

Targeting DNA Topoisomerase II in Antifungal Chemotherapy


Abstract
1. Introduction
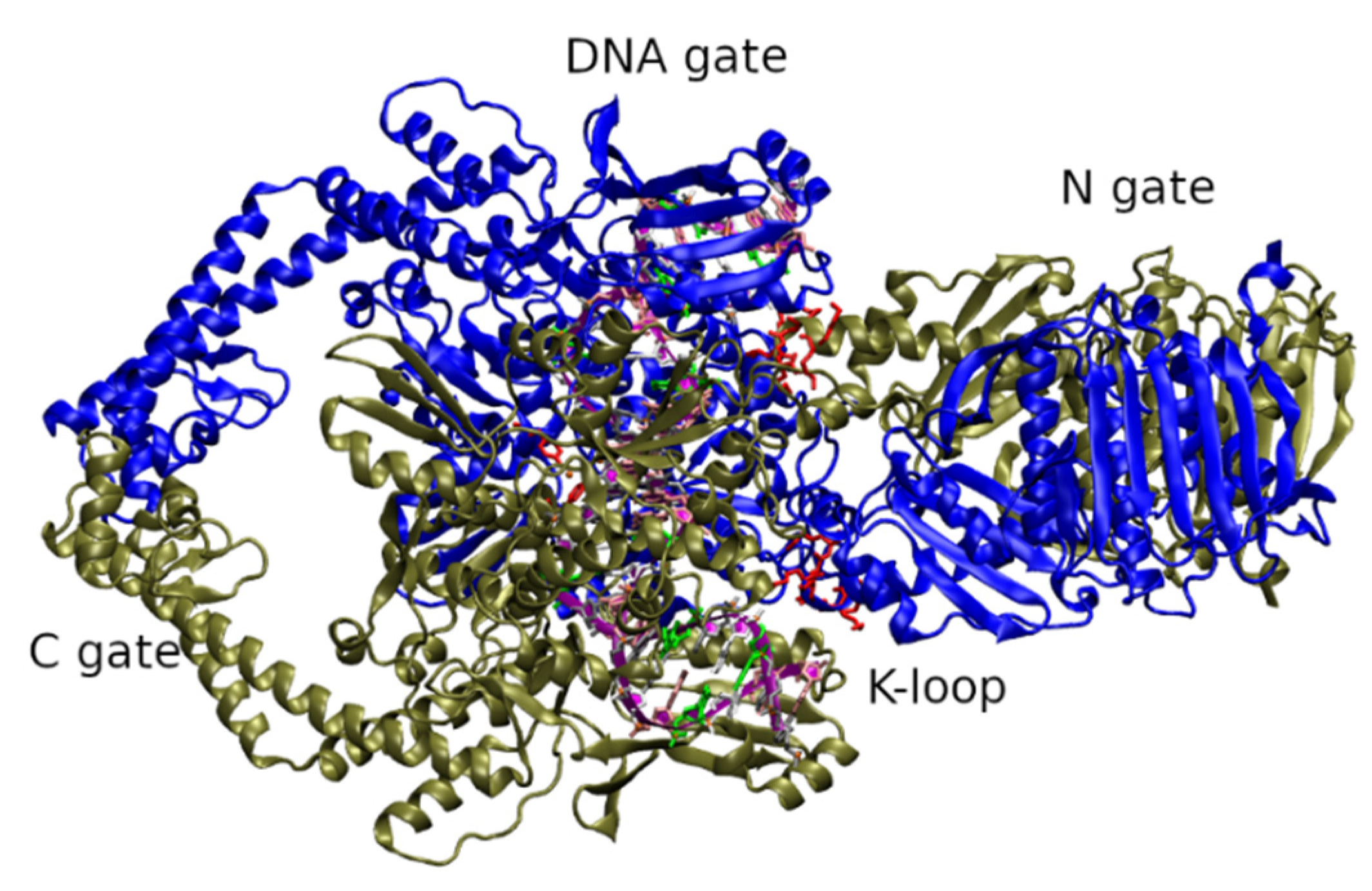
2. Role and Structure of Fungal Topoisomerase II
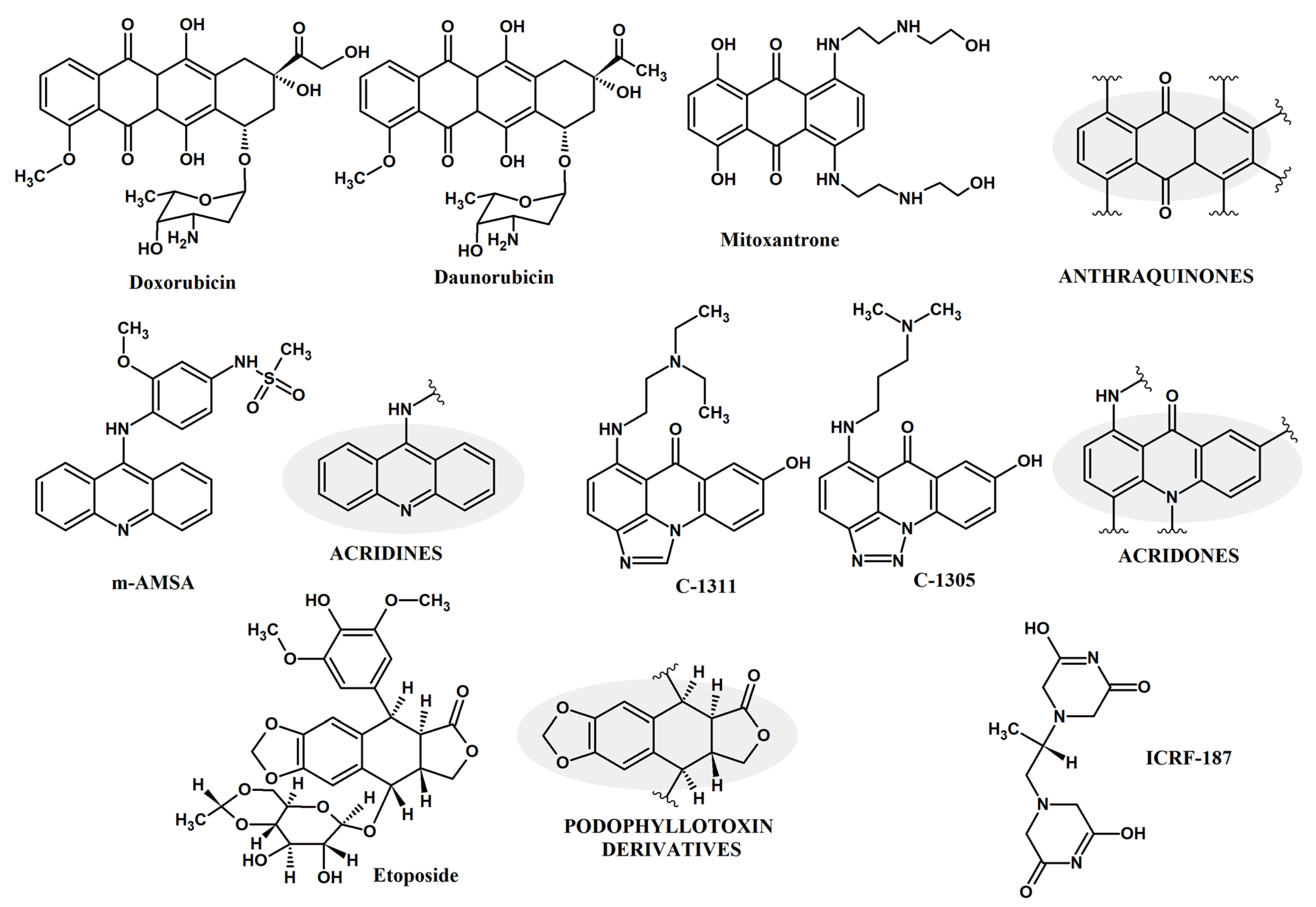
2.1. Fungal Topoisomerase II Inhibition
- At the ATP domain.
- After 1st ATP hydrolysis and before 2nd ATP hydrolysis.
- Inhibition of DNA DSBs re-ligation at the catalytic domain.
- De novo duplications at the DSBs using Non-Homologous End Joining (NHEJ) repair pathway.
- Blocking active catalytic tyrosine.
- Preventing sister chromatids segregation at termination of DNA replication.
2.2. Molecular Basis of Differential Sensitivity of Fungal Topoisomerase II to Anticancer Drugs
2.3. The Effect of Known Human Topoisomerase Inhibitors on the Growth of Fungal Cells
3. Conclusion and Future Perspective
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palmieri, F.; Koutsokera, A.; Bernasconi, E.; Junier, P.; von Garnier, C.; Ubags, N. Recent Advances in Fungal Infections: From Lung Ecology to Therapeutic Strategies with a Focus on Aspergillus spp. Front. Med. 2022, 9, 832510. [Google Scholar] [CrossRef]
- Bongomin, F.; Gago, S.; Oladele, R.O.; Denning, D.W. Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J. Fungi 2017, 3, 57. [Google Scholar] [CrossRef]
- Farmakiotis, D.; Kontoyiannis, D.P. Epidemiology of Antifungal Resistance in Human Pathogenic Yeasts: Current Viewpoint and Practical Recommendations for Management. Int. J. Antimicrob. Agents 2017, 50, 318–324. [Google Scholar] [CrossRef]
- Hawser, S.; Islam, K. Comparisons of the Effects of Fungicidal and Fungistatic Antifungal Agents on the Morphogenetic Transformation of Candida Albicans. J. Antimicrob. Chemother. 1999, 43, 411–413. [Google Scholar] [CrossRef]
- Meletiadis, J.; Antachopoulos, C.; Stergiopoulou, T.; Pournaras, S.; Roilides, E.; Walsh, T.J. Differential Fungicidal Activities of Amphotericin B and Voriconazole against Aspergillus Species Determined by Microbroth Methodology. Antimicrob. Agents Chemother. 2007, 51, 3329–3337. [Google Scholar] [CrossRef]
- Gabriel, I.; Vetter, N.D.; Palmer, D.R.J.; Milewska, M.J.; Wojciechowski, M.; Milewski, S. Homoisocitrate Dehydrogenase from Candida Albicans: Properties, Inhibition, and Targeting by an Antifungal pro-Drug. FEMS Yeast Res. 2013, 13, 143–155. [Google Scholar] [CrossRef]
- Milewska, M.J.; Prokop, M.; Gabriel, I.; Wojciechowski, M.; Milewski, S. Antifungal Activity of Homoaconitate and Homoisocitrate Analogs. Molecules 2012, 17, 14022–14036. [Google Scholar] [CrossRef]
- Liu, J.; Bolstad, D.B.; Smith, A.E.; Priestley, N.D.; Wright, D.L.; Anderson, A.C. Structure-Guided Development of Efficacious Antifungal Agents Targeting Candida Glabrata Dihydrofolate Reductase. Chem. Biol. 2008, 15, 990–996. [Google Scholar] [CrossRef]
- Casalini, G.; Giacomelli, A.; Ridolfo, A.; Gervasoni, C.; Antinori, S. Invasive Fungal Infections Complicating COVID-19: A Narrative Review. J. Fungi 2021, 7, 921. [Google Scholar] [CrossRef]
- Holm, C.; Stearns, T.; Botstein, D. DNA Topoisomerase II Must Act at Mitosis to Prevent Nondisjunction and Chromosome Breakage. Mol. Cell. Biol. 1989, 9, 159–168. [Google Scholar] [CrossRef]
- Shen, L.L.; Fostel, J.M. DNA Topoisomerase Inhibitors as Antifungal Agents. Adv. Pharmacol. 1994, 29, 227–244. [Google Scholar]
