
Effect of Process Parameters on the Graphite Expansion Produced by a Green Modification of the Hummers Method

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results
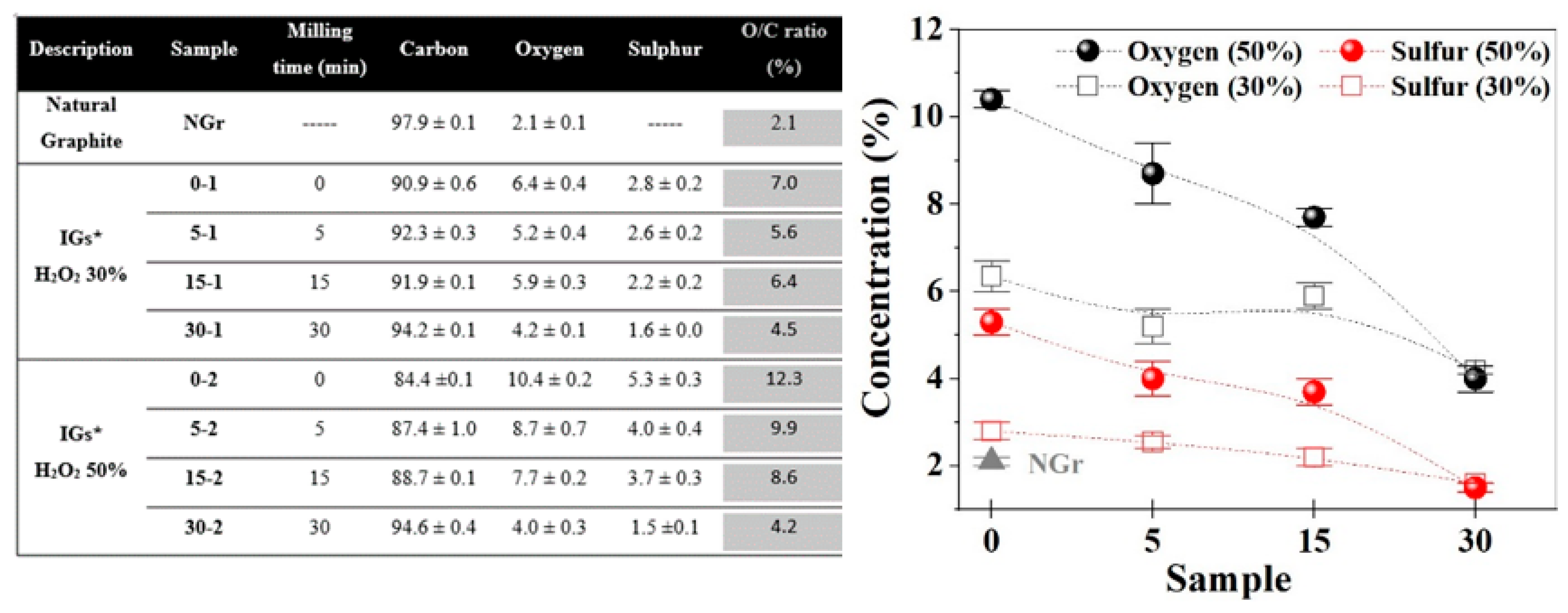
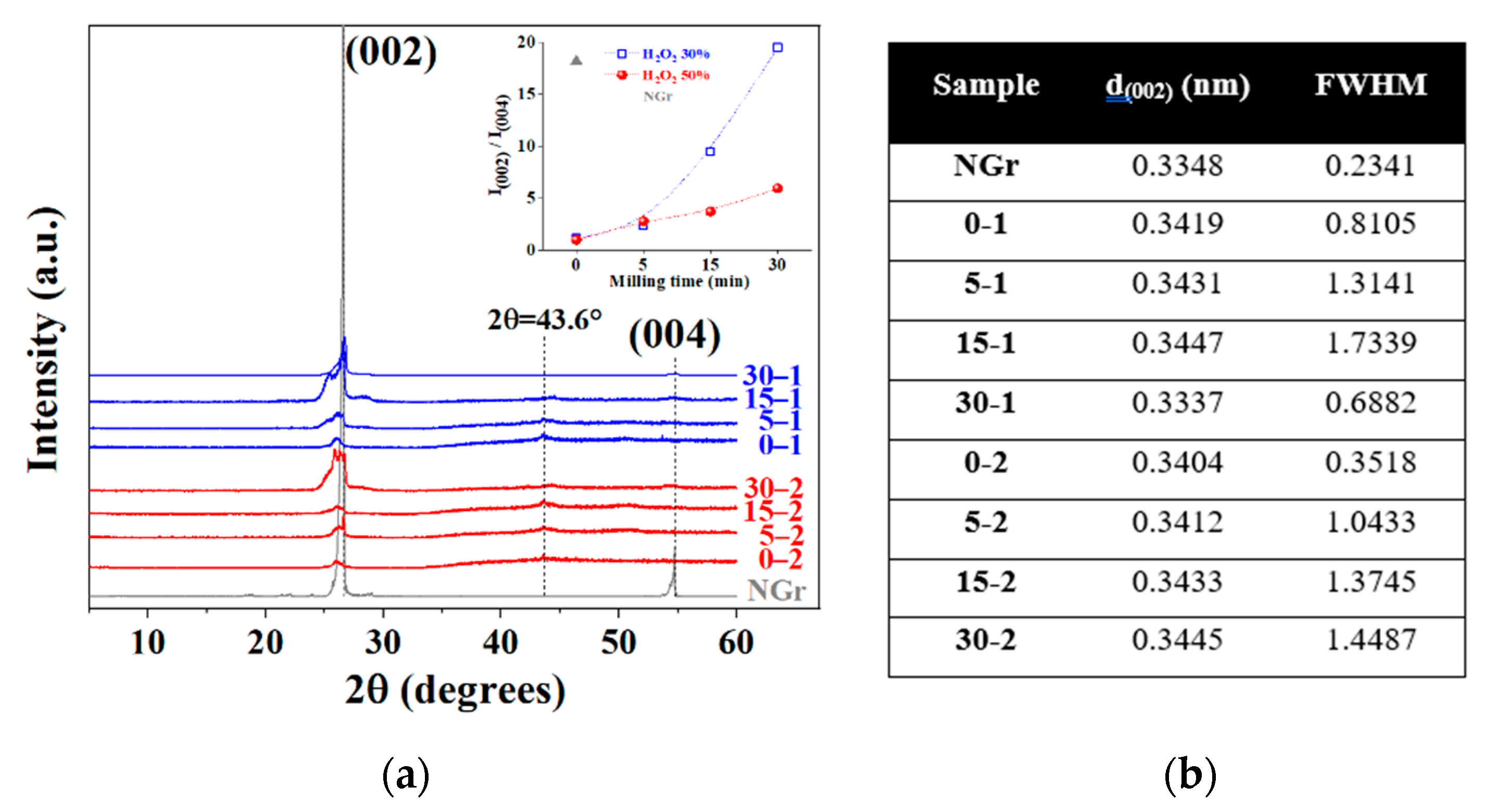
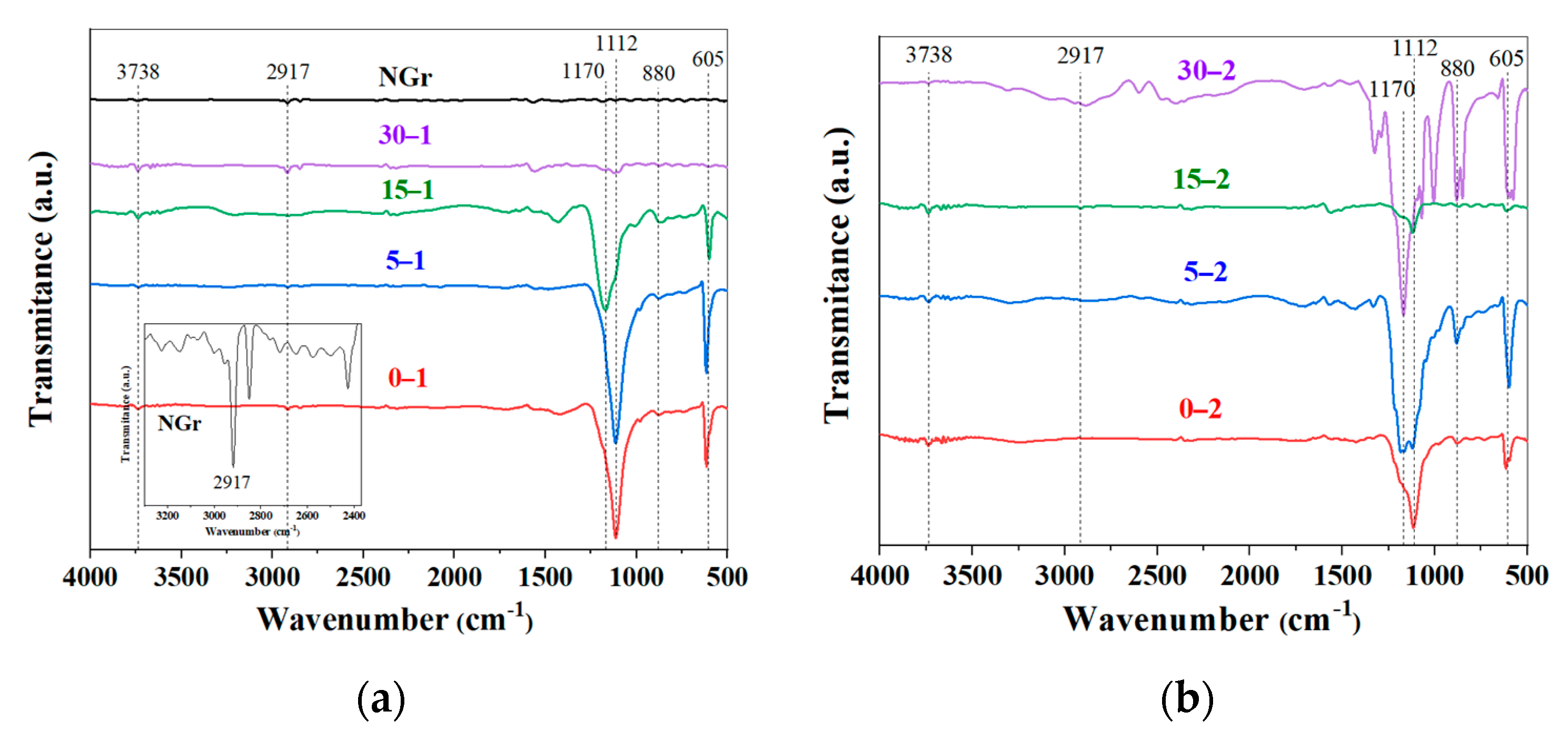
