Abstract
Janus-type triskelion-shaped fluorophores comprising coumarins bearing various electron-donating substituents (1aad, 1add, 1ccd, and 1cdd) were successfully synthesized via an intramolecular Ullmann coupling. Density functional theory (DFT) calculations indicated that all the compounds presented two different molecular surfaces, similar to Janus-type molecules. The absorption and fluorescence spectra of asymmetrical derivatives 1aad, 1add, 1ccd, and 1cdd exhibited a bathochromic shift due to their narrow highest occupied molecular orbital (HOMO) –lowest unoccupied molecular orbital (LUMO) gap. Natural transition orbital (NTO) analysis indicated that the excited state orbital overlaps differ among the C3 symmetrical and asymmetrical dyes. These triskelion-shaped fluorophores were found to form molecular nanoaggregates in THF/H2O mixtures and demonstrated aggregation-induced emission (AIE) enhancement characteristics as a result of restricting their molecular inversion. These results indicate that Janus-type AIE fluorophores are potentially applicable as solid-state fluorescent chiral materials, which can be optimized by controlling their molecular rearrangement in the solid state.
1. Introduction
Aggregation-induced emission (AIE) is an important characteristic of some molecular luminescent materials, such as 1,1,2,3,4,5-hexaphenylsilole (HPS) [1] and tetraphenylethene (TPE) [2]. These derivatives and their analogs have been extensively applied in bio-imaging, chemical sensing, organic light-emitting diodes (OLEDs), and circularly polarized luminescence (CPL) because they exhibit strong emission upon the formation of nanoaggregates under appropriate concentration conditions [3]. Nanoaggregated molecules can be in close proximity to each other while maintaining an appropriate spatial distance. Therefore, this unusual behavior is elicited by restricting molecular rotations, vibrations, and motions, which are the major causes of non-radiative decay. Conversely, reducing intermolecular interactions, such as π···π interactions in nanoaggregates, is among the strategies for enhancing luminous efficiency. For instance, it is possible to mitigate intermolecular forces by introducing bulky substituents into the target molecules. This chemical modification is also efficient in suppressing the rotation of HPS and TPE [4,5]. Furthermore, 1,4,5,8,9,12-hexamethyltriphenylene (HMTP, Figure 1a) adopts a twisted structure as a result of intramolecular steric repulsion of the adjacent methyl groups [6]. This twisting of HMTP inhibits intermolecular π-stacking, resulting in highly efficient emission in the solid state [7]. Thus, molecular twisting plays a vital role in controlling molecular association. We focused on the enhanced emission intensity caused by molecular twisting and hypothesized that this phenomenon applies to distorted π-conjugated molecules. Based on these insights and ideas, we initiated the development of distorted fluorophores as components of novel AIE luminogens (AIE-gens).
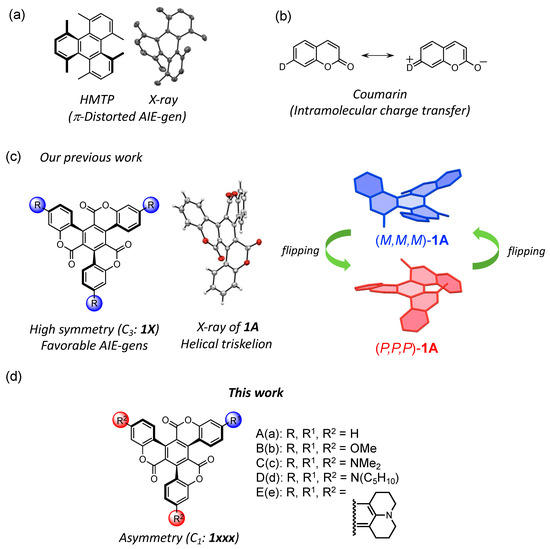
Figure 1.
(a) Chemical structures of 1,4,5,8,9,12-hexamethyltriphenylene (HMTP). The X-ray structure of HMPT is shown [4,5]; (b) resonance structure of coumarin; (c) chemical structure of C3 symmetric triskelion 1 and molecular structure of 1A; (d) chemical structures of asymmetric triskelion-shaped target compounds.
The molecular framework of 1,3,5-triphenylbenzene (TPB) is suitable for this purpose and has often been used as a building block in optical devices [8]. Furthermore, bridging between the central benzene ring and the outer phenyl groups enhances rigidity, bending, and symmetry. TPB is bridged via a sterically crowded lactone chain to form the triskelion-shaped molecules bearing coumarin units (1). This triskelion-shaped framework is expected to adopt a distorted geometry owing to the intramolecular repulsion between the oxygen atoms of the carbonyl groups and the hydrogen atoms of the outer phenyl units.
Coumarins with electron-donating groups at the 7-position exhibit strong fluorescence emission due to intramolecular charge transfer (ICT, Figure 1b) [9,10]. Their photophysical properties are promising for laser dyes [9,10,11], molecular sensors [12,13,14], and solar cells [15]. Furthermore, numerous extended coumarin dyes have been synthesized, and the photophysical properties based on their rigid and planar structures have been described [16,17,18,19,20,21,22,23,24]. Conversely, unusual coumarin dyes with helical geometries have also been reported; these compounds represented a platform for the development of molecular chiral dopants [25,26,27]. Thus, the design of novel coumarin dyes utilizing both their emission characteristics and topologies is required for advancing next-generation optical materials.
Our group has previously succeeded in synthesizing C3 symmetric triskelion-shaped coumarin dye (1A) and its donor-substituted derivatives (1B–1E, Figure 1c). Their molecular structures and photophysical properties have been reported [28,29]. X-ray crystallography revealed that the molecular framework of these compounds presented a helical geometry arising from intramolecular steric repulsion. Furthermore, in the crystal structures of 1A–1D, the left-handed (M,M,M-1) and right-handed (P,P,P-1) structural isomers were presented in a 1:1 ratio. Theoretical calculation indicated that these molecules rapidly underwent racemization due to a low helical inversion barrier. Interestingly, these distorted coumarin dyes exhibited AIE characteristics owing to the formation of the nanoaggregates in the THF/H2O mixture. That is, we achieved control of conformational changes in the internal conversion process by aggregating twisted molecules. Thus, we have found the potential of this coumarin-based framework as AIE fluorophores.
As compound 1 possesses three coumarin units, different electron-donating groups can be introduced synthetically into the coumarin units. Such a chemical modification with various combinations of the substituents is effective with respect to tuning molecular orbital levels and introducing asymmetry, and it is an interesting effect on the luminescence property. There, we designed novel asymmetric triskelion-shaped coumarin dyes with several donor groups (1aad, 1add, 1ccd, and 1cdd, Figure 1d) and investigated their electronic structures and photophysical properties based on the asymmetric distorted structures. Herein, we describe the synthesis and AIE properties of asymmetrical triskelion-shaped fluorophores 1aad, 1add, 1ccd, and 1cdd. To better understand these coumarin-bearing triskelions, they were compared with C3 symmetric triskelions 1A–1E.