- Merino, A.; Madden, K.R.; Lane, W.S.; Champoux, J.J.; Reinberg, D. DNA Topoisomerase I Is Involved in Both Repression and Activation of Transcription. Nature 1993, 365, 227–232. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.C.; Liu, L.F. Involvement of DNA Topoisomerase I in Transcription of Human Ribosomal RNA Genes. Proc. Natl. Acad. Sci. USA 1988, 85, 1060–1064. [Google Scholar] [CrossRef]
- Earnshaw, W.C.; Heck, M.M. Localization of Topoisomerase II in Mitotic Chromosomes. J. Cell Biol. 1985, 100, 1716–1725. [Google Scholar] [CrossRef]
- Earnshaw, W.C.; Halligan, B.; Cooke, C.A.; Heck, M.M.; Liu, L.F. Topoisomerase II Is a Structural Component of Mitotic Chromosome Scaffolds. J. Cell Biol. 1985, 100, 1706–1715. [Google Scholar] [CrossRef]
- Baxter, J.; Diffley, J.F.X. Topoisomerase II Inactivation Prevents the Completion of DNA Replication in Budding Yeast. Mol. Cell 2008, 30, 790–802. [Google Scholar] [CrossRef]
- Martinez-Garcia, M.; White, C.I.; Franklin, F.C.H.; Sanchez-Moran, E. The Role of Topoisomerase II in DNA Repair and Recombination in Arabidopsis Thaliana. Int. J. Mol. Sci. 2021, 22, 13115. [Google Scholar] [CrossRef]
- Catapano, C.V.; Carbone, G.M.; Pisani, F.; Qiu, J.; Fernandes, D.J. Arrest of Replication Fork Progression at Sites of Topoisomerase II-Mediated DNA Cleavage in Human Leukemia CEM Cells Incubated with VM-26. Biochemistry 1997, 36, 5739–5748. [Google Scholar] [CrossRef]
- McKie, S.J.; Neuman, K.C.; Maxwell, A. DNA Topoisomerases: Advances in Understanding of Cellular Roles and Multi-Protein Complexes via Structure-Function Analysis. BioEssays 2021, 43, 2000286. [Google Scholar] [CrossRef]
- Corbett, K.D. DNA Topology. By Andrew D Bates and Anthony Maxwell. Q. Rev. Biol. 2006, 81, 58–59. [Google Scholar] [CrossRef]
- Buzun, K.; Bielawska, A.; Bielawski, K.; Gornowicz, A. DNA Topoisomerases as Molecular Targets for Anticancer Drugs. J. Enzyme Inhib. Med. Chem. 2020, 35, 1781–1799. [Google Scholar] [CrossRef]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as Anticancer Targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Jaswal, S.; Nehra, B.; Kumar, S.; Monga, V. Recent Advancements in the Medicinal Chemistry of Bacterial Type II Topoisomerase Inhibitors. Bioorg. Chem. 2020, 104, 104266. [Google Scholar] [CrossRef]
- Skladanowski, A.; Plisov, S.Y.; Konopa, J.; Larsen, A.K. Inhibition of DNA Topoisomerase II by Imidazoacridinones, New Antineoplastic Agents with Strong Activity against Solid Tumors. Mol. Pharmacol. 1996, 49, 772–780. [Google Scholar]
- Nitiss, J.L. Targeting DNA Topoisomerase II in Cancer Chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Hevener, K.E.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent Developments in Topoisomerase-Targeted Cancer Chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Jain, C.; Majumder, H.; Roychoudhury, S. Natural Compounds as Anticancer Agents Targeting DNA Topoisomerases. Curr. Genom. 2016, 18, 75–92. [Google Scholar] [CrossRef]
- Skok, Ž.; Zidar, N.; Kikelj, D.; Ilaš, J. Dual Inhibitors of Human DNA Topoisomerase II and Other Cancer-Related Targets. J. Med. Chem. 2020, 63, 884–904. [Google Scholar] [CrossRef]
