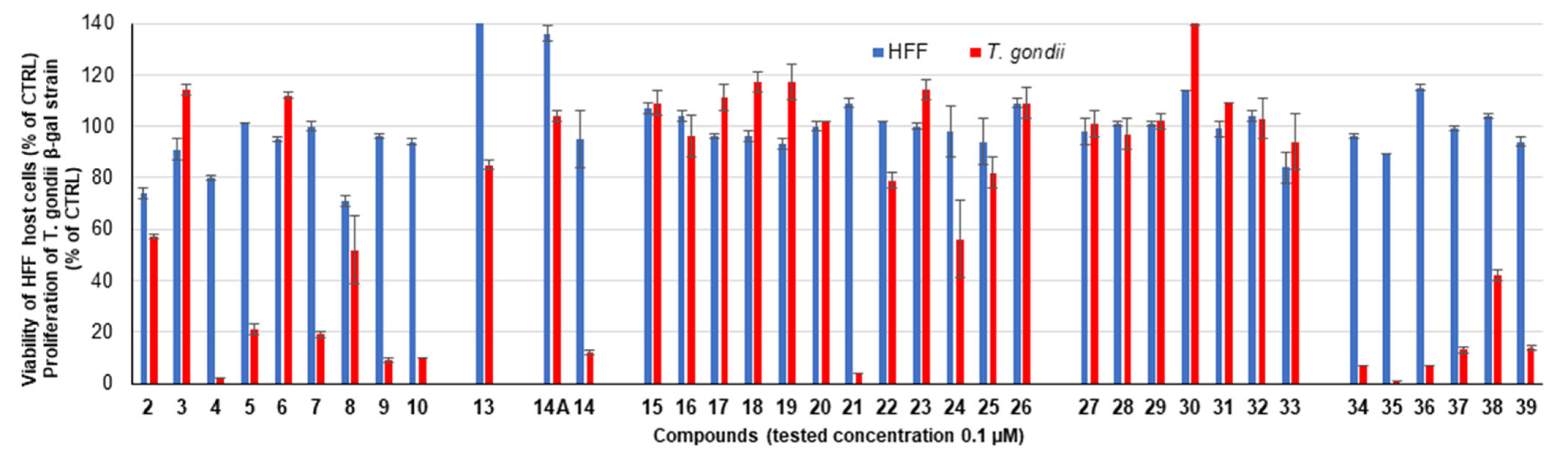
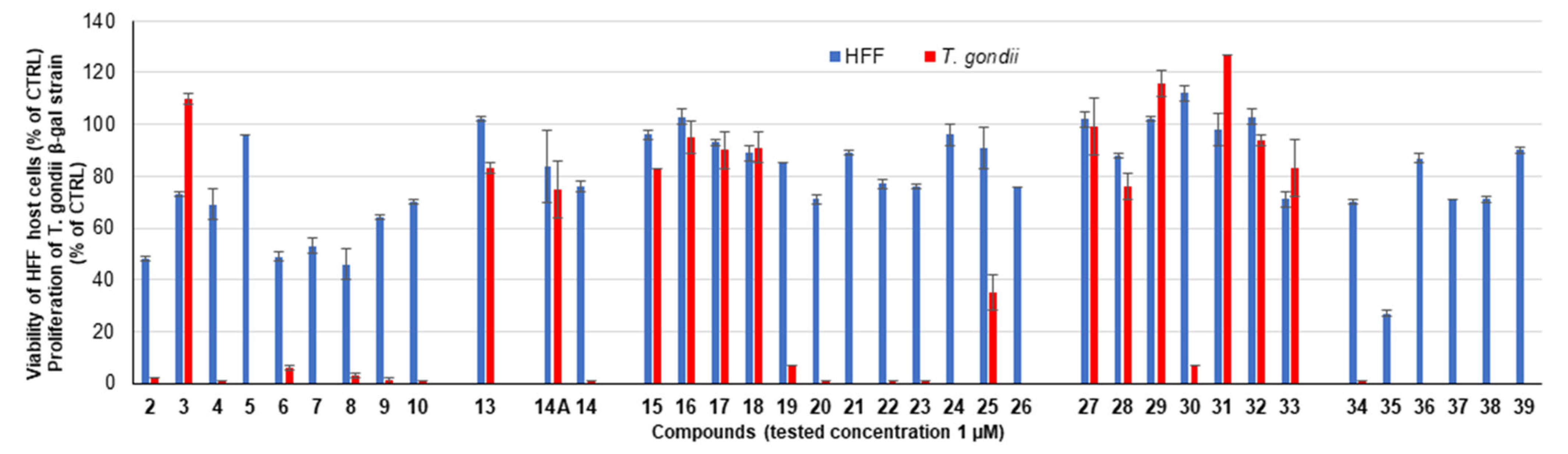
2.2.1. Primary Screening
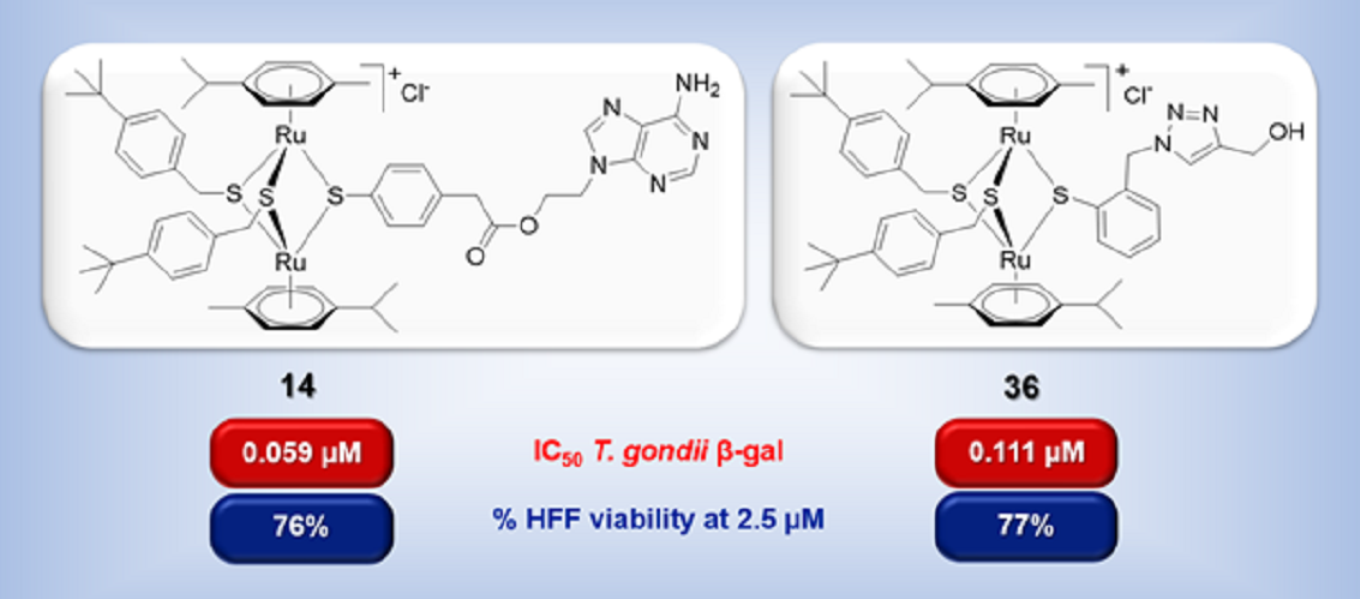
2.2.2. IC50 Values against T. gondii β-Gal Tachyzoites and HFF Toxicity at 2.5 µM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R | IC50 (µM) | [LS; LI] c | SE d | HFF Viability (%) e | SD f |
|---|---|---|---|---|---|---|
| Diruthenium intermediates | ||||||
| 4 b |  | 0.038 | [0.060; 0.023] | 0.110 | 4 | 2 |
| 5 |  | 0.038 | [0.050; 0.029] | 0.063 | 34 | 1 |
| 6 |  | 0.288 | [0.348; 0.238] | 0.188 | 17 | 1 |
| 7 |  | 0.289 | [0.363; 0.230] | 0.229 | 51 | 3 |
| 9 |  | 0.048 | [0.058; 0.040] | 0.139 | 11 | 1 |
| 10 |  | 0.064 | [0.080; 0.051] | 0.050 | 38 | 1 |
| Family 2 | ||||||
| 14 |  | 0.059 | [0.085; 0.040] | 0.037 | 76 | 3 |
| Family 3 | ||||||
| 19 |  | 0.460 | [0.626; 0.338] | 0.307 | 50 | 0 |
| 20 |  | 0.363 | [0.371; 0.354] | 0.023 | 39 | 1 |
| 21 |  | 0.046 | [0.058; 0.037] | 0.048 | 38 | 1 |
| 22 |  | 0.108 | [0.141; 0.083] | 0.066 | 52 | 1 |
| 23 |  | 0.426 | [0.553; 0.328] | 0.260 | 64 | 1 |
| 24 |  | 0.357 | [0.418; 0.305] | 0.156 | 85 | 4 |
| 26 |  | 0.178 | [0.226; 0.140] | 0.061 | 45 | 2 |
| Family 4 | ||||||
| 30 |  | 0.659 | [0.684; 0.635] | 0.037 | 97 | 1 |
| Family 5 | ||||||
| 34 |  | 0.092 | [0.108; 0.078] | 0.038 | 3 | 0 |
| 36 |  | 0.111 | [0.135; 0.090] | 0.039 | 77 | 1 |
| 37 |  | 0.096 | [0.122; 0.076] | 0.057 | 17 | 1 |
| 38 |  | 0.128 | [0.164; 0.099] | 0.058 | 59 | 1 |
| 39 |  | 0.075 | [0.091; 0.061] | 0.043 | 1 | 0 |
| Pyrimethamine a | 0.326 | [0.396; 0.288] | 0.051 | 99 | 6 | |
3. Materials and Methods
3.1. Chemistry
3.2. Biological Evaluation
3.2.1. Cell and Parasite Culture
3.2.2. In Vitro Assessment against T. gondii Tachyzoites and Human Foreskin Fibroblasts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torgerson, P.R.; Devleesschauwer, B.; Praet, N.; Speybroeck, N.; Willingham, A.L.; Kasuga, F.; Rokni, M.B.; Zhou, X.N.; Fevre, E.M.; Sripa, B.; et al. World Health Organization estimates of the global and regional disease burden of 11 foodborne parasitic diseases, 2010: A data synthesis. PLoS Med. 2015, 12, e1001920. [Google Scholar] [CrossRef] [PubMed]
- Meireles, L.R.; Ekman, C.C.; Andrade Jr, H.F.; Luna, E.J. Human toxoplasmosis outbreaks and the agent infecting form. Findings from a systematic review. Rev. Inst. Med. Trop. Sao Paulo 2015, 57, 369–376. [Google Scholar] [CrossRef]
- Neville, A.J.; Zach, S.J.; Wang, X.; Larson, J.J.; Judge, A.K.; Davis, L.A.; Vennerstrom, J.L.; Davis, P.H. Clinically available medicines demonstrating anti-Toxoplasma activity. Antimicrob. Agents Chemother. 2015, 59, 7161–7169. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Villavicencio, J.A.; Besne-Merida, A.; Correa, D. Vertical transmission and fetal damage in animal models of congenital toxoplasmosis: A systematic review. Vet. Parasitol. 2016, 223, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Alday, P.H.; Doggett, J.S. Drugs in development for toxoplasmosis: Advances, challenges, and current status. Drug Des. Dev. Ther. 2017, 11, 273–293. [Google Scholar] [CrossRef]
- Konstantinovic, N.; Guegan, H.; Stajner, T.; Belaz, S.; Robert-Gangneux, F. Treatment of toxoplasmosis: Current options and future perspectives. Food Waterborne Parasitol. 2019, 15, e00036. [Google Scholar] [CrossRef]
- Sibley, L.D. Toxoplasma gondii: Perfecting an intracellular life style. Traffic 2003, 4, 581–586. [Google Scholar] [CrossRef]
- Coppens, I. Exploitation of auxotrophies and metabolic defects in Toxoplasma as therapeutic approaches. Int. J. Parasitol. 2014, 44, 109–120. [Google Scholar] [CrossRef]
- Krug, E.C.; Marr, J.J.; Berens, R.L. Purine metabolism in Toxoplasma gondii. J. Biol. Chem. 1989, 264, 10601–10607. [Google Scholar] [CrossRef]
- Chaudhary, K.; Darling, J.A.; Fohl, L.M.; Sullivan, W.J., Jr.; Donald, R.G.; Pfefferkorn, E.R.; Ullman, B.; Roos, D.S. Purine salvage pathways in the apicomplexan parasite Toxoplasma gondii. J. Biol. Chem. 2004, 279, 31221–31227. [Google Scholar] [CrossRef]
- Perrotto, J.; Keister, D.B.; Gelderman, A.H. Incorporation of precursors into Toxoplasma DNA. J. Protozool. 1971, 18, 470–473. [Google Scholar] [CrossRef]
- Fox, B.A.; Bzik, D.J. Biochemistry and metabolism of Toxoplasma gondii: Purine and pyrimidine acquisition in Toxoplasma gondii and other Apicomplexa. In Toxoplasma gondii. The Model Apicomplexan—Perspectives and Methods, 3rd ed.; Weiss, L.M., Kim, K., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 397–449. [Google Scholar]
- Fox, B.A.; Bzik, D.J. De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii. Nature 2002, 415, 926–929. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.A.; Bzik, D.J. Avirulent uracil auxotrophs based on disruption of orotidine-5′-monophosphate decarboxylase elicit protective immunity to Toxoplasma gondii. Infect. Immun. 2010, 78, 3744–3752. [Google Scholar] [CrossRef] [PubMed]