2. Materials and Methods
All reagents were commercially sourced. Mesitylene, iron powder, bromine, N-bromosuccinimide, benzoyl peroxide, sodium acetate, thionyl chloride, potassium carbonate, 1-bromo-3-chloropropane, m-aminophenol, 1,5-dibromopentane, N-ethyldiisopropylamine, copper powder, and organic solvents were purchased from FUJIFILM Wako pure Chemical Co., Ltd. (Osaka, Japan). 2-Bromophenol and 3-methoxyphenol were purchased from Tokyo Chemical Industries Co., Ltd. (Tokyo, Japan). Melting points were determined using a Yanaco MP-500P micro-melting point apparatus. 1H (600 or 400 MHz) and 13C (150 or 100 MHz) nuclear magnetic resonance (NMR) spectra were recorded using a Bruker AVANCE instrument. The following abbreviations were used to describe the multiplicities: singlet (s), doublet (d), triplet (t), doublet of doublets (dd), and multiplet (m). Absorption spectra were recorded using a JASCO V-560 instrument. High-resolution mass spectrometry (HRMS) spectra were recorded on a Thermo Fisher Scientific (Tokyo, Japan), Exactive Plus Orbitrap mass spectrometer for ionization. Only relatively intense peaks and structurally diagnostic mass spectral fragment ion peaks are reported. All quantum chemical calculations were performed using the Gaussian 16 program (Revision C.01) [30]. The optimized structures of symmetrical and asymmetrical triskelion-shaped molecules were determined using density functional theory (DFT) calculations. All DFT calculations were performed using the ωB97-XD long-range corrected hybrid functional and 6-31G(d) basis set. Geometry optimizations were performed for the symmetrical derivatives while maintaining the C3 symmetry. Normal mode analysis calculations were conducted at the same level of theory to ensure that local minima optimized structures were obtained. Electronic excitation energies and natural transition orbitals [31] were estimated for all triskelion-shaped derivatives using time-dependent (TD)-DFT calculations. Fluorescence spectra were collected using a HITACHI F-4500 fluorescence spectrometer and a JASCO FP-8550 spectrofluorometer. Relative fluorescence quantum yields (FF) were determined using rhodamine B in EtOH as the standard (FF = 0.9). Dynamic light scattering (DLS) measurements were recorded using a Sysmex Zetasizer NanoZS instrument.
The syntheses of 1A–E and 8A–E have been described in our previous reports [28,29].
Synthesis of 9. To a 50 mL recovery flask equipped with a reflux condenser were added K2CO3 (28 mg, 0.20 mmol), 7d (50 mg, 0.20 mmol), 6 (102 mg, 0.20 mmol), and acetone (15 mL). The reaction mixture was refluxed overnight. After cooling to ambient temperature, residual solids were removed by filtration and washed with CHCl3. The filtrate was then evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (CH2Cl2: hexane = 4:1 (v/v)) to obtain 9 (81 mg, 0.11 mmol) as yellow block crystals in 17% yield. Mp = 155 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 8.8 Hz, 1H), 6.88 (d, J = 2.8 Hz, 1H), 6.76 (dd, J = 2.8 and 8.8 Hz, 1H), 3.17 (t, J = 5.6 Hz, 4H), 1.70 (m, 4H), 1.62–1.58 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 165.2, 161.5, 152.5, 147.9, 141.8, 138.2, 133.7, 117.6, 116.0, 114.7, 110.2, 102.6, 50.0, 25.6, 24.2. UV-vis (CH2Cl2, c = 1.0 × 10−5 M) λmax (ε) 261 (17,300) nm. HRMS (ESI, positive mode): m/z calcd for C20H14Br4Cl2NO4: [M + H]+ 721.6987; found: 721.6986.
Synthesis of 10. To a 50 mL recovery flask equipped with a reflux condenser were added K2CO3 (83 mg, 0.60 mmol), 7d (152 mg, 0.60 mmol), 6 (150 mg, 0.30 mmol), and acetone (15 mL). The reaction mixture was refluxed overnight. After cooling to ambient temperature, the residual solids were removed by filtration and washed with CHCl3. The filtrate was then evaporated under reduced pressure and the residue was purified by column chromatography on silica gel (CH2Cl2: hexane = 4:1 (v/v)) to obtain 10 (140 mg, 0.15 mmol) as yellow block crystals in 50% yield. Mp = 185 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ 7.45–7.43 (m, 2H), 6.92–6.91 (m, 2H), 6.77–6.74 (m, 2H), 3.19–3.16 (m, 8H), 1.73–1.68 (m, 8H), 1.62–1.58 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 165.5, 161.9, 152.5, 147.9, 141.7, 138.0, 133.7, 120.0, 117.1, 116.0, 110.3, 102.8, 50.1, 25.6, 24.2. UV-vis (CH2Cl2, c = 1.0 × 10–5 M) λmax (ε) 316 (4400), 263 (34,100) nm. HRMS (ESI, positive mode): m/z calcd for C31H26Br5ClN2O5: [M + H]+ 942.7459; found: 942.7455.
Synthesis of 8aad. To a 50 mL recovery flask equipped with a reflux condenser were added K2CO3 (39 mg, 0.28 mmol), 7a (30 μL, 0.28 mmol), 9 (100 mg, 0.14 mmol), and acetone (15 mL). The reaction mixture was refluxed overnight. After cooling to ambient temperature, the residual solids were removed by filtration and washed with CHCl3. The filtrate was then evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (CH2Cl2: hexane = 4:1 (v/v)) to obtain 8aad (103 mg, 0.10 mmol) as yellow block crystals in 74% yield. Mp = 98 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ 7.69 (dd, J = 1.6 and 8.0 Hz, 2H), 7.50 (dd, J = 1.6 and 8.0 Hz, 2H), 7.45 (d, J = 8.8 Hz, 2H), 7.41 (dd, J = 1.6 and 8.0 Hz, 1H), 3.18 (t, J = 5.6 Hz, 4H), 1.73–1.68 (m, 4H), 1.62–1.58 (m,2H). 13C NMR (100 MHz, CDCl3) δ 162.2, 162.1, 152.5, 148.0, 147.6, 137.9, 137.7, 134.1, 133.7, 128.7, 128.3, 123.5, 119.6, 119.5, 116.0, 115.6, 110.4, 102.8, 50.1, 25.6, 24.2. UV-vis (CH2Cl2, c = 1.0 × 10–5 M) λmax (ε) 266 (15,500) nm. HRMS (ESI, positive mode): m/z calcd for C32H21Br6NO6: [M + H]+ 995.6481; found: 995.6480.
Synthesis of 8add. To a 50 mL recovery flask equipped with a reflux condenser were added K2CO3 (15 mg, 0.11 mmol), 7a (11 μL, 0.11 mmol), 10 (101 mg, 0.11 mmol), and acetone (15 mL). The reaction mixture was refluxed overnight. After cooling to ambient temperature, the residual solids were removed by filtration and washed with CHCl3. The filtrate was then evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (CH2Cl2: hexane = 4:1 (v/v)) to obtain 8add (66 mg, 0.06 mmol) as colorless block crystals in 58% yield. Mp = 268 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ 7.70–7.68 (m, 1H), 7.52–7.49 (m, 1H), 7.46–7.45 (m, 2H), 7.44–7.40 (m, 1H), 7.24–7.20 (m, 1H), 6.96–6.95 (m, 1H), 6.77–6.74 (m, 2H), 3.20–3.17 (m, 8H), 1.74–1.69 (m, 8H), 1.63–1.59 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 162.3, 162.2, 152.5, 148.1, 147.7, 138.0, 134.2, 133.7, 128.7, 128.3, 123.5, 119.7, 119.5, 116.0, 115.6, 110.5, 102.9, 50.2, 25.7, 24.2. UV-vis (CH2Cl2, c = 1.0 × 10–5 M) λmax (ε) 264 (31,700) nm. HRMS (ESI, positive mode): m/z calcd for C37H30Br6N2O6: [M + H]+ 1078.7216; found: 1078.7217.