- Elsea, S.H.; Westergaard, M.; Burden, D.A.; Lomenick, J.P.; Osheroff, N. Quinolones Share a Common Interaction Domain on Topoisomerase II with Other DNA Cleavage-Enhancing Antineoplastic Drugs. Biochemistry 1997, 36, 2919–2924. [Google Scholar] [CrossRef]
- Singh, S.; Pandey, V.P.; Yadav, K.; Yadav, A.; Dwivedi, U.N. Natural Products as Anti-Cancerous Therapeutic Molecules Targeted towards Topoisomerases. Curr. Protein Pept. Sci. 2020, 21, 1103–1142. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Analysis of FDA Approved Anticancer Drugs Reveals the Future of Cancer Therapy. Cell Cycle 2004, 3, 1033–1040. [Google Scholar] [CrossRef]
- Sabourin, M.; Byl, J.A.W.; Hannah, S.E.; Nitiss, J.L.; Osheroff, N. A Mutant Yeast Topoisomerase II (Top2G437S) with Differential Sensitivity to Anticancer Drugs in the Presence and Absence of ATP. J. Biol. Chem. 1998, 273, 29086–29092. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Classen, S.; Olland, S.; Berger, J.M. Structure of the Topoisomerase II ATPase Region and Its Mechanism of Inhibition by the Chemotherapeutic Agent ICRF-187. Proc. Natl. Acad. Sci. USA 2003, 100, 10629–10634. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Marians, K.J. Escherichia Coli Topoisomerase IV. Purification, Characterization, Subunit Structure, and Subunit Interactions. J. Biol. Chem. 1993, 268, 24481–24490. [Google Scholar] [CrossRef]
- Lynn, R.; Giaever, G.; Swanberg, S.L.; Wang, J.C. Tandem Regions of Yeast DNA Topoisomerase II Share Homology with Different Subunits of Bacterial Gyrase. Science 1986, 233, 647–649. [Google Scholar] [CrossRef]
- DiNardo, S.; Voelkel, K.; Sternglanz, R. DNA Topoisomerase II Mutant of Saccharomyces Cerevisiae: Topoisomerase II Is Required for Segregation of Daughter Molecules at the Termination of DNA Replication. Proc. Natl. Acad. Sci. USA 1984, 81, 2616–2620. [Google Scholar] [CrossRef]
- Rogojina, A.; Gajewski, S.; Bahmed, K.; Osheroff, N.; Nitiss, J.L. Topoisomerase II Inhibitors: Chemical Biology. In DNA Topoisomerases and Cancer. Cancer Drug Discovery and Development; Springer: New York, NY, USA, 2012; pp. 211–243. [Google Scholar] [CrossRef]
- Lemke, K.; Wojciechowski, M.; Laine, W.; Bailly, C.; Colson, P.; Baginski, M.; Larsen, A.K.; Skladanowski, A. Induction of Unique Structural Changes in Guanine-Rich DNA Regions by the Triazoloacridone C-1305, a Topoisomerase II Inhibitor with Antitumor Activities. Nucleic Acids Res. 2005, 33, 6034–6047. [Google Scholar] [CrossRef]
- Węsierska-Gądek, J.; Schloffer, D.; Gueorguieva, M.; Uhl, M.; Skladanowski, A. Increased Susceptibility of Poly(ADP-Ribose) Polymerase-1 Knockout Cells to Antitumor Triazoloacridone C-1305 Is Associated with Permanent G2 Cell Cycle Arrest. Cancer Res. 2004, 64, 4487–4497. [Google Scholar] [CrossRef][Green Version]
- Cholody, W.M.; Martelli, S.; Paradziej-Lukowicz, J.; Konopa, J. 5-[(Aminoalkyl)Amino]Imidazo [4,5,1-de]Acridin-6-Ones as a Novel Class of Antineoplastic Agents. Synthesis and Biological Activity. J. Med. Chem. 1990, 33, 49–52. [Google Scholar] [CrossRef]
- Mazerska, Z.; Sowiński, P.; Konopa, J. Molecular Mechanism of the Enzymatic Oxidation Investigated for Imidazoacridinone Antitumor Drug, C-1311. Biochem. Pharmacol. 2003, 66, 1727–1736. [Google Scholar] [CrossRef]