- Schwartzman, J.D.; Pfefferkorn, E.R. Pyrimidine synthesis by intracellular Toxoplasma gondii. J. Parasitol. 1981, 67, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Iltzsch, M.H. Pyrimidine salvage pathways in Toxoplasma gondii. J. Eukaryot Microbiol. 1993, 40, 24–28. [Google Scholar] [CrossRef]
- Hajj, R.E.; Tawk, L.; Itani, S.; Hamie, M.; Ezzeddine, J.; El Sabban, M.; El Hajj, H. Toxoplasmosis: Current and emerging parasite druggable targets. Microorganisms 2021, 9, 531. [Google Scholar] [CrossRef]
- Wu, R.Z.; Zhou, H.Y.; Song, J.F.; Xia, Q.H.; Hu, W.; Mou, X.D.; Li, X. Chemotherapeutics for Toxoplasma gondii: Molecular biotargets, binding modes, and structure-activity relationship investigations. J. Med. Chem. 2021, 64, 17627–17655. [Google Scholar] [CrossRef]
- Cajazeiro, D.C.; Toledo, P.P.M.; de Sousa, N.F.; Scotti, M.T.; Reimao, J.Q. Drug repurposing based on protozoan proteome: In vitro evaluation of in silico screened compounds against Toxoplasma gondii. Pharmaceutics 2022, 14, 1634. [Google Scholar] [CrossRef]
- Silva, M.D.; Teixeira, C.; Gomes, P.; Borges, M. Promising drug targets and compounds with anti-Toxoplasma gondii activity. Microorganisms 2021, 9, 1960. [Google Scholar] [CrossRef]
- Hopper, A.T.; Brockman, A.; Wise, A.; Gould, J.; Barks, J.; Radke, J.B.; Sibley, L.D.; Zou, Y.; Thomas, S. Discovery of Selective Toxoplasma gondii Dihydrofolate Reductase Inhibitors for the Treatment of Toxoplasmosis. J. Med. Chem. 2019, 62, 1562–1576. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, B.; Hou, S.; Su, T.; Fan, Q.; Alyafeai, E.; Tang, Y.; Wu, M.; Liu, X.; Li, J.; et al. Discovery and evaluation of chalcone derivatives as novel potential anti-Toxoplasma gondii agents. Eur. J. Med. Chem. 2022, 234, 114244. [Google Scholar] [CrossRef]
- Sadeghi, M.; Sarvi, S.; Emami, S.; Khalilian, A.; Hosseini, S.A.; Montazeri, M.; Shahdin, S.; Nayeri, T.; Daryani, A. Evaluation of anti-parasitic activities of new quinolones containing nitrofuran moiety against Toxoplasma gondii. Exp. Parasitol. 2022, 240, 108344. [Google Scholar] [CrossRef] [PubMed]
- Weglinska, L.; Bekier, A.; Trotsko, N.; Kapron, B.; Plech, T.; Dzitko, K.; Paneth, A. Inhibition of Toxoplasma gondii by 1,2,4-triazole-based compounds: Marked improvement in selectivity relative to the standard therapy pyrimethamine and sulfadiazine. J. Enzyme Inhib. Med. Chem. 2022, 37, 2621–2634. [Google Scholar] [CrossRef]
- Bekier, A.; Gatkowska, J.; Chyb, M.; Sokolowska, J.; Chwatko, G.; Glowacki, R.; Paneth, A.; Dzitko, K. 4-Arylthiosemicarbazide derivatives—Pharmacokinetics, toxicity and anti-Toxoplasma gondii activity in vivo. Eur. J. Med. Chem. 2022, 244, 114812. [Google Scholar] [CrossRef] [PubMed]
- Molina, D.A.; Ramos, G.A.; Zamora-Velez, A.; Gallego-Lopez, G.M.; Rocha-Roa, C.; Gomez-Marin, J.E.; Cortes, E. In vitro evaluation of new 4-thiazolidinones on invasion and growth of Toxoplasma gondii. Int. J. Parasitol. Drugs Drug Resist. 2021, 16, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Araujo-Silva, C.A.; De Souza, W.; Martins-Duarte, E.S.; Vommaro, R.C. HDAC inhibitors Tubastatin A and SAHA affect parasite cell division and are potential anti-Toxoplasma gondii chemotherapeutics. Int. J. Parasitol. Drugs Drug Resist. 2021, 15, 25–35. [Google Scholar] [CrossRef]
- Imhof, D.; Anghel, N.; Winzer, P.; Balmer, V.; Ramseier, J.; Hanggeli, K.; Choi, R.; Hulverson, M.A.; Whitman, G.R.; Arnold, S.L.M.; et al. In vitro activity, safety and in vivo efficacy of the novel bumped kinase inhibitor BKI-1748 in non-pregnant and pregnant mice experimentally infected with Neospora caninum tachyzoites and Toxoplasma gondii oocysts. Int. J. Parasitol. Drugs Drug Resist. 2021, 16, 90–101. [Google Scholar] [CrossRef]
- Doggett, J.S.; Schultz, T.; Miller, A.J.; Bruzual, I.; Pou, S.; Winter, R.; Dodean, R.; Zakharov, L.N.; Nilsen, A.; Riscoe, M.K.; et al. Orally bioavailable endochin-like quinolone carbonate ester prodrug reduces Toxoplasma gondii brain cysts. Antimicrob. Agents Chemother. 2020, 64, e00535-20. [Google Scholar] [CrossRef]
- Muller, J.; Boubaker, G.; Imhof, D.; Hanggeli, K.; Haudenschild, N.; Uldry, A.C.; Braga-Lagache, S.; Heller, M.; Ortega-Mora, L.M.; Hemphill, A. Differential affinity chromatography coupled to mass spectrometry: A suitable tool to identify common binding proteins of a broad-range antimicrobial peptide derived from leucinostatin. Biomedicines 2022, 10, 2675. [Google Scholar] [CrossRef]