Synthesis of 8ccd. To a 50 mL recovery flask equipped with a reflux condenser were added K2CO3 (40 mg, 0.28 mmol), 7c (70 mg, 0.28 mmol), 9 (101 mg, 0.14 mmol), and acetone (15 mL). The reaction mixture was refluxed overnight. After cooling to ambient temperature, the residual solids were removed by filtration and washed with CHCl3. The filtrate was then evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (CH2Cl2) to obtain 8ccd (78 mg, 0.07 mmol) as yellow block crystals in 52% yield. Mp = 197 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ 7.45–7.42 (m, 3H), 6.96–6.95 (m, 1H), 6.76–6.73 (m, 3H), 6.55–6.52 (m, 2H), 3.19–3.16 (m, 4H), 2.97 (s, 12H), 1.73–1.68 (m, 4H), 1.62–1.58 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 162.3, 162.3, 152.5, 150.8, 148.1, 137.9, 137.9, 133.6, 119.5, 119.5, 115.9, 112.4, 110.5, 106.7, 102.9, 100.4, 50.1, 40.6, 25.7, 24.2. UV-vis (CH2Cl2, c = 1.2 × 10–5 M) λmax (ε) 311 (7400), 266 (38,300), 261 (38,100), 255 (31,400) nm. HRMS (ESI, positive mode): m/z calcd for C36H32Br6N3O6: [M + H]+ 1081.7325; found: 1078.7327.
Synthesis of 8cdd. To a 50 mL recovery flask equipped with a reflux condenser were added K2CO3 (20 mg, 0.14 mmol), 7c (30 mg, 0.14 mmol), 10 (131 mg, 0.14 mmol), and acetone (20 mL). The reaction mixture was refluxed overnight. After cooling to ambient temperature, the residual solids were removed by filtration and washed with CHCl3. The filtrate was then evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (CH2Cl2) to obtain 8cdd (78 mg, 0.07 mmol) as white block crystals in 67% yield. Mp = 197 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ 7.46–7.42 (m, 3H), 6.96–6.95 (m, 2H), 6.77–6.73 (m, 3H), 6.55–6.53 (m, 1H), 3.19–3.17 (m, 8H), 2.97 (s, 6H), 1.74–1.68 (m, 8H), 1.62–1.58 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 162.3, 162.3, 152.5, 150.8, 148.1, 148.1, 137.9, 137.9, 133.7, 119.5, 119.5, 115.9, 112.4, 110.5, 106.7, 102.9, 100.4, 50.1, 40.6, 25.7, 24.4. UV-vis (CH2Cl2, c = 1.0 × 10–5 M) λmax (ε) 319 (7300), 266 (48,100), 259 (42,300), 254 (28,600). HRMS (ESI, positive mode): m/z calcd for C39H36Br6N3O6: [M + H]+ 1121.7638; found: 1121.7638.
Synthesis of 1aad. To a 50 mL Schlenk tube was added 8aad (100 mg, 0.10 mmol), excess activated Cu Powder (1410 mg, 22.0 mmol), and dry DMF (30 mL) under argon atmosphere. The reaction mixture was refluxed for 18 h. After cooling to ambient temperature, the reaction mixture was filtered through Celite and washed with EtOAc. The organic phase was then washed with H2O and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified by flash column chromatography on Al2O3 (CH2Cl2: EtOAc = 30:1 (v/v)) to give 1aad (24 mg, 0.05 mmol) as a reddish solid in 47% yield. Mp = 150 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ 8.01–7.99 (m, 2H), 7.79–7.77 (m, 1H), 7.60–7.55 (m, 2H), 7.40–7.38 (m, 2H), 7.28–7.24 (m, 2H), 6.77–6.74 (m, 1H), 6.60–6.65 (m, 1H), 3.50–3.44 (m, 4H), 1.74–1.68 (m, 4H), 1.58–1.54 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 159.1, 158.8, 158.5, 154.7, 154.5, 151.6, 151.6, 146.7, 146.7, 146.5, 133.1, 133.0, 131.8, 131.6, 129.0, 123.8, 123.7, 117.3, 117.3, 117.1, 117.0, 115.3, 113.8, 113.1, 110.5, 106.4, 98.9, 48.4, 25.5, 24.5. UV-vis (CH2Cl2, c = 1.1 × 10–5 M) λmax (ε) 472 (20,600), 335 (19,200) nm. HRMS (ESI, positive mode): m/z calcd for C32H21NO6 [M]+ 515.1364; found: 515.1363.
Synthesis of 1add. To a 50 mL Schlenk tube was added 8add (100 mg, 0.09 mmol), excess activated Cu Powder (1300 mg, 20.5 mmol), and dry DMF (28 mL) under argon atmosphere. The reaction mixture was refluxed for 18 h. After cooling to ambient temperature, the reaction mixture was filtered through Celite and washed with EtOAc. The organic phase was then washed with H2O and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified by flash column chromatography on Al2O3 (CH2Cl2: EtOAc = 30:1 (v/v)) to give 1add (26 mg, 0.04 mmol) as a reddish solid in 47% yield. Mp = 115 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ 8.98–7.96 (m, 1H), 7.78–7.76 (m, 2H), 7.55–7.51 (m, 1H), 7.37–7.35 (m, 1H), 7.25–7.21 (m, 1H), 6.76–6.72 (m, 2H), 6.65–6.64 (m, 2H), 3.49–3.37 (m, 8H), 1.77–1.64 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 159.6, 159.3, 159.0, 154.5, 154.5, 154.3, 154.3, 151.5, 147.2, 146.9, 146.9, 132.6, 131.7, 131.7, 130.7, 123.5, 117.6, 116.8, 113.0, 112.2, 111.2, 110.4, 106.7, 106.6, 99.1, 99.0, 48.5, 25.5, 24.5. UV-vis (CH2Cl2, c = 1.1 × 10–5 M) λmax (ε) 463 (44,900), 357 (14,400) nm. HRMS (ESI, positive mode): m/z calcd for C37H30N2O6: [M + H]+ 599.2177; found: 599.2176.