- Denny, W.A. Acridine Derivatives as Chemotherapeutic Agents. Curr. Med. Chem. 2002, 9, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Belmont, P.; Bosson, J.; Godet, T.; Tiano, M. Acridine and Acridone Derivatives, Anticancer Properties and Synthetic Methods: Where Are We Now? Anti-Cancer Agents Med. Chem. 2007, 7, 139–169. [Google Scholar] [CrossRef]
- Holm, C.; Goto, T.; Wang, J.C.; Botstein, D. DNA Topoisomerase II Is Required at the Time of Mitosis in Yeast. Cell 1985, 41, 553–563. [Google Scholar] [CrossRef]
- Goto, T.; Wang, J.C. Cloning of Yeast TOP1, the Gene Encoding DNA Topoisomerase I, and Construction of Mutants Defective in Both DNA Topoisomerase I and DNA Topoisomerase II. Proc. Natl. Acad. Sci. USA 1985, 82, 7178–7182. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Ohkura, H.; Adachi, Y.; Morino, K.; Shiozaki, K.; Yanagida, M. DNA Topoisomerase II Is Required for Condensation and Separation of Mitotic Chromosomes in S. Pombe. Cell 1987, 50, 917–925. [Google Scholar] [CrossRef]
- Dyson, S.; Segura, J.; Martínez-García, B.; Valdés, A.; Roca, J. Condensin Minimizes Topoisomerase II-Mediated Entanglements of DNA In Vivo. EMBO J. 2021, 40, e105393. [Google Scholar] [CrossRef]
- Nikolaou, C.; Bermúdez, I.; Manichanh, C.; García-Martinez, J.; Guigó, R.; Pérez-Ortín, J.E.; Roca, J. Topoisomerase II Regulates Yeast Genes with Singular Chromatin Architectures. Nucleic Acids Res. 2013, 41, 9243–9256. [Google Scholar] [CrossRef]
- Joshi, R.S.; Nikolaou, C.; Roca, J. Structure and Chromosomal Organization of Yeast Genes Regulated by Topoisomerase II. Int. J. Mol. Sci. 2018, 19, 134. [Google Scholar] [CrossRef]
- Schmidt, B.H.; Osheroff, N.; Berger, J.M. Structure of a Topoisomerase II-DNA-Nucleotide Complex Reveals a New Control Mechanism for ATPase Activity. Nat. Struct. Mol. Biol. 2012, 19, 1147–1154. [Google Scholar] [CrossRef]
- Dong, K.C.; Berger, J.M. Structural Basis for Gate-DNA Recognition and Bending by Type IIA Topoisomerases. Nature 2007, 450, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.H.; Burgin, A.B.; Deweese, J.E.; Osheroff, N.; Berger, J.M. A Novel and Unified Two-Metal Mechanism for DNA Cleavage by Type II and IA Topoisomerases. Nature 2010, 465, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.M.; Gamblin, S.J.; Harrison, S.C.; Wang, J.C. Structure and Mechanism of DNA Topoisomerase II. Nature 1996, 379, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Fass, D.; Bogden, C.E.; Berger, J.M. Quaternary Changes in Topoisomerase II May Direct Orthogonal Movement of Two DNA Strands. Nat. Struct. Biol. 1999, 6, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Deweese, J.E.; Osheroff, N. The DNA Cleavage Reaction of Topoisomerase II: Wolf in Sheep’s Clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Roca, J.; Berger, J.M.; Harrison, S.C.; Wang, J.C. DNA Transport by a Type II Topoisomerase: Direct Evidence for a Two-Gate Mechanism. Proc. Natl. Acad. Sci. USA 1996, 93, 4057–4062. [Google Scholar] [CrossRef]
- Williams, N.L.; Maxwell, A. Probing the Two-Gate Mechanism of DNA Gyrase Using Cysteine Cross-Linking. Biochemistry 1999, 38, 13502–13511. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Ruthenburg, A.J.; Bechis, S.K.; Verdine, G.L. Nucleotide-Dependent Domain Movement in the ATPase Domain of a Human Type IIA DNA Topoisomerase. J. Biol. Chem. 2005, 280, 37041–37047. [Google Scholar] [CrossRef]