- Martynowicz, J.; Doggett, J.S.; Sullivan, W.J., Jr. Efficacy of Guanabenz combination therapy against chronic toxoplasmosis across multiple mouse strains. Antimicrob. Agents Chemother. 2020, 64, e00539-20. [Google Scholar] [CrossRef]
- Hammarton, T.C.; Mottram, J.C.; Doerig, C. The cell cycle of parasitic protozoa: Potential for chemotherapeutic exploitation. Prog. Cell. Cycle Res. 2003, 5, 91–101. [Google Scholar] [PubMed]
- Mukherjee, S.; Mukherjee, N.; Gayen, P.; Roy, P.; Babu, S.P. Metabolic inhibitors as antiparasitic drugs: Pharmacological, biochemical and molecular perspectives Curr. Drug. Metab. 2016, 17, 937–970. [Google Scholar] [CrossRef] [PubMed]
- de Koning, H.P.; Bridges, D.J.; Burchmore, R.J. Purine and pyrimidine transport in pathogenic protozoa: From biology to therapy. FEMS Microbiol. Rev. 2005, 29, 987–1020. [Google Scholar] [CrossRef] [PubMed]
- el Kouni, M.H. Potential chemotherapeutic targets in the purine metabolism of parasites. Pharmacol. Ther. 2003, 99, 283–309. [Google Scholar] [CrossRef] [PubMed]
- el Kouni, M.H. Adenosine metabolism in Toxoplasma gondii: Potential targets for chemotherapy. Curr. Pharm. Des. 2007, 13, 581–597. [Google Scholar] [CrossRef]
- Wang, Y.; Karnataki, A.; Parsons, M.; Weiss, L.M.; Orlofsky, A. 3-Methyladenine blocks Toxoplasma gondii division prior to centrosome replication. Mol. Biochem. Parasitol. 2010, 173, 142–153. [Google Scholar] [CrossRef][Green Version]
- Kim, Y.A.; Rawal, R.K.; Yoo, J.; Sharon, A.; Jha, A.K.; Chu, C.K.; Rais, R.H.; Al Safarjalani, O.N.; Naguib, F.N.; El Kouni, M.H. Structure-activity relationships of carbocyclic 6-benzylthioinosine analogues as subversive substrates of Toxoplasma gondii adenosine kinase. Bioorg. Med. Chem. 2010, 18, 3403–3412. [Google Scholar] [CrossRef]
- Kim, Y.A.; Sharon, A.; Chu, C.K.; Rais, R.H.; Al Safarjalani, O.N.; Naguib, F.N.; el Kouni, M.H. Synthesis, biological evaluation and molecular modeling studies of N6-benzyladenosine analogues as potential anti-toxoplasma agents. Biochem. Pharmacol. 2007, 73, 1558–1572. [Google Scholar] [CrossRef]
- Gherardi, A.; Sarciron, M.E. Molecules targeting the purine salvage pathway in apicomplexan parasites. Trends Parasitol. 2007, 23, 384–389. [Google Scholar] [CrossRef]
- Sullivan, W.J., Jr.; Dixon, S.E.; Li, C.; Striepen, B.; Queener, S.F. IMP dehydrogenase from the protozoan parasite Toxoplasma gondii. Antimicrob. Agents Chemother. 2005, 49, 2172–2179. [Google Scholar] [CrossRef]
- Al Safarjalani, O.N.; Naguib, F.N.; El Kouni, M.H. Uptake of nitrobenzylthioinosine and purine beta-L-nucleosides by intracellular Toxoplasma gondii. Antimicrob. Agents Chemother. 2003, 47, 3247–3251. [Google Scholar] [CrossRef] [PubMed]
- Azzouz, S.; Lawton, P. In vitro effects of purine and pyrimidine analogues on Leishmania donovani and Leishmania infantum promastigotes and intracellular amastigotes. Acta Parasitol. 2017, 62, 582–588. [Google Scholar] [CrossRef]
- Al Safarjalani, O.N.; Rais, R.H.; Kim, Y.A.; Chu, C.K.; Naguib, F.N.; El Kouni, M.H. Carbocyclic 6-benzylthioinosine analogues as subversive substrates of Toxoplasma gondii adenosine kinase: Biological activities and selective toxicities. Biochem. Pharmacol. 2010, 80, 955–963. [Google Scholar] [CrossRef]
- Donaldson, T.M.; Cassera, M.B.; Ho, M.C.; Zhan, C.; Merino, E.F.; Evans, G.B.; Tyler, P.C.; Almo, S.C.; Schramm, V.L.; Kim, K. Inhibition and structure of Toxoplasma gondii purine nucleoside phosphorylase. Eukaryot Cell 2014, 13, 572–579. [Google Scholar] [CrossRef]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs are unique: Opportunities and challenges of discovery and development Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef] [PubMed]
- Loginova, N.V.; Harbatsevich, H.I.; Osipovich, N.P.; Ksendzova, G.A.; Koval’chuk, T.V.; Polozov, G.I. Metal complexes as promising agents for biomedical applications. Curr. Med. Chem. 2020, 27, 5213–5249. [Google Scholar] [CrossRef] [PubMed]
- Ong, Y.C.; Gasser, G. Organometallic compounds in drug discovery: Past, present and future. Drug Discov. Today Technol. 2020, 37, 117–124. [Google Scholar] [CrossRef]
- Konkankit, C.C.; Marker, S.C.; Knopf, K.M.; Wilson, J.J. Anticancer activity of complexes of the third row transition metals, rhenium, osmium, and iridium. Dalton Trans. 2018, 47, 9934–9974. [Google Scholar] [CrossRef]
- Simpson, P.V.; Desai, N.M.; Casari, I.; Massi, M.; Falasca, M. Metal-based antitumor compounds: Beyond cisplatin. Future Med. Chem. 2019, 11, 119–135. [Google Scholar] [CrossRef]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic anticancer compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef]
- Deng, Y.; Wu, T.; Zhai, S.Q.; Li, C.H. Recent progress on anti-Toxoplasma drugs discovery: Design, synthesis and screening. Eur. J. Med. Chem. 2019, 183, 111711. [Google Scholar] [CrossRef] [PubMed]