Synthesis of 1ccd. To a 50 mL Schlenk tube was added 8ccd (100 mg, 0.09 mmol), excess activated Cu Powder (1330 mg, 20.5 mmol), and dry DMF (28 mL) under argon atmosphere. The reaction mixture was refluxed for 18 h. After cooling to ambient temperature, the reaction mixture was filtered through Celite and washed with EtOAc. The organic phase was then washed with H2O and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified by flash column chromatography on Al2O3 (CH2Cl2: EtOAc = 20:1 (v/v)) to give 1ccd (36 mg, 0.06 mmol) as a reddish solid in 64% yield. Mp = 222 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ 7.79–7.77 (m, 3H), 6.75–6.72 (m, 1H), 6.64–6.63 (m, 1H), 6.60–6.57 (m, 2H), 6.48–6.47 (m, 2H), 3.45–3.39 (m, 4H), 3.10 (s, 12H), 1.72–1.66 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 159.9, 159.9, 159.9, 154.3, 154.2, 154.1, 153.6, 1477, 147.6, 147.4, 131.6, 110.3, 110.2, 110.1, 108.4, 107.1, 106.2, 99.2, 97.2, 77.5, 77.2, 76.8, 48.5, 40.2, 25.5, 24.5. UV-vis (CH2Cl2, c = 1.1 × 10–5 M) λmax (ε) 453 (62,900) nm. HRMS (ESI, positive mode): m/z calcd for C36H31N3O6: [M]+ 601.2207; found: 601.2208.
Synthesis of 1cdd. 50 mL Schlenk tube was added 8cdd (101 mg, 0.09 mmol), excess activated Cu Powder (1300 mg, 20.5 mmol), and dry DMF (27 mL) under argon atmosphere. The reaction mixture was refluxed for 18 h. After cooling to ambient temperature, the reaction mixture was filtered through Celite and washed with EtOAc. The organic phase was washed with H2O and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified by flash column chromatography on Al2O3 (CH2Cl2: EtOAc = 20:1 (v/v)) to give 1cdd (28 mg, 0.04 mmol) as a reddish solid in 49% yield. Mp = 125 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ 7.79–7.77 (m, 3H), 6.74–6.72 (m, 2H), 6.64–6.63 (m, 2H), 6.59–6.57 (m, 1H), 6.48–6.47 (m, 1H), 3.44–3.40 (m, 8H), 3.10 (s, 6H), 1.72–1.67 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 159.9, 159.9, 159.8, 154.3, 154.2, 154.1, 153.6, 147.7, 147.4, 147.4, 131.6, 110.4, 110.4, 110.3, 110.2, 108.4, 107.1, 106.2, 99.2, 97.2, 48.5, 40.2, 25.5, 24.5. UV-vis (CH2Cl2, c = 1.1 × 10–5 M) λmax (ε) 453 (55,400) nm. HRMS (ESI, positive mode): m/z calcd for C39H36N3O6: [M + H]+ 642.2599; found: 642.2598.
3. Results and Discussion
The synthetic route to C3 symmetrical compounds 1A–E is depicted in Scheme 1. The central benzene core 6 was efficiently prepared from mesitylene (2) in five steps, according to the literature procedures [32,33,34,35]. Compound 6 was reacted with the corresponding 2-bromophenol derivatives in the presence of K2CO3 in acetone to afford precursors 8A–E in good yields. Finally, the targeted molecular triskelions 1A–E were obtained via an intramolecular Ullmann coupling reaction. The C3 symmetric derivatives were fully characterized using 1H and 13C NMR spectroscopy and high-resolution mass spectrometry (HRMS) [28,29].
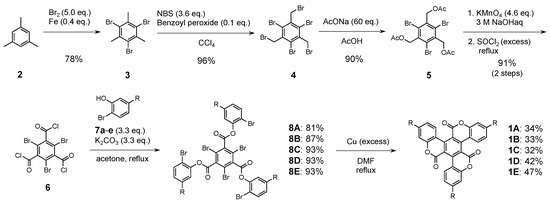
Scheme 1.
Synthetic route for 1A–E.
Subsequently, we employed the synthetic routes for 1A–E (Scheme 2) for the synthesis of asymmetric triskelions. First, 6 and 1.0 or 2.0 eq. of 7d were treated with K2CO3 in acetone to give mono-substituted 9 and di-substituted 10 in 19% and 50% yield, respectively. Next, asymmetric precursors 8aad, 8add, 8ccd, and 8cdd were obtained in 52–74% yield using 2.0 eq. of 7a or 7c for 9, and 1.0 eq. of 7a or 7c for 10. Owing to the nucleophilic reactivity of 6, it was difficult to introduce three different-type phenyl units into the central benzene core. The intramolecular Ullmann coupling of 8aad, 8add, 8ccd, and 8cdd in the presence of excess copper in DMF afforded the desired asymmetric triskelions 1aad, 1add, 1ccd, and 1cdd, which were analogous to 1A–E. Although the 1H and 13C NMR spectra of these derivatives were highly complex due to their C1 symmetry, the asymmetric triskelions structures were identified (Figures S13–S20). In this coupling condition, several debrominated compounds of 8, mono- and bis-coupling products were observed as the byproducts, however, they could not be isolated.
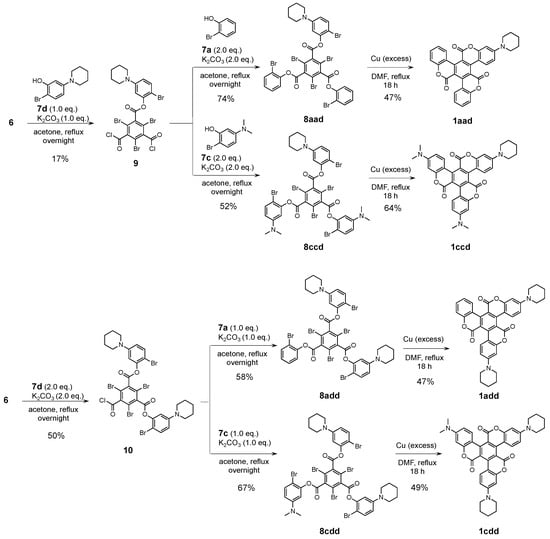
Scheme 2.
Synthetic route for 1aad, 1add, 1ccd, and 1cdd.
The molecular structures of 1A–D were confirmed using X-ray crystallography [28,29]. As expected, the molecular framework of 1 presented a helical geometry arising from intramolecular steric repulsion. This result was in good agreement with the corresponding optimized structures derived from quantum chemical calculations. Similarly, 1aad, 1add, 1ccd, and 1cdd were optimized as twisted propeller-shaped structures (Figure 2). Furthermore, in the case of these asymmetric triskelions, left-handed M,M,M- and right-handed P,P,P-formers were the most stable conformation regardless of the substituted groups.
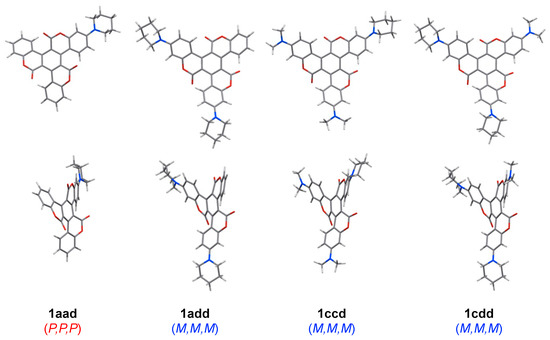
Figure 2.
Optimized structures of 1aad, 1add, 1ccd, and 1cdd at the RB3LYP/6-31G level.