- Stanger, F.V.; Dehio, C.; Schirmer, T. Structure of the N-Terminal Gyrase B Fragment in Complex with ADP⋅Pi Reveals Rigid-Body Motion Induced by ATP Hydrolysis. PLoS ONE 2014, 9, e107289. [Google Scholar] [CrossRef]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The Structure of DNA-Bound Human Topoisomerase II Alpha: Conformational Mechanisms for Coordinating Inter-Subunit Interactions with DNA Cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef]
- Wu, C.-C.; Li, T.-K.; Farh, L.; Lin, L.-Y.; Lin, T.-S.; Yu, Y.-J.; Yen, T.-J.; Chiang, C.-W.; Chan, N.-L. Structural Basis of Type II Topoisomerase Inhibition by the Anticancer Drug Etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, J.C. Identification of Active Site Residues in the “GyrA” Half of Yeast DNA Topoisomerase II. J. Biol. Chem. 1998, 273, 20252–20260. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Antoci, V.; Oniciuc, L.; Amariucai-mantu, D.; Moldoveanu, C.; Mangalagiu, V.; Zbancioc, G. Benzoquinoline Derivatives: A Straightforward and Efficient Route to Antibacterial and Antifungal Agents. Pharmaceuticals 2021, 14, 335. [Google Scholar] [CrossRef] [PubMed]
- Brighenti, V.; Iseppi, R.; Pinzi, L.; Mincuzzi, A.; Ippolito, A.; Messi, P.; Sanzani, S.M.; Rastelli, G.; Pellati, F. Antifungal Activity and DNA Topoisomerase Inhibition of Hydrolysable Tannins from Punica granatum L. Int. J. Mol. Sci. 2021, 22, 4175. [Google Scholar] [CrossRef]
- Shen, L.L.; Baranowski, J.; Fostel, J.; Montgomery, D.A.; Lartey, P.A. DNA Topoisomerases from Pathogenic Fungi: Targets for the Discovery of Antifungal Drugs. Antimicrob. Agents Chemother. 1992, 36, 2778–2784. [Google Scholar] [CrossRef]
- Lee, J.H.; Wendorff, T.J.; Berger, J.M. Resveratrol: A Novel Type of Topoisomerase II Inhibitor. J. Biol. Chem. 2017, 292, 21011–21022. [Google Scholar] [CrossRef]
- Rząd, K.; Paluszkiewicz, E.; Neubauer, D.; Olszewski, M.; Kozłowska-Tylingo, K.; Kamysz, W.; Gabriel, I. The Effect of Conjugation with Octaarginine, a Cell-Penetrating Peptide on Antifungal Activity of Imidazoacridinone Derivative. Int. J. Mol. Sci. 2021, 22, 13190. [Google Scholar] [CrossRef]
- Kwok, S.C.; Schelenz, S.; Wang, X.; Steverding, D. In Vitro Effect of DNA Topoisomerase Inhibitors on Candida Albicans. Med. Mycol. 2010, 48, 155–160. [Google Scholar] [CrossRef]
- Khan, S.I.; Nimrod, A.C.; Mehrpooya, M.; Nitiss, J.L.; Walker, L.A.; Clark, A.M. Antifungal Activity of Eupolauridine and Its Action on DNA Topoisomerases. Antimicrob. Agents Chemother. 2002, 46, 1785–1792. [Google Scholar] [CrossRef]
- Nitiss, J.L. Using Yeast to Study Resistance to Topoisomerase II-Targeting Drugs. Cancer Chemother. Pharmacol. 1994, 34, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Elsea, S.H.; Hsiung, Y.; Nitiss, J.L.; Osheroff, N. A Yeast Type II Topoisomerase Selected for Resistance to Quinolones. Mutation of Histidine 1012 to Tyrosine Confers Resistance to Nonintercalative Drugs but Hypersensitivity to Ellipticine. J. Biol. Chem. 1995, 270, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Stantial, N.; Rogojina, A.; Gilbertson, M.; Sun, Y.; Miles, H.; Shaltz, S.; Berger, J.; Nitiss, K.C.; Jinks-Robertson, S.; Nitiss, J.L. Trapped Topoisomerase II Initiates Formation of de Novo Duplications via the Nonhomologous End-Joining Pathway in Yeast. Proc. Natl. Acad. Sci. USA 2020, 117, 26876–26884. [Google Scholar] [CrossRef] [PubMed]