- Mbaba, M.; Golding, T.M.; Smith, G.S. Recent advances in the biological investigation of organometallic platinum-group metal (Ir, Ru, Rh, Os, Pd, Pt) complexes as antimalarial agents. Molecules 2020, 25, 5276. [Google Scholar] [CrossRef]
- Gambino, D.; Otero, L. Design of prospective antiparasitic metal-based compounds including selected organometallic cores. Inorg. Chim. Acta 2018, 472, 58–75. [Google Scholar] [CrossRef]
- Navarro, M.; Justo, R.M.S.; Delgado, G.Y.S.; Visbal, G. Metallodrugs for the treatment of Trypanosomatid diseases: Recent advances and new insights. Curr. Pharm. Des. 2021, 27, 1763–1789. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.W.; Hyland, C.J.T. Medicinal organometallic chemistry—An emerging strategy for the treatment of neglected tropical diseases Med. Chem. Commun. 2015, 6, 1230–1243. [Google Scholar] [CrossRef]
- Ong, Y.C.; Roy, S.; Andrews, P.C.; Gasser, G. Metal compounds against neglected tropical diseases. Chem. Rev. 2019, 119, 730–796. [Google Scholar] [CrossRef] [PubMed]
- Coverdale, J.P.C.; Laroiya-McCarron, T.; Romero-Canelón, I. Designing ruthenium anticancer drugs: What have we learnt from the key drug candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, C.Y.; Nam, T.G. Ruthenium complexes as anticancer agents: A brief history and perspectives. Drug Des. Dev. Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef] [PubMed]
- Barna, F.; Debache, K.; Vock, C.A.; Kuster, T.; Hemphill, A. In vitro effects of novel ruthenium complexes in Neospora caninum and Toxoplasma gondii tachyzoites. Antimicrob. Agents Chemother. 2013, 57, 5747–5754. [Google Scholar] [CrossRef] [PubMed]
- Gambino, D.; Otero, L. Perspectives on what ruthenium-based compounds could offer in the development of potential antiparasitic drugs. Inorg. Chim. Acta 2012, 393, 103–114. [Google Scholar] [CrossRef]
- Adams, M.; Li, Y.; Khot, H.; De Kock, C.; Smith, P.J.; Land, K.; Chibale, K.; Smith, G.S. The synthesis and antiparasitic activity of aryl- and ferrocenyl-derived thiosemicarbazone ruthenium(II)-arene complexes. Dalton Trans. 2013, 42, 4677–4685. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Arce, E.R.; Sarniguet, C.; Morais, T.S.; Tomaz, A.I.; Azar, C.O.; Figueroa, R.; Diego Maya, J.; Medeiros, A.; Comini, M.; et al. Novel ruthenium(II) cyclopentadienyl thiosemicarbazone compounds with antiproliferative activity on pathogenic trypanosomatid parasites. J. Inorg. Biochem. 2015, 153, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Stringer, T.; Quintero, M.A.S.; Wiesner, L.; Smith, G.S.; Nordlander, E. Evaluation of PTA-derived ruthenium(II) and iridium(III) quinoline complexes against chloroquine-sensitive and resistant strains of the Plasmodium falciparum malaria parasite. J. Inorg. Biochem. 2019, 191, 164–173. [Google Scholar] [CrossRef]
- Fandzloch, M.; Jedrzejewski, T.; Dobrzanska, L.; Esteban-Parra, G.M.; Wisniewska, J.; Paneth, A.; Paneth, P.; Sitkowski, J. New organometallic ruthenium(II) complexes with purine analogs—A wide perspective on their biological application. Dalton Trans. 2021, 50, 5557–5573. [Google Scholar] [CrossRef]
- Wang, X.; Guo, Z. Targeting and delivery of platinum-based anticancer drugs. Chem. Soc. Rev. 2013, 42, 202–224. [Google Scholar] [CrossRef]
- Kenny, R.G.; Marmion, C.J. Toward multi-targeted platinum and ruthenium drugs—A new paradigm in cancer drug treatment regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kang, Y.; Xu, F.; Zheng, W.; Luo, Q.; Zhang, Y.; Jia, F.; Wang, F. Pharmacophore conjugation strategy for multi-targeting metal-based anticancer complexes. In Advances in Inorganic Chemistry; Sadler, P.J., van Eldik, R., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 75, pp. 257–285. [Google Scholar]
- Kowalski, K. Organometallic nucleosides—Synthesis, transformations, and applications Coord. Chem. Rev. 2021, 432, 213705. [Google Scholar] [CrossRef]
- Kowalski, K. Ferrocenyl-nucleobase complexes: Synthesis, chemistry and applications. Coord. Chem. Rev. 2016, 317, 132–156. [Google Scholar] [CrossRef]
- Singh, A.; Lumb, I.; Mehra, V.; Kumar, V. Ferrocene-appended pharmacophores: An exciting approach for modulating the biological potential of organic scaffolds. Dalton Trans. 2019, 48, 2840–2860. [Google Scholar] [CrossRef]
- Daniluk, M.; Buchowicz, W.; Koszytkowska-Stawińska, M.; Jarząbek, K.; Jarzembska, K.N.; Kamiński, R.; Piszcz, M.; Laudy, A.E.; Tyski, S. Ferrocene amino acid ester uracil conjugates: Synthesis, structure, electrochemistry and antimicrobial evaluation. ChemistrySelect 2019, 4, 11130–11135. [Google Scholar] [CrossRef]
- James, P.; Neudorfl, J.; Eissmann, M.; Jesse, P.; Prokop, A.; Schmalz, H.G. Enantioselective synthesis of ferrocenyl nucleoside analogues with apoptosis-inducing activity. Org. Lett. 2006, 8, 2763–2766. [Google Scholar] [CrossRef] [PubMed]