The maximum transition state energy for the isomerization between (P,P,P) and (M,M,M)-1A was estimated to be 9.46 kcal/mol at the ωB97-XD/6-31G(d) level of theory. Even in the case of 1C, bearing N-containing donor groups, the corresponding energy was 8.7 kcal/mol. This indicates that these derivatives underwent continuous flipping in diluted solutions. Furthermore, all triskelion-shaped compounds adopted a Janus-type structure and presented two distinct molecular π-surfaces (Figure 3). Based on the helical triskelion-shaped framework, the electrostatic potential mapping of these compounds demonstrated that the outer phenyl groups on the front surface of 1 were positively charged (blue) and the three electron-withdrawing carbonyl groups on the back face were negatively charged (red), regardless of the molecular symmetry. Thus, these molecules exhibit out-of-plane anisotropy via the central benzene ring from the positive area on the front to the negative area on the back face. The estimated dipole moments of the derivatives bearing donor groups reached 7.83 Debye, which is higher than those of bowl-shaped sumanene [36] and Janus-type subphthalocyanine [37]. Interestingly, the dipole moments of asymmetrical derivatives 1aad, 1add, 1ccd, and 1cdd were also showed large owing to the triskelion-shaped framework. We believe that this Janus-type anisotropy is beneficial for nanomaterials applicable in molecular recognition that operates through molecular rearrangement control [38].
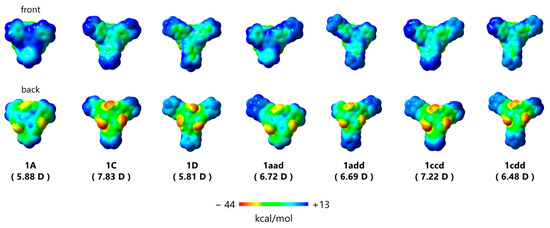
Figure 3.
Electrostatic potential surfaces of 1A, 1C, 1D, 1aad, 1add, 1ccd, and 1cdd estimated employing quantum chemical calculations. Calculated values of the corresponding dipole moments [D = Debye] are shown in parentheses.
The absorption and fluorescence spectra of asymmetrical triskelion-shaped coumarin dyes 1aad, 1add, 1ccd, and 1cdd were recorded in toluene to investigate their photophysical properties and compare them with those of C3 symmetrical dyes 1A, 1C, and 1D (Figure 4). As shown in Figure 4a, in the absorption spectra of the symmetrical dyes, a single peak was observed at 342 nm for 1A and at 443 nm for 1C and 1D. In contrast, two peaks were observed in the absorption spectra of 1aad (335 and 472 nm) and 1add (357 and 463 nm). On the other hand, the absorption spectra of 1ccd and 1cdd only contained one peak at 443 nm, similar to the results for the symmetrical dyes. These results indicated that the absorption maxima of 1aad (λmax: 472 nm) and 1add (λmax: 463 nm) exhibited a bathochromic shift compared to that of 1A (λmax: 342 nm). This shift stems from the narrow gap between the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) resulting from reduced molecular symmetry (Figure 5). Conversely, the absorption bands of 1ccd and 1cdd at 443 nm resembled that of 1C (λmax: 443 nm). Although there is not much difference among 1aad, 1add, 1ccd, and 1cdd, the HOMO and LUMO levels of 1ccd and 1cdd were similar to those of N-substituted C3 triskelions due to the presence of strong donor groups, despite their C1 molecular symmetry.
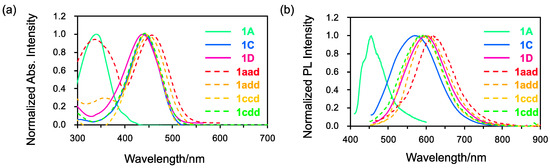
Figure 4.
(a) Normalized absorption and (b) fluorescence spectra of 1A, 1C, 1D, 1aad, 1add, 1ccd, and 1cdd in toluene (c = 1.0 × 10−5 M).
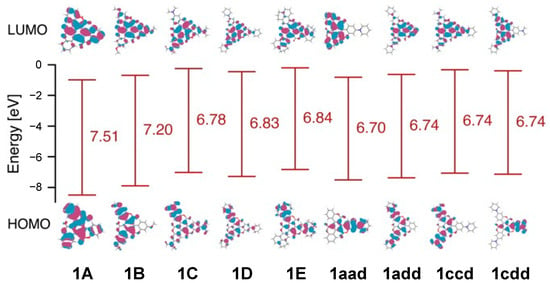
Figure 5.
Highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of 1A, 1B, 1C, 1D, 1E, 1aad, 1add, 1ccd, and 1cdd, and corresponding HOMO-LUMO gaps energies estimated applying quantum chemical calculations.
To further elucidate the photochemical behavior of the asymmetrical triskelion-shaped coumarin dyes, their excited states were examined using quantum chemical calculations. Figure 6 shows the absorption spectra simulated using quantum chemical calculations and the main components of the natural transition orbitals (NTOs) for each electronic excitation. The simulated absorption spectra are in good agreement with the experimental results (Figure 4a), wherein two peaks were observed in the spectra of 1aad and 1add, and a single peak for 1ccd and 1cdd. In the case of 1aad, the NTO analysis shown in Figure 6 indicates that the S0 → S2 transition, the peak of which is observed in the longer-wavelength region, mainly involves the N-substituted moiety in the molecule (d unit), whereas the S0 → S4 transition, the peak of which appears in the shorter wavelength region, mainly involves two unsubstituted moieties (a units). Similarly, for 1add, the S0 → S3 transition with a peak in the long-wavelength region involves two N-substituted moieties (d units), whereas the S0 → S4 transition with a peak in the short wavelength region involves one non-substituent moiety (a unit). On the other hand, in the cases of 1ccd and 1cdd, the S0 → Sn (n = 2, 3, and 4) transitions all involve intramolecular N-substituted moieties, and their transition energies do not differ significantly; thus, they are observed as a single peak in the long wavelength region of the absorption spectra. Another interesting point here is that the oscillation intensity of S0 → S4 transition is smaller than that of S0 → S2 in the case of 1aad and 1add, but larger in the case of 1ccd and 1cdd. In general, the transition probability depends on the overlap between the molecular orbitals involved in the electronic excitation. Indeed, as shown in Figure 6, the overlap of the hole-particle pairs of NTOs for the S0 → S4 transition is small in the case of 1aad and 1add, but large in the cases of 1ccd and 1cdd. That is, due to the combination of the substitute positions, this framework was found to be designable for controlling the frontier orbitals and electronic transitions. In contrast, the NTO analysis of C3 symmetrical derivatives 1A–1E suggested that their corresponding S0 → S2 or S0 → S3 transitions were composed of two pairs with large eigenvalues (Figure S24). This is due to the degenerated molecular orbitals resulting from their C3 symmetry. Therefore, distribution and overlapping from the hole to particle cover the whole of the molecular framework.
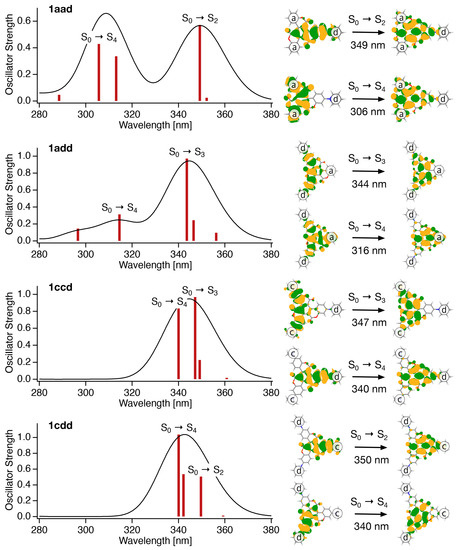
Figure 6.