- Breaks, S.; Rogojina, A.T.; Nitiss, J.L. Isolation and Characterization of MAMSA-Hypersensitive Mutants Cytotoxicity of Top2 Covalent Complexes Containing DNA Single. J. Biol. Chem. 2008, 283, 29239–29250. [Google Scholar] [CrossRef]
- Dong, J.; Walker, J.; Nitiss, J.L. A Mutation in Yeast Topoisomerase II That Confers Hypersensitivity to Multiple Classes of Topoisomerase II Poisons. J. Biol. Chem. 2000, 275, 7980–7987. [Google Scholar] [CrossRef]
- Wainwright, M. Acridine—A Neglected Antibacterial Chromophore. J. Antimicrob. Chemother. 2001, 47, 1–13. [Google Scholar] [CrossRef]
- Puri, S.C.; Nazir, A.; Chawla, R.; Arora, R.; Riyaz-Ul-Hasan, S.; Amna, T.; Ahmed, B.; Verma, V.; Singh, S.; Sagar, R.; et al. The Endophytic Fungus Trametes Hirsuta as a Novel Alternative Source of Podophyllotoxin and Related Aryl Tetralin Lignans. J. Biotechnol. 2006, 122, 494–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, X.; Li, B.; Gao, C.; Jiang, Y. Acridine and Its Derivatives: A Patent Review (2009–2013). Expert Opin. Ther. Pat. 2014, 24, 647–664. [Google Scholar] [CrossRef]
- Plšíková, J.; Kašpárková, J.; Brabec, V.; Jendz, R.; Janoc, J.; Hamul, S.; Fedoroc, P. European Journal of Pharmaceutical Sciences Inhibition of DNA Topoisomerases I and II and Growth Inhibition of HL-60 Cells by Novel Acridine-Based Compounds. Eur. J. Pharm. Sci. 2015, 76, 192–202. [Google Scholar] [CrossRef]
- Gabriel, I. “Acridines” as New Horizons in Antifungal Treatment. Molecules 2020, 25, 1480. [Google Scholar] [CrossRef]
- Kaya, M.; Yıldırır, Y.; Çelik, G.Y. Synthesis, Characterization, and In Vitro Antimicrobial and Antifungal Activity of Novel Acridines. Pharm. Chem. J. 2015, 48, 722–726. [Google Scholar] [CrossRef]
- Markovich, Y.D.; Kudryavtseva, T.N.; Bogatyrev, K.V.; Sysoev, P.I.; Klimova, L.G.; Nazarov, G.V. Synthesis of 2-(4-Methyl-1,3-Thiazol-5-Yl)Ethyl Esters of Acridone Carboxylic Acids and Evaluation of Their Antibacterial Activity. Russ. Chem. Bull. 2014, 63, 1153–1158. [Google Scholar] [CrossRef]
- Chen, R.; Huo, L.; Jaiswal, Y.; Huang, J.; Zhong, Z.; Zhong, J.; Williams, L.; Xia, X.; Liang, Y.; Yan, Z. Design, Synthesis, Antimicrobial, and Anticancer Activities of Acridine Thiosemicarbazides Derivatives. Molecules 2019, 24, 2065. [Google Scholar] [CrossRef] [PubMed]
- Kulkarny, V.V.; Chavez-Dozal, A.; Rane, H.S.; Jahng, M.; Bernardo, S.M.; Parra, K.J.; Lee, S.A. Quinacrine Inhibits Candida Albicans Growth and Filamentation at Neutral PH. Antimicrob. Agents Chemother. 2014, 58, 7501–7509. [Google Scholar] [CrossRef]
- Gabriel, I.; Rząd, K.; Paluszkiewicz, E.; Kozłowska-Tylingo, K. Antifungal Activity of Capridine β as a Consequence of Its Biotransformation into Metabolite Affecting Yeast Topoisomerase II Activity. Pathogens 2021, 10, 189. [Google Scholar] [CrossRef]
- Taraszkiewicz, A.; Grinholc, M.; Bielawski, K.P.; Kawiak, A.; Nakonieczna, J. Imidazoacridinone Derivatives as Efficient Sensitizers in Photoantimicrobial Chemotherapy. Appl. Environ. Microbiol. 2013, 79, 3692–3702. [Google Scholar] [CrossRef]