- Florindo, P.R.; Pereira, D.M.; Borralho, P.M.; Piedade, M.F.M.; Conceição Oliveira, M.; Dias, A.M.; Rodrigues, C.M.P.; Fernandes, A.C. Organoruthenium(II) nucleoside conjugates as colon cytotoxic agents. New J. Chem. 2019, 43, 1195–1205. [Google Scholar] [CrossRef]
- Leitao, M.I.P.S.; Herrera, F.; Petronilho, A. N-Heterocyclic Carbenes Derived from Guanosine: Synthesis and Evidences of Their Antiproliferative Activity. ACS Omega 2018, 3, 15653–15656. [Google Scholar] [CrossRef] [PubMed]
- Collado, A.; Gómez-Gallego, M.; Sierra, M.A. Nucleobases having M–C bonds: An emerging bio-organometallic field. Eur. J. Org. Chem. 2018, 2018, 1617–1623. [Google Scholar] [CrossRef]
- Kaczmarek, R.; Korczynski, D.; Krolewska-Golinska, K.; Wheeler, K.A.; Chavez, F.A.; Mikus, A.; Dembinski, R. Organometallic nucleosides: Synthesis and biological evaluation of substituted dicobalt hexacarbonyl 2′-deoxy-5-oxopropynyluridines. ChemistryOpen 2018, 7, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.V.; Sallustrau, A.; Balzarini, J.; Bedford, M.R.; Eden, J.C.; Georgousi, N.; Hodges, N.J.; Kedge, J.; Mehellou, Y.; Tselepis, C.; et al. Organometallic nucleoside analogues with ferrocenyl linker groups: Synthesis and cancer cell line studies. J. Med. Chem. 2014, 57, 5817–5822. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.K.; Khan, Z.; Rana, M.; Horswell, S.L.; Male, L.; Nguyen, H.V.; Perotti, A.; Romero-Canelon, I.; Wilkinson, E.A.; Hodges, N.J.; et al. Effect of regiochemistry and methylation on the anticancer activity of a ferrocene-containing organometallic nucleoside analogue. ChemBioChem 2020, 21, 2487–2494. [Google Scholar] [CrossRef]
- Skiba, J.; Kowalski, K.; Prochnicka, A.; Ott, I.; Solecka, J.; Rajnisz, A.; Therrien, B. Metallocene-uracil conjugates: Synthesis and biological evaluation of novel mono-, di- and tri-nuclear systems. J. Organomet. Chem. 2015, 782, 52–61. [Google Scholar] [CrossRef]
- Singh, A.; Biot, C.; Viljoen, A.; Dupont, C.; Kremer, L.; Kumar, K.; Kumar, V. 1H-1,2,3-triazole-tethered uracil-ferrocene and uracil-ferrocenylchalcone conjugates: Synthesis and antitubercular evaluation. Chem. Biol. Drug Des. 2017, 89, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Rep, V.; Piskor, M.; Simek, H.; Misetic, P.; Grbcic, P.; Padovan, J.; Gabelica Markovic, V.; Jadresko, D.; Pavelic, K.; Kraljevic Pavelic, S.; et al. Purine and purine isostere derivatives of ferrocene: An evaluation of ADME, antitumor and electrochemical properties. Molecules 2020, 25, 1570. [Google Scholar] [CrossRef]
- Djaković, S.; Glavaš-Obrovac, L.; Lapić, J.; Maračić, S.; Kirchofer, J.; Knežević, M.; Jukić, M.; Raić-Malić, S. Synthesis and biological evaluations of mono- and bis-ferrocene uracil derivatives. Appl. Organomet. Chem. 2020, 35, e6052. [Google Scholar] [CrossRef]
- Kowalski, K.; Szczupak, L.; Saloman, S.; Steverding, D.; Jablonski, A.; Vrcek, V.; Hildebrandt, A.; Lang, H.; Rybarczyk-Pirek, A. Cymantrene, cyrhetrene and ferrocene nucleobase conjugates: Synthesis, structure, computational study, electrochemistry and antitrypanosomal activity. ChemPlusChem 2017, 82, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.-G.; Cai, X.-Q.; Li, X.-C.; Qin, D.-A.; Hu, M.-L. Arene-ruthenium(II) complexes containing 5-fluorouracil-1-methyl isonicotinate: Synthesis and characterization of their anticancer activity. Inorg. Chim. Acta 2012, 388, 78–83. [Google Scholar] [CrossRef]
- Li, Z.J.; Hou, Y.; Qin, D.A.; Jin, Z.M.; Hu, M.L. Two half-sandwiched ruthenium (II) compounds containing 5-fluorouracil derivatives: Synthesis and study of DNA intercalation. PLoS ONE 2015, 10, e0120211. [Google Scholar] [CrossRef] [PubMed]
- Florindo, P.R.; Pereira, D.M.; Borralho, P.M.; Costa, P.J.M.; Rodrigues, C.M.P.; Piedade, M.F.M.; Fernandes, A.C. New [(η5-C5H5)Ru(N–N)(PPh3)][PF6] compounds: Colon anticancer activity and GLUT-mediated cellular uptake of carbohydrate-appended complexes. Dalton Trans. 2016, 45, 11926–11930. [Google Scholar] [CrossRef]
- Furrer, J.; Süss-Fink, G. Thiolato-bridged dinuclear arene ruthenium complexes and their potential as anticancer drugs. Coord. Chem. Rev. 2016, 309, 36–50. [Google Scholar] [CrossRef]
- Basto, A.P.; Anghel, N.; Rubbiani, R.; Muller, J.; Stibal, D.; Giannini, F.; Suss-Fink, G.; Balmer, V.; Gasser, G.; Furrer, J.; et al. Targeting of the mitochondrion by dinuclear thiolato-bridged arene ruthenium complexes in cancer cells and in the apicomplexan parasite Neospora caninum. Metallomics 2019, 11, 462–474. [Google Scholar] [CrossRef]