Absorption spectra simulated by quantum chemical calculations, and the main components of the natural transition orbitals (NTOs) for each electronic excitation. Absorption spectra were simulated by the Gaussian-shaped bands’ half-width at half maximum of 1000 cm−1.
In the fluorescence spectra of 1aad, 1add, 1ccd, and 1cdd in toluene, the emission maxima were observed at 619, 606, 593, and 596 nm, respectively, with relatively large Stokes shifts of 5700–5800 cm−1 (Figure 4b). These emission maxima were as high as that of 1D (599 nm) or slightly bathochromically shifted. The fluorescence quantum yields of these compounds were low (ΦF = 0.003–0.007). This inefficiency is possibly caused by thermal non-radiative transitions arising from their helical inversion, in addition to the internal conversion from Sn (n = 2~4) to S1. These values are lower than those of obtained for the C3 symmetrical compounds 1C (ΦF = 0.14) and 1D (ΦF = 0.13) in toluene. The orbital degeneracy can be resolved by varying the substitution style on the molecular triskelion. As a result, the orbital overlap between the ground and excited states of the asymmetric derivatives would be reduced owing to their partial localization as shown in NTO analysis.
Next, we investigated the solvent dependence of the photophysical properties of 1aad, 1add, 1ccd, and 1cdd (Figure S25 and Table S1). All of the compounds demonstrated a gradual bathochromic shift in their absorption spectra depending on the solvent polarity. Conversely, the asymmetrical dyes, expect for 1ccd, exhibited a blue-shift in their fluorescence spectra in highly polar solvents such as DMF and DMSO. The THF and EtOAc solutions of these compounds gave rise to broad emission bands containing shoulder peaks. This tendency was observed in the case of 1C and 1D [29]. This indicates that asymmetrical compounds undergo at least two radiative decay processes. Intriguingly, 1ccd exhibited a redshift in highly polar solvents. For triskelion-shaped scaffolds, the charge separation structures in the excited states may be controllable depending on the combination of the three substituents. Based on the photophysical data, we evaluated the intramolecular charge transfer (ICT) characteristics of 1aad, 1add, 1ccd, and 1cdd by using Lippert-Mataga plots (Figure 7) [39,40]. Compounds 1aad, 1add, and 1cdd exhibited a negative correlation, whereas 1ccd showed a minimally increasing trend. However, linearity was poor in all cases, indicating that the ICT characteristics of these asymmetrical derivatives depend minimally on solvent polarity, in contract to typical coumarin dyes. It is considered that the resonance structures of 1 in the excited state are destabilized with the loss of aromaticity of the central benzene ring. The fluorescence quantum yields in the tested solvents were considerably low. The cause of low efficiency under diluted solutions is presumed to occur due to the dominant non-radiative process resulting from the helical inversion. We reasoned that fluorescence efficiency can be improved by suppression of molecular inversion in the aggregates, as in the case of AIE-gens [3].
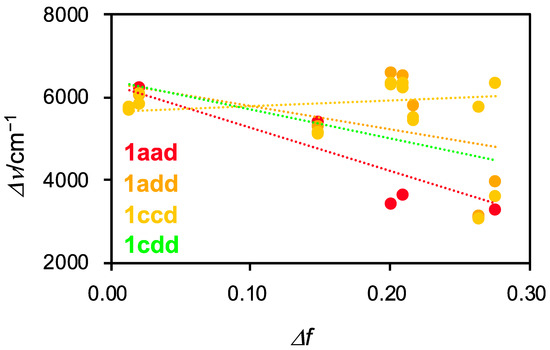
Figure 7.
Lippert-Mataga plots for 1aad, 1add, 1ccd, and 1cdd in various solvents. Δν (Stokes shift) = νabs − νfl, Δf (Orientation polarizability) = (εr − 1/2εr + 1) – (n2 − 1/2n2 + 1).
To investigate the AIE enhancement (AIEE) properties of 1aad, 1add, 1ccd, and 1cdd, we first prepared THF/H2O solutions (water fraction (Fw): 10–80%) of 1aad, 1add, 1ccd, and 1cdd, and measured their absorption (Figure S26). For all the compounds, the absorption maxima gradually decreased as the Fw increased to 50–60%. At Fws of ≥60%, the absorbance decreased drastically, and scattering absorption bands were observed in the long-wavelength region. This result indicated that these compounds formed nanoaggregates in the THF/H2O mixture. Moreover, the DLS results indicated the existence of nanoaggregates with constant particle sizes in the range of 2~700 nm (Figure S27). As shown in Figure 8, the fluorescence spectral peaks of 1aad, 1add, 1ccd, and 1cdd at Fws of 10–50% were very low-intensity. Conversely, as the Fw increased to ≥60%, the intensity increased drastically owing to the formation of nanoaggregates. The maximum intensity was observed at Fw = 70%. This suggests that the AIEE characteristics of these asymmetrical triskelions arise due to molecular motions, such as vibrations and inversion, which are restricted in the nanoaggregates. Interestingly, these compounds exhibited a bathochromic shift of approximately 650 nm. Newly formed nanoaggregates of 1aad, 1add, 1ccd, and 1cdd may be affected by the reorientation of H2O in their excited state. However, at an Fw of 80%, the intensity decreased owing to reprecipitation. Thus, these asymmetrical derivatives are potentially applicable as components of effective AIE-gens owing to their twisted geometry, similar to C3 symmetrical triskelion-shaped dyes. The introduction of several substituents to the triskelion scaffold enabled the tuning of their orbital levels and molecular packing. It is interested in their emission behavior in the solid-state, such as powder, crystalline, and thin films.
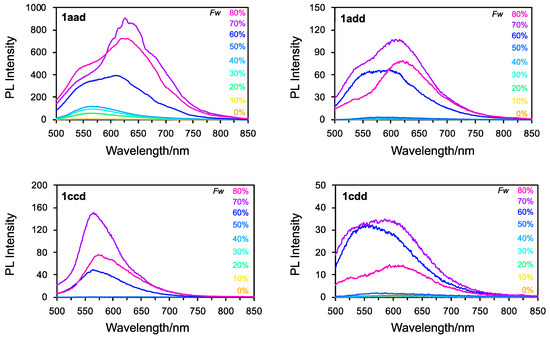
Figure 8.
Fluorescence spectra of 1aad, 1add, 1ccd, and 1cdd recorded in various THF/H2O mixtures.