- Taraszkiewicz, A.; Szewczyk, G.; Sarna, T.; Bielawski, K.P.; Nakonieczna, J. Photodynamic Inactivation of Candida Albicans with Imidazoacridinones: Influence of Irradiance, Photosensitizer Uptake and Reactive Oxygen Species Generation. PLoS ONE 2015, 10, e0129301. [Google Scholar] [CrossRef][Green Version]
- Felip-León, C.; Martínez-Arroyo, O.; Díaz-Oltra, S.; Miravet, J.F.; Apostolova, N.; Galindo, F. Synthesis, Spectroscopic Studies and Biological Evaluation of Acridine Derivatives: The Role of Aggregation on the Photodynamic Efficiency. Bioorg. Med. Chem. Lett. 2018, 28, 869–874. [Google Scholar] [CrossRef]
- Bailly, C. Contemporary Challenges in the Design of Topoisomerase II Inhibitors for Cancer Chemotherapy. Chem. Rev. 2012, 112, 3611–3640. [Google Scholar] [CrossRef]
- Wynn, J.E.; Zhang, W.; Falkinham, J.O.; Santos, W.L. Branched Peptides: Acridine and Boronic Acid Derivatives as Antimicrobial Agents. ACS Med. Chem. Lett. 2017, 8, 820–823. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, X.; Song, J.; Zheng, X.; Chen, J.; Ma, P.; Zhang, B.; Wang, R. Conjugation with Acridines Turns Nuclear Localization Sequence into Highly Active Antimicrobial Peptide. Engineering 2015, 1, 500–505. [Google Scholar] [CrossRef]
- Rząd, K.; Paluszkiewicz, E.; Gabriel, I. A New 1-Nitro-9-Aminoacridine Derivative Targeting Yeast Topoisomerase II Able to Overcome Fluconazole-Resistance. Bioorganic Med. Chem. Lett. 2021, 35, 127815. [Google Scholar] [CrossRef] [PubMed]
- Prasher, P.; Sharma, M. Medicinal Chemistry of Acridine and Its Analogues. Med. Chem. Commun. 2018, 9, 1589–1618. [Google Scholar] [CrossRef]
- De Oliveira, D.B.C.; Silva, L.B.; da Silva, B.V.; Borges, T.C.; Marques, B.C.; dos Santos, M.B.; de Oliveira, L.F.; Bolzani, V.S.; Rodrigues, A.R.A.; Regasini, L.O.; et al. A New Acridone with Antifungal Properties against Candida spp. and Dermatophytes, and Antibiofilm Activity against C. albicans. J. Appl. Microbiol. 2019, 127, 1362–1372. [Google Scholar] [CrossRef]
- Pawlak, K.; Pawlak, J.W.; Konopa, J. Cytotoxic and Antitumor Activity of 1-Nitroacridines as an Aftereffect of Their Interstrand DNA Cross-Linking. Cancer Res. 1984, 44, 4289–4296. [Google Scholar] [PubMed]
- Wiśniewska, A.; Niemira, M.; Jagiełło, K.; Potęga, A.; Świst, M.; Henderson, C.; Skwarska, A.; Augustin, E.; Konopa, J.; Mazerska, Z. Diminished Toxicity of C-1748, 4-Methyl-9-Hydroxyethylamino-1-Nitroacridine, Compared with Its Demethyl Analog, C-857, Corresponds to Its Resistance to Metabolism in HepG2 Cells. Biochem. Pharmacol. 2012, 84, 30–42. [Google Scholar] [CrossRef]
- Woynarowski, J.M.; McNamee, H.; Szmigiero, L.; Beerman, T.A.; Konopa, J. Induction of DNA-Protein Crosslinks by Antitumor 1-Nitro-9-Aminoacridines in L1210 Leukemia Cells. Biochem. Pharmacol. 1989, 38, 4095–4101. [Google Scholar] [CrossRef]
- Steverding, D.; Evans, P.; Msika, L.; Riley, B.; Wallington, J.; Schelenz, S. In Vitro Antifungal Activity of DNA Topoisomerase Inhibitors. Med. Mycol. 2012, 50, 333–336. [Google Scholar] [CrossRef]
- Futuro, D.O.; Ferreira, P.G.; Nicoletti, C.D.; Borba-Santos, L.P.; Da Silva, F.C.; Rozental, S.; Ferreira, V.F. The Antifungal Activity of Naphthoquinones: An Integrative Review. An. Acad. Bras. Cienc. 2018, 90, 1187–1214. [Google Scholar] [CrossRef]