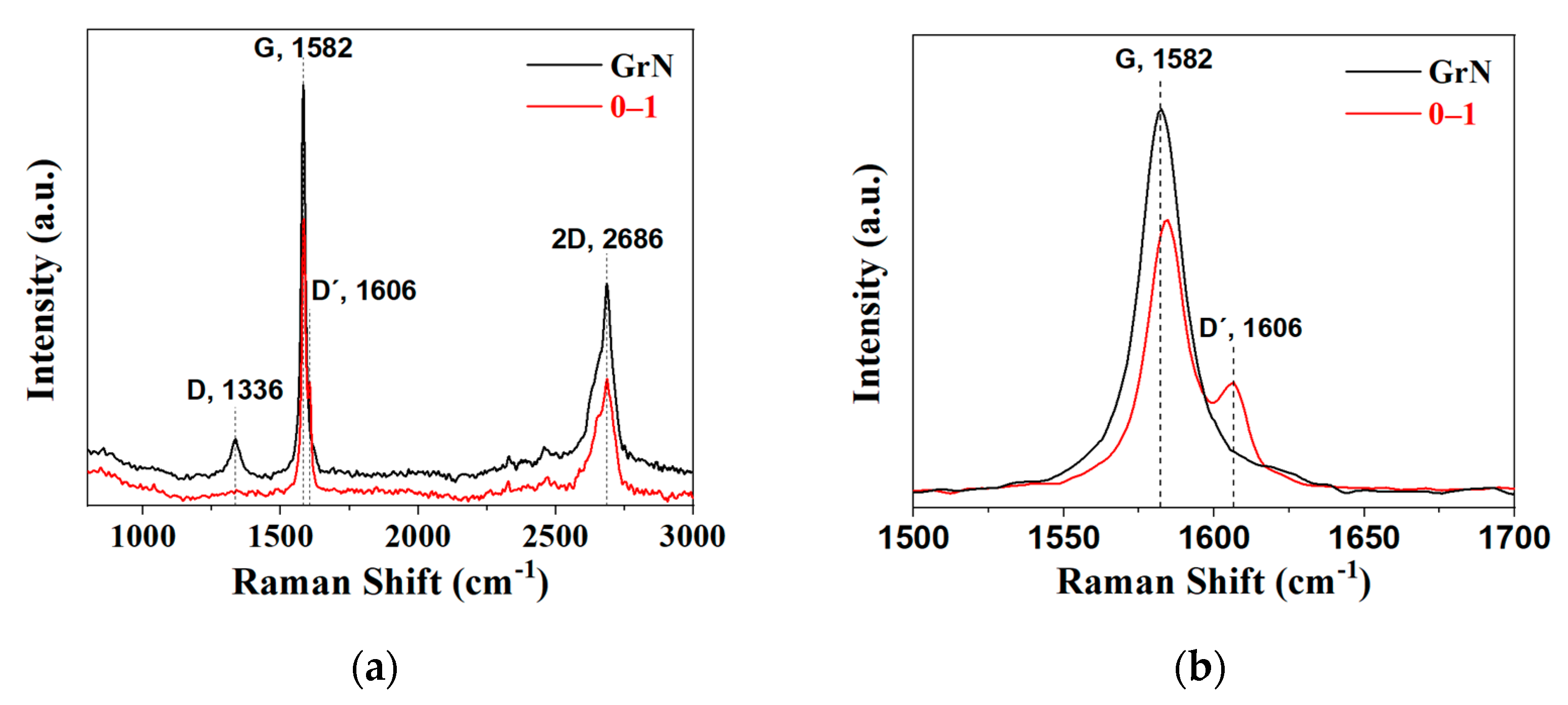
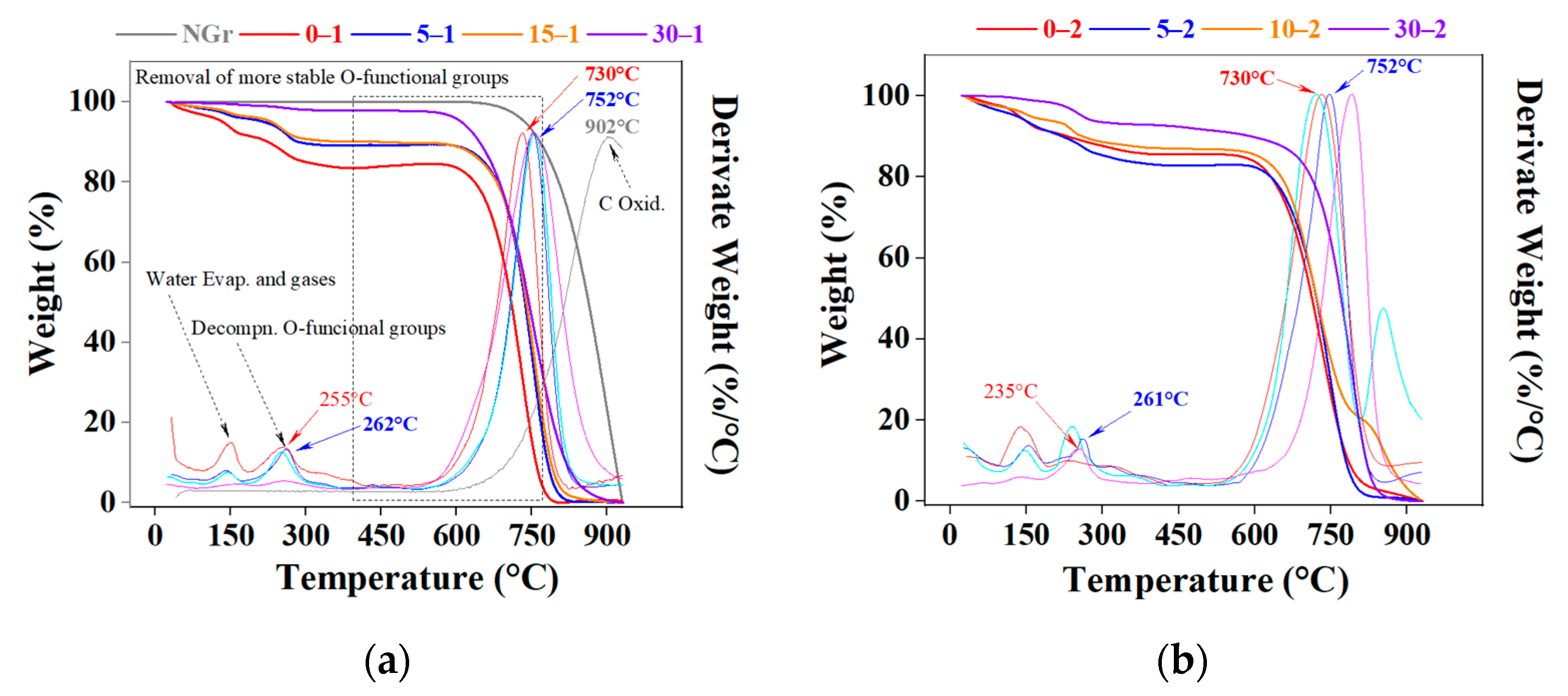