- Basto, A.P.; Muller, J.; Rubbiani, R.; Stibal, D.; Giannini, F.; Suss-Fink, G.; Balmer, V.; Hemphill, A.; Gasser, G.; Furrer, J. Characterization of the activities of dinuclear thiolato-bridged arene ruthenium complexes against Toxoplasma gondii. Antimicrob. Agents Chemother. 2017, 61, e01017–e01031. [Google Scholar] [CrossRef]
- Jelk, J.; Balmer, V.; Stibal, D.; Giannini, F.; Suss-Fink, G.; Butikofer, P.; Furrer, J.; Hemphill, A. Anti-parasitic dinuclear thiolato-bridged arene ruthenium complexes alter the mitochondrial ultrastructure and membrane potential in Trypanosoma brucei bloodstream forms. Exp. Parasitol. 2019, 205, 107753. [Google Scholar] [CrossRef]
- Giannini, F.; Bartoloni, M.; Paul, L.E.H.; Süss-Fink, G.; Reymond, J.-L.; Furrer, J. Cytotoxic peptide conjugates of dinuclear areneruthenium trithiolato complexes. Med. Chem. Commun. 2015, 6, 347–350. [Google Scholar] [CrossRef]
- Desiatkina, O.; Paunescu, E.; Mosching, M.; Anghel, N.; Boubaker, G.; Amdouni, Y.; Hemphill, A.; Furrer, J. Coumarin-tagged dinuclear trithiolato-bridged ruthenium(II)-arene complexes: Photophysical properties and antiparasitic activity. ChemBioChem 2020, 21, 2818–2835. [Google Scholar] [CrossRef] [PubMed]
- Desiatkina, O.; Boubaker, G.; Anghel, N.; Amdouni, Y.; Hemphill, A.; Furrer, J.; Paunescu, E. Synthesis, photophysical properties and biological evaluation of new conjugates BODIPY—Dinuclear trithiolato-bridged ruthenium(II)-arene complexes. ChemBioChem, 2022; in press. [Google Scholar] [CrossRef] [PubMed]
- Stibal, D.; Therrien, B.; Suss-Fink, G.; Nowak-Sliwinska, P.; Dyson, P.J.; Cermakova, E.; Rezacova, M.; Tomsik, P. Chlorambucil conjugates of dinuclear p-cymene ruthenium trithiolato complexes: Synthesis, characterization and cytotoxicity study in vitro and in vivo. J. Biol. Inorg. Chem. 2016, 21, 443–452. [Google Scholar] [CrossRef]
- Desiatkina, O.; Johns, S.K.; Anghel, N.; Boubaker, G.; Hemphill, A.; Furrer, J.; Păunescu, E. Synthesis and antiparasitic activity of new conjugates-organic drugs tethered to trithiolato-bridged dinuclear ruthenium(II)–arene complexes. Inorganics 2021, 9, 59. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Meldal, M.; Tornoe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Zhang, W.Y.; Banerjee, S.; Imberti, C.; Clarkson, G.J.; Wang, Q.; Zhong, Q.; Young, L.S.; Romero-Canelon, I.; Zeng, M.; Habtemariam, A.; et al. Strategies for conjugating iridium(III) anticancer complexes to targeting peptides via copper-free click chemistry. Inorg. Chim. Acta 2020, 503, 119396. [Google Scholar] [CrossRef] [PubMed]
- Farrer, N.J.; Griffith, D.M. Exploiting azide-alkyne click chemistry in the synthesis, tracking and targeting of platinum anticancer complexes. Curr. Opin. Chem. Biol. 2020, 55, 59–68. [Google Scholar] [CrossRef]
- Zabarska, N.; Stumper, A.; Rau, S. CuAAC click reactions for the design of multifunctional luminescent ruthenium complexes. Dalton Trans. 2016, 45, 2338–2351. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, M.; Gao, F.; Wei, W.; Qian, Y.; Liu, H.K.; Zhao, J. Imaging of a clickable anticancer iridium catalyst. J. Inorg. Biochem. 2018, 180, 179–185. [Google Scholar] [CrossRef]
- Ganesh, V.; Sudhir, V.S.; Kundu, T.; Chandrasekaran, S. 10 years of click chemistry: Synthesis and applications of ferrocene-derived triazoles. Chem. Asian J. 2011, 6, 2670–2694. [Google Scholar] [CrossRef]
- Singh, G.; Arora, A.; Kalra, P.; Maurya, I.K.; Ruizc, C.E.; Estebanc, M.A.; Sinha, S.; Goyal, K.; Sehgal, R. A strategic approach to the synthesis of ferrocene appended chalcone linked triazole allied organosilatranes: Antibacterial, antifungal, antiparasitic and antioxidant studies. Bioorg. Med. Chem. 2019, 27, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Carrere-Kremer, S.; Kremer, L.; Guerardel, Y.; Biot, C.; Kumar, V. Azide-alkyne cycloaddition en route towards 1H-1,2,3-triazole-tethered beta-lactam-ferrocene and beta-lactam-ferrocenylchalcone conjugates: Synthesis and in vitro anti-tubercular evaluation. Dalton Trans. 2013, 42, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Viljoen, A.; Kremer, L.; Kumar, V. Azide-alkyne cycloaddition en route to 4-aminoquinoline-ferrocenylchalcone conjugates: Synthesis and anti-TB evaluation. Future Med. Chem. 2017, 9, 1701–1708. [Google Scholar] [CrossRef]
- Kroll, A.; Monczak, K.; Sorsche, D.; Rau, S. A luminescent ruthenium azide complex as a substrate for copper-catalyzed click reactions. Eur. J. Inorg. Chem. 2014, 2014, 3462–3466. [Google Scholar] [CrossRef]
- Karmis, R.E.; Carrara, S.; Baxter, A.A.; Hogan, C.F.; Hulett, M.D.; Barnard, P.J. Luminescent iridium(III) complexes of N-heterocyclic carbene ligands prepared using the ‘click reaction’. Dalton Trans. 2019, 48, 9998–10010. [Google Scholar] [CrossRef]