4. Conclusions
We successfully synthesized Janus-type triskelion-shaped fluorophores comprising variably substituted coumarins (1A–E, 1aad, 1add, 1ccd, and 1cdd) via the copper-mediated intramolecular transannulation Ullmann reaction. Based on the optimized structures, we reasonably assumed that asymmetrical triskelions 1aad, 1add, 1ccd, and 1cdd would exhibit similar distorted structures of C3 symmetric triskelions 1A–1E. All of the compounds presented out-of-plane anisotropy with large dipole moments (5.81–7.83 Debye) owing to the Janus-type structure, which presents two different π-surfaces. The absorption and emission maxima of the asymmetrical compounds 1aad, 1add, 1ccd, and 1cdd were redshifted compared to those of C3 symmetrical compounds 1A, 1C, and 1D, owing to the narrow HOMO-LUMO gaps of the former. The emission behavior of the asymmetrical compounds, with the exception of 1ccd, was weakly associated with solvent polarity. The quantum yields of 1aad, 1add, 1ccd, and 1cdd were considerably lower than those of the C3 symmetrical derivatives. NTO analysis suggested that the molecular orbitals of the C3 symmetrical compounds in the excited state are delocalized over the triskelion framework in the S0 → Sn (n = 2, 3, and 4) electronic transition. Conversely, those of the asymmetrical compounds are located on one or two coumarin units through the central benzene ring. Therefore, we reasoned that the low emission efficiencies of 1aad, 1add, 1ccd, and 1cdd resulted from orbital distribution overlap. All the compounds formed nanoaggregates in THF/H2O mixtures and demonstrated AIEE characteristics arising from their distorted geometry. Thus, Janus-type triskelion-shaped AIE fluorophores are potentially promising candidates for the development of solid-state fluorescent and chiral materials, which can be optimized by controlling their molecular rearrangement in the solid state.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27217450/s1, Figures S1–20: The copies of 1H and 13C NMR charts; Figure S21: Molecular orbitals of 1aad, 1add, 1ccd, and 1cdd; Figure S22: Calculated spectra and the main components of the natural transition orbitals (NTOs) for each electronic excitation of 1aad, 1add, 1ccd, and 1cdd; Figure S23: Emission spectra of 1aad, 1add, 1ccd, and 1cdd in several solvents; Figure S24: Absorption spectra of 1aad, 1add, 1ccd, and 1cdd in THF/H2O mixtures; Figure S25: The size distribution in THF/H2O mixtures based on DLS data for 1aad, 1add, 1ccd, and 1cdd. Table S1: Photophysical properties of 1aad, 1add, 1ccd, and 1cdd in various solvents.
Author Contributions
Conceptualization, M.U. and Y.M.; Synthesis, Measurement, and Analysis: M.U. and M.K.; Theoretical calculation and Computational analysis: M.U., N.Y. (Nao Yanagi) and N.Y. (Norifumi Yamamoto); Writing: M.U., N.Y. (Norifumi Yamamoto) and Y.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Kitasato University Research Grant for Young Researchers and JSPS KAKENHI Grant Numbers JP21K14615 and JP22K05025.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We are grateful to Takahiro Tsuchiya and Masashi Hasegawa (Kitasato University) for their discussions and support. All the quantum chemical calculations were performed at the Research Center for Computational Science, Okazaki, Japan (21-IMS-C188 and 22-IMS-C029).
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Chen, J.; Law, C.C.W.; Lam, J.W.Y.; Dong, Y.; Lo, S.M.F.; Williams, I.D.; Zhu, D.; Tang, B.Z. Synthesis, Light Emission, Nanoaggregation, and Restricted Intramolecular Rotation of 1,1-Substituted 2,3,4,5-Tetraphenylsiloles. Chem. Mater. 2003, 15, 1535–1546. [Google Scholar] [CrossRef]
- Dong, Y.; Lam, J.W.Y.; Qin, A.; Liu, J.; Li, Z.; Tang, B.Z. Aggregation-induced emissions of tetraphenylethene derivatives and their utilities as chemical vapor sensors and in organic light-emitting diodes. Appl. Phys. Lett. 2007, 91, 011111. [Google Scholar] [CrossRef]
- Mei, J.; Leung, N.L.C.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Aggregation-Induced Emission: Together We Shine, United We Soar! Chem. Rev. 2015, 115, 11718–11940. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, Y.; Mi, B.; Tang, Y.; Häussler, M.; Tong, H.; Dong, Y.; Lam, J.W.Y.; Ren, Y.; Sung, H.H.Y.; et al. Structural Control of the Photoluminescence of Silole Regioisomers and Their Utility as Sensitive Regiodiscriminating Chemosensors and Efficient Electroluminescent Materials. J. Phys. Chem. B 2005, 109, 10061–10066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-F.; Chen, Z.-Q.; Aldred, M.P.; Hu, Z.; Chen, T.; Huang, Z.; Meng, X.; Zhu, M.-Q. Direct validation of the restriction of intramolecular rotation hypothesis via the synthesis of novel ortho-methyl substituted tetraphenylethenes and their application in cell imaging. Chem. Commun. 2014, 50, 12058–12060. [Google Scholar] [CrossRef]
- Wang, Y.; Stretton, A.D.; McConnell, M.C.; Wood, P.A.; Parsons, S.; Henry, J.B.; Mount, A.R.; Galow, T.H. 1,4,5,8,9,12-Hexamethyltriphenylene. A Molecule with a Flipping Twist. J. Am. Chem. Soc. 2007, 129, 13193–13200. [Google Scholar] [CrossRef]
- Levell, J.W.; Ruseckas, A.; Henry, J.B.; Wang, Y.; Stretton, A.D.; Mount, A.R.; Galow, T.H.; Samuel, I.D.W. Fluorescence Enhancement by Symmetry Breaking in a Twisted Triphenylene Derivative. J. Phys. Chem. A 2010, 114, 13291–13295. [Google Scholar] [CrossRef]
- Schmidt, B.; Rinke, M.; Güsten, H. PhotoPhysical properties of 1,3,5-Tris-(p-oligophenylene)-benzenes. J. Photochem. PhotoBiol. A Chem. 1989, 49, 131–135. [Google Scholar] [CrossRef]
- Drexhage, K.H. Structure and Properties of Laser Dyes (Chap. 4). In Dye Lasers; Schäfer, F.P., Ed.; Springer: Berlin/Heidelberg, Germany, 1973; pp. 144–193. [Google Scholar]
- Kim, J.; Oh, J.H.; Kim, D. Recent advances in single-benzene-based fluorophores: Physicochemical properties and applications. Org. Biomol. Chem. 2021, 19, 933–946. [Google Scholar] [CrossRef]
- Runack, K.; Spieles, M. Fluorescence Quantum Yields of a Series of Red and Near-Infrared Dyes Emitting at 600–1000 nm. Anal. Chem. 2011, 83, 1232–1242. [Google Scholar]