- Andrade-Pavón, D.; Gómez-García, O. Etoposide and Camptothecin Reduce Growth, Viability, the Generation of Petite Mutants, and Recognize the Active Site of DNA Topoisomerase I and II Enzymes in Candida Glabrata. Indian J. Microbiol. 2021, 61, 306–314. [Google Scholar] [CrossRef]
- Tagle-Olmedo, T.; Andrade-Pavón, D.; Martínez-Gamboa, A.; Gómez-García, O.; García-Sierra, F.; Hernández-Rodríguez, C.; Villa-Tanaca, L. Inhibitors of DNA Topoisomerases I and II Applied to Candida Dubliniensis Reduce Growth, Viability, the Generation of Petite Mutants and Toxicity, While Acting Synergistically with Fluconazole. FEMS Yeast Res. 2021, 21, foab023. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, A.; Bansode, B.; Phule, D.; Shelar, A.; Patil, R.; Gade, W.; Kharat, K.; Karuppayil, S.M. The Antibacterial Agent, Moxifloxacin Inhibits Virulence Factors of Candida Albicans through Multitargeting. World J. Microbiol. Biotechnol. 2017, 33, 96. [Google Scholar] [CrossRef] [PubMed]
- Ptaszyńska, N.; Gucwa, K.; Olkiewicz, K.; Heldt, M.; Serocki, M.; Stupak, A.; Martynow, D.; Dębowski, D.; Gitlin-Domagalska, A.; Lica, J.; et al. Conjugates of Ciprofloxacin and Levofloxacin with Cell-Penetrating Peptide Exhibit Antifungal Activity and Mammalian Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 4696. [Google Scholar] [CrossRef] [PubMed]
- Miłosz Wieczór, Adam Hospital, Genis Bayarri, Jacek Czub, Modesto Orozco, Molywood: Streamlining the design and rendering of molecular movies. Bioinformatics 2020, 36, 4660–4661. [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Key Feature | ScTopo II Uniprot P06786 | HsTopo Iiα Uniprot P11388 | HsTopo Iiβ Uniprot Q02880 |
|---|---|---|---|
| ATP Binding site | N70, N99 [34] | N91, N120 [59,60] | N112, N141 |
| Magnesium 1 binding, catalytic | E449, D526 [52] | E461, D541 [61] | E482, D562 |
| Magnesium 2 binding | D526, D528 [52] | D541, D543 [61] | D562, D564 [62] |
| Transition state stabilizer | R781 [53] | R804 | R825 |
| Active site | Y782 [53,63] | Y805 | Y826 [62] |
| Intercalates into and bends DNA | I833 [52] | I856 | I877 |
| ATP nucleotide binding | SSN 127–129 GRNGYGAK 140–147 GTK 365–367 [34] | SSN 148–150 GRNGYGAK 161–168 GTK 376–378 [59,60] | SSN 169–171 GRNGYGAK 182–189 GTK 397–399 |
| TOPRIM domain * | C----L 443–557 | C----E 455–572 | C----E 476–593 |
| A part of K-loop * | KKK 333–336 [51] | KKK 342–344 [51] | KKK 363–365 |
| Interaction with DNA * | K----N 965–974 [53] | K----S 990–999 [61] | K----S 1011–1020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondaka, K.; Gabriel, I. Targeting DNA Topoisomerase II in Antifungal Chemotherapy. Molecules 2022, 27, 7768. https://doi.org/10.3390/molecules27227768
Kondaka K, Gabriel I. Targeting DNA Topoisomerase II in Antifungal Chemotherapy. Molecules. 2022; 27(22):7768. https://doi.org/10.3390/molecules27227768
Chicago/Turabian StyleKondaka, Kavya, and Iwona Gabriel. 2022. "Targeting DNA Topoisomerase II in Antifungal Chemotherapy" Molecules 27, no. 22: 7768. https://doi.org/10.3390/molecules27227768
APA StyleKondaka, K., & Gabriel, I. (2022). Targeting DNA Topoisomerase II in Antifungal Chemotherapy. Molecules, 27(22), 7768. https://doi.org/10.3390/molecules27227768