- Mandal, S.; Das, R.; Gupta, P.; Mukhopadhyay, B. Synthesis of a sugar-functionalized iridium complex and its application as a fluorescent lectin sensor. Tetrahedron Lett. 2012, 53, 3915–3918. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Păunescu, E.; Boubaker, G.; Desiatkina, O.; Anghel, N.; Amdouni, Y.; Hemphill, A.; Furrer, J. The quest of the best—A SAR study of trithiolato-bridged dinuclear ruthenium(II)-arene compounds presenting antiparasitic properties. Eur. J. Med. Chem. 2021, 222, 113610. [Google Scholar] [CrossRef]
- Păunescu, E.; Louise, L.; Jean, L.; Romieu, A.; Renard, P.-Y. A versatile access to new halogenated 7-azidocoumarins for photoaffinity labeling: Synthesis and photophysical properties. Dyes Pigm. 2011, 91, 427–434. [Google Scholar] [CrossRef]
- Kitamura, M.; Kato, S.; Yano, M.; Tashiro, N.; Shiratake, Y.; Sando, M.; Okauchi, T. A reagent for safe and efficient diazo-transfer to primary amines: 2-azido-1,3-dimethylimidazolinium hexafluorophosphate. Org. Biomol. Chem. 2014, 12, 4397–4406. [Google Scholar] [CrossRef]
- Beckmann, H.S.; Wittmann, V. One-pot procedure for diazo transfer and azide-alkyne cycloaddition: Triazole linkages from amines. Org. Lett. 2007, 9, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Dutta, S.; Choudhary, A.; Basak, A.; Dasgupta, S. Design, synthesis and RNase A inhibition activity of catechin and epicatechin and nucleobase chimeric molecules. Bioorg. Med. Chem. Lett. 2008, 18, 5411–5414. [Google Scholar] [CrossRef] [PubMed]
- Vo, D.D.; Duca, M. Design of Multimodal Small Molecules Targeting miRNAs Biogenesis: Synthesis and In Vitro Evaluation; Schmidt, M.F., Ed.; Springer: New York, NY, USA, 2017. [Google Scholar]
- Krim, J.; Taourirte, M.; Engels, J.W. Synthesis of 1,4-disubstituted mono and bis-triazolocarbo-acyclonucleoside analogues of 9-(4-hydroxybutyl)guanine by Cu(I)-catalyzed click azide-alkyne cycloaddition. Molecules 2011, 17, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Lolk, L.; Pohlsgaard, J.; Jepsen, A.S.; Hansen, L.H.; Nielsen, H.; Steffansen, S.I.; Sparving, L.; Nielsen, A.B.; Vester, B.; Nielsen, P. A click chemistry approach to pleuromutilin conjugates with nucleosides or acyclic nucleoside derivatives and their binding to the bacterial ribosome. J. Med. Chem. 2008, 51, 4957–4967. [Google Scholar] [CrossRef]
- Krim, J.; Sillahi, B.; Taourirte, M.; Rakib, E.M.; Engels, J.W. Microwave-assisted click chemistry: Synthesis of mono and bis-1,2,3-triazole acyclonucleoside analogues of Acyclovir via copper(I)-catalyzed cycloaddition. ARKIVOC 2009, 2009, 142–152. [Google Scholar] [CrossRef]
- Lindsell, W.E.; Murray, C.; Preston, P.N.; Woodman, T.A.J. Synthesis of 1,3-diynes in the purine, pyrimidine, 1,3,5-triazine and acridine series. Tetrahedron 2000, 56, 1233–1245. [Google Scholar] [CrossRef]
- Rocha, D.H.A.; Machado, C.M.; Sousa, V.; Sousa, C.F.V.; Silva, V.L.M.; Silva, A.M.S.; Borges, J.; Mano, J.F. Customizable and regioselective one-pot N−H Functionalization of DNA nucleobases to create a library of nucleobase derivatives for biomedical applications. Eur. J. Org. Chem. 2021, 2021, 4423–4433. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, P.; Zhu, L. Structural determinants of alkyne reactivity in copper-catalyzed azide-alkyne cycloadditions. Molecules 2016, 21, 1697. [Google Scholar] [CrossRef]
- Kokina, A.; Ozolina, Z.; Liepins, J. Purine auxotrophy: Possible applications beyond genetic marker. Yeast 2019, 36, 649–656. [Google Scholar] [CrossRef]
- Giannini, F.; Furrer, J.; Ibao, A.F.; Suss-Fink, G.; Therrien, B.; Zava, O.; Baquie, M.; Dyson, P.J.; Stepnicka, P. Highly cytotoxic trithiophenolatodiruthenium complexes of the type [(eta6-p-MeC6H4Pri)2Ru2(SC6H4-p-X)3]+: Synthesis, molecular structure, electrochemistry, cytotoxicity, and glutathione oxidation potential. J. Biol. Inorg. Chem. 2012, 17, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Giannini, F.; Suss-Fink, G.; Furrer, J. Efficient oxidation of cysteine and glutathione catalyzed by a dinuclear areneruthenium trithiolato anticancer complex. Inorg. Chem. 2011, 50, 10552–10554. [Google Scholar] [CrossRef][Green Version]
- Anghel, N.; Muller, J.; Serricchio, M.; Jelk, J.; Butikofer, P.; Boubaker, G.; Imhof, D.; Ramseier, J.; Desiatkina, O.; Paunescu, E.; et al. Cellular and molecular targets of nucleotide-tagged trithiolato-bridged arene ruthenium complexes in the protozoan parasites Toxoplasma gondii and Trypanosoma brucei. Int. J. Mol. Sci. 2021, 22, 10787. [Google Scholar] [CrossRef] [PubMed]
- McFadden, D.C.; Seeber, F.; Boothroyd, J.C. Use of Toxoplasma gondii expressing beta-galactosidase for colorimetric assessment of drug activity in vitro. Antimicrob. Agents Chemother. 1997, 41, 1849–1853. [Google Scholar] [CrossRef]