- Cao, D.; Liu, Z.; Verwilst, P.; Koo, S.; Jangjili, P.; Kim, J.S.; Lin, W. Coumarin-Based Small-Molecule Fluorescent Chemosensors. Chem. Rev. 2019, 119, 10403–10519. [Google Scholar] [CrossRef] [PubMed]
- Lang, W.; Yuan, C.; Zhu, L.; Du, S.; Qian, L.; Ge, J.; Yao, S.Q. Recent advances in construction of small molecule-based fluorophore-drug conjugates. J. Pherma. Anal. 2020, 10, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Panda, B. The Recent Developments and Applications of Photoremovable Protecting Groups in Organic Chemistry. Curt. Chin. Chem. 2022, 2, e020222200770. [Google Scholar] [CrossRef]
- Hara, K.; Sayama, K.; Ohga, Y.; Shinpo, A.; Suga, S.; Arakawa, H. A coumarin-derivative dye sensitized nanocrystalline TiO2 solar cell having a high solar-energy conversion efficiency up to 5.6%. Chem. Commun. 2001, 6, 569–570. [Google Scholar] [CrossRef]
- Kim, I.; Kim, T.-H.; Kang, Y.; Lim, Y.-b. BBr3-promoted cyclization to produce ladder-type conjugated polymer. Tetrahedron Lett. 2006, 47, 8689–8692. [Google Scholar] [CrossRef]
- Węcławski, M.K.; Tasior, M.; Hammann, T.; Cywiński, P.J.; Gryko, D.T. From p-expanded coumarins to p-expanded pentacenes. Chem. Commun. 2014, 50, 9105–9108. [Google Scholar] [CrossRef]
- Nazir, R.; Stasyuk, A.J.; Gryko, D.T. Vertically p-Expanded Coumarins: The Synthesis and Optical Properties. J. Org. Chem. 2016, 22, 11104–11114. [Google Scholar] [CrossRef]
- Hintz, H.A.; Sortedahl, N.; Meyer, S.M.; Decato, D.A.; Dahl, B.J. The synthesis of lactone-bridged 1,3,5-triphenylbenzene derivatives as pi-expanded coumarin triskelions. Tetrahedron Lett. 2017, 58, 4703–4708. [Google Scholar] [CrossRef]
- Nitisha; Venkatakrishnan, P. Accessing [g]-Face p-Expanded Fluorescent Coumarin by Scholl Cyclization. J. Org. Chem. 2019, 84, 10679–10689. [Google Scholar] [CrossRef]
- Xue, W.; Wang, D.; Li, C.; Zhai, Z.; Wang, T.; Liang, Y.; Zhang, Z. p-Expanded Coumarins: One-Pot Photo Synthesis of 5H-benzo[12.1]tetrapheno[7,6,5-cde]chromen-5-ones and Photophysical Properties. J. Org. Chem. 2020, 85, 3689–3698. [Google Scholar] [CrossRef]
- Kumar, A.; Rajpoot, A.; Imroze, F.; Maddala, S.; Dutta, S.; Venkatakrishnan, P. Linear Coumarinacenes Beyoud Benzo[g]coumarins: Synthesis and Promising Characteristics. Eur. J. Org. Chem. 2020, 2020, 6976–6980. [Google Scholar] [CrossRef]
- Nitisha; Chetti, P. Parthasarathy, V. Coronene-embedded ‘super’coumarins. Chem. Commun. 2022, 58, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Kielesiński, Ł.; Deperasińska, I.; Morawski, O.; Vygrannenko, K.V.; Ouellette, E.T.; Gryko, D.T. Polarized, V-Shaped, and Conjoined Biscoumarins: From Lack of Dipole Moment Alignment to High Brightness. J. Org. Chem. 2022, 87, 5961–5975. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Hossen, T.; Ghosh, I.; Koner, A.L.; Nau, W.M.; Sahu, K.; Moorthy, J.N. Helicity-Dependent Regiodifferentiation in the Excited-State Quenching and Chiroptical Properties of Inward/Outward Helical Coumarins. Chem. Eur. J. 2017, 23, 14797–14805. [Google Scholar] [CrossRef] [PubMed]
- Usui, K.; Yamamoto, K.; Ueno, Y.; Igawa, K.; Hagihara, R.; Masuda, T.; Ojida, A.; Karasawa, S.; Tomooka, K.; Hirai, G.; et al. Internal-Edge-Substituted Coumarin-Fused [6]Helicenes: Asymmetric Synthesis, Structural Features, and Control of Self-Assembly. Chem. Eur. J. 2018, 24, 14617–14621. [Google Scholar] [CrossRef]
- Mukhopadhway, A.; Jana, K.; Hossen, T.; Sahu, K.; Moorthy, J.N. Coumarin-Annealed Regioisomeric Heptahelicenes: Influence of Helicity on Excited-State Properties and Chiroptical Properties. J. Org. Chem. 2019, 84, 10658–10668. [Google Scholar] [CrossRef]
- Ueda, M.; Kokubun, M.; Mazaki, Y. Triskelion-shaped p-Luminophores Bearing Coumarin: Syntheses, Structures, and Luminescence Properties. ChemPhotoChem 2020, 4, 5159–5167. [Google Scholar] [CrossRef]
- Ueda, M.; Kokubn, M.; Mazaki, Y. Synthesis, Structures, and Properties of Triskelion-shaped Fluorophores Bearing Coumarins with Nitrogen-Containing Donor Group. Bull. Chem. Soc. Jpn. 2021, 94, 2906–2913. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Peterson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C. 01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Anthonu, J.E.; Khan, S.I.; Rubin, Y. 1,3,5/2,4,6-Differentiated hexaalkynylbenzenes: Absorption and fluorescence properties of a D3h-symmetric donor-substituted system. Tetrahedron Lett. 1997, 38, 3499–3502. [Google Scholar] [CrossRef]
- Bruns, D.; Miura, H.; Vollhardt, K.P.C.; Stanger, A. En Route to Archimedene: Total Synthesis of C3h-Symmetric [7]Phenylene. Org. Lett. 2003, 5, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Ni, B. Intramolecular Triple Heck Reaction. An Efficient Entry to Fused Tetracycles with a Benzene Core. J. Org. Chem. 2002, 67, 8280–8283. [Google Scholar] [CrossRef] [PubMed]
- Bushey, M.L.; Nguyen, T.-C.; Nuckolls, C. Synthesis, Self-Assembly, and Switching of One-Dimensional Nanostructures from New Crowded Aromatics. J. Am. Chem. Soc. 2003, 125, 8264–8269. [Google Scholar] [CrossRef]
- Sakurai, H.; Daiko, T.; Hirano, T. A Synthesis of Sumanene, a Fullerene Fragment. Science 2003, 301, 1878. [Google Scholar] [CrossRef]
- Del Rey, B.; Keller, U.; Torres, T.; Rojo, G.; Agulló-López, F.; Nonell, S.; Martí, C.; Brasselet, S.; Ledoux, I.; Zyss, J. Synthesis and Nonlinear Optical Photophysical, and Electrochemical Properties of Subphthalocyanines. J. Am. Chem. Soc. 1998, 120, 12808–12817. [Google Scholar] [CrossRef]
- Furukawa, S.; Suda, Y.; Kobayashi, J.; Kawashima, T.; Tada, T.; Fujii, S.; Kiguchi, M.; Saito, M. Triphosphasumanene Trisulfide: High Out-of-Plane Anisotropy and Janus-Type p-Surfaces. J. Am. Chem. Soc. 2017, 139, 5787–5792. [Google Scholar] [CrossRef] [PubMed]
- Mataga, N.; Kaifu, Y.; Koizumi, M. Solvent Effects upon Fluorescence Spectra and the Dipolemoments of Excited Molecules. Bull. Chem. Soc. Jpn. 1956, 29, 465–470. [Google Scholar] [CrossRef]
- Lippert, E.V. Spektroskopische bestimmung des dipolmomentes aromatischer verbindungen im ersten angeregten singuulettzustand. Z. Elektrochem. 1957, 61, 962–975. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).