- Studer, V.; Anghel, N.; Desiatkina, O.; Felder, T.; Boubaker, G.; Amdouni, Y.; Ramseier, J.; Hungerbuhler, M.; Kempf, C.; Heverhagen, J.T.; et al. Conjugates containing two and three trithiolato-bridged dinuclear ruthenium(II)-arene units as in vitro antiparasitic and anticancer agents. Pharmaceuticals 2020, 13, 471. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Aguado-Martinez, A.; Manser, V.; Balmer, V.; Winzer, P.; Ritler, D.; Hostettler, I.; Arranz-Solis, D.; Ortega-Mora, L.; Hemphill, A. Buparvaquone is active against Neospora caninum in vitro and in experimentally infected mice. Int. J. Parasitol. Drugs Drug. Resist. 2015, 5, 16–25. [Google Scholar] [CrossRef]
- Vo, D.D.; Duca, M. Drug Target miRNA: Methods and Protocols; Schmidt, E.M.F., Ed.; Springer: New York, NY, USA, 2017; pp. 137–154. [Google Scholar]
- Giannini, F.; Furrer, J.; Süss-Fink, G.; Clavel, C.M.; Dyson, P.J. Synthesis, characterization and in vitro anticancer activity of highly cytotoxic trithiolato diruthenium complexes of the type [(η6-p-MeC6H4iPr)2Ru2(μ2-SR1)2(μ2-SR2)]+ containing different thiolato bridges. J. Organomet. Chem. 2013, 744, 41–48. [Google Scholar] [CrossRef]
- Jansa, P.; Špaček, P.; Votruba, I.; Břehová, P.; Dračínský, M.; Klepetářová, B.; Janeba, Z. Efficient one-pot synthesis of polysubstituted 6-[(1H-1,2,3-triazol-1-yl)methyl]uracils through the “click” protocol. Collect. Czech. Chem. Commun. 2011, 76, 1121–1131. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]













| Compound | R | HFF Viability (%) | T. gondii β-gal Growth (%) | ||
|---|---|---|---|---|---|
| 0.1 µM | 1 µM | 0.1 µM | 1 µM | ||
| Diruthenium intermediates | |||||
| 2 a |  | 74 ± 2 | 48 ± 1 | 57 ± 1 | 2 ± 0 |
| 3 a |  | 91 ± 4 | 73 ± 1 | 114 ± 2 | 110 ± 2 |
| 4 a |  | 80 ± 1 | 69 ± 6 | 2 ± 0 | 1 ± 0 |
| 5 |  | 101 ± 0 | 96 ± 0 | 21 ± 2 | 0 ± 0 |
| 6 |  | 95 ± 1 | 49 ± 2 | 112 ± 1 | 6 ± 1 |
| 7 |  | 100 ± 2 | 53 ± 3 | 19 ± 1 | 0 ± 0 |
| 8 a |  | 71 ± 2 | 46 ± 6 | 52 ± 13 | 3 ± 1 |
| 9 |  | 96 ± 1 | 64 ± 1 | 9 ± 1 | 1 ± 1 |
| 10 |  | 94 ± 1 | 70 ± 1 | 10 ± 0 | 1 ± 0 |
| Family 1 | |||||
| 13 |  | 156 ± 1 | 102 ± 1 | 85 ± 2 | 83 ± 2 |
| Family 2 | |||||
| 14 |  | 95 ± 11 | 76 ± 2 | 12 ± 1 | 1 ± 0 |
| Family 3 | |||||
| 19 |  | 93 ± 2 | 85 ± 0 | 117 ± 7 | 7 ± 0 |
| 20 |  | 100 ± 2 | 71 ± 2 | 102 ± 0 | 1 ± 0 |
| 21 |  | 109 ± 2 | 89 ± 1 | 4 ± 0 | 0 ± 0 |
| 22 |  | 102 ± 0 | 77 ± 2 | 79 ± 3 | 1 ± 0 |
| 23 |  | 100 ± 1 | 76 ± 1 | 114 ± 4 | 1 ± 0 |
| 24 |  | 98 ± 10 | 96 ± 4 | 56 ± 15 | 0 ± 0 |
| 25 |  | 94 ± 9 | 91 ± 8 | 82 ± 6 | 35 ± 7 |
| 26 |  | 109 ± 2 | 76 ± 0 | 109 ± 6 | 0 ± 0 |
| Family 4 | |||||
| 28 |  | 101 ± 1 | 88 ± 1 | 97 ± 6 | 76 ± 5 |
| 29 |  | 101 ± 1 | 102 ± 1 | 102 ± 3 | 116 ± 5 |
| 30 |  | 114 ± 0 | 112 ± 3 | 142 ± 3 | 7 ± 0 |
| 31 |  | 99 ± 3 | 98 ± 6 | 109 ± 0 | 127 ± 0 |
| 33 |  | 84 ± 6 | 71 ± 3 | 94 ± 11 | 83 ± 11 |
| Family 5 | |||||
| 34 |  | 96 ± 1 | 70 ± 1 | 7 ± 0 | 1 ± 0 |
| 35 |  | 89 ± 0 | 27 ± 1 | 1 ± 0 | 0 ± 0 |
| 36 |  | 115 ± 1 | 87 ± 2 | 7 ± 0 | 0 ± 0 |
| 37 |  | 99 ± 1 | 71 ± 0 | 13 ± 1 | 0 ± 0 |
| 38 |  | 104 ± 1 | 71 ± 1 | 42 ± 2 | 0 ± 0 |
| 39 |  | 94 ± 2 | 90 ± 1 | 14 ± 1 | 0 ± 0 |
| Nucleobase intermediates | |||||
| 14A |  | 136 ± 3 | 84 ± 14 | 104 ± 2 | 75 ± 11 |
| 15 |  | 107 ± 2 | 96 ± 2 | 109 ± 5 | 83 ± 0 |
| 16 |  | 104 ± 2 | 103 ± 3 | 96 ± 8 | 95 ± 6 |
| 17 |  | 96 ± 1 | 93 ± 1 | 111 ± 5 | 90 ± 7 |
| 18 |  | 96 ± 2 | 89 ± 3 | 117 ± 4 | 91 ± 6 |
| 27 |  | 98 ± 5 | 102 ± 3 | 101 ± 5 | 99 ± 11 |
| 32 |  | 104 ± 2 | 103 ± 3 | 103 ± 8 | 94 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desiatkina, O.; Mösching, M.; Anghel, N.; Boubaker, G.; Amdouni, Y.; Hemphill, A.; Furrer, J.; Păunescu, E. New Nucleic Base-Tethered Trithiolato-Bridged Dinuclear Ruthenium(II)-Arene Compounds: Synthesis and Antiparasitic Activity. Molecules 2022, 27, 8173. https://doi.org/10.3390/molecules27238173
Desiatkina O, Mösching M, Anghel N, Boubaker G, Amdouni Y, Hemphill A, Furrer J, Păunescu E. New Nucleic Base-Tethered Trithiolato-Bridged Dinuclear Ruthenium(II)-Arene Compounds: Synthesis and Antiparasitic Activity. Molecules. 2022; 27(23):8173. https://doi.org/10.3390/molecules27238173
Chicago/Turabian StyleDesiatkina, Oksana, Martin Mösching, Nicoleta Anghel, Ghalia Boubaker, Yosra Amdouni, Andrew Hemphill, Julien Furrer, and Emilia Păunescu. 2022. "New Nucleic Base-Tethered Trithiolato-Bridged Dinuclear Ruthenium(II)-Arene Compounds: Synthesis and Antiparasitic Activity" Molecules 27, no. 23: 8173. https://doi.org/10.3390/molecules27238173
APA StyleDesiatkina, O., Mösching, M., Anghel, N., Boubaker, G., Amdouni, Y., Hemphill, A., Furrer, J., & Păunescu, E. (2022). New Nucleic Base-Tethered Trithiolato-Bridged Dinuclear Ruthenium(II)-Arene Compounds: Synthesis and Antiparasitic Activity. Molecules, 27(23), 8173. https://doi.org/10.3390/molecules27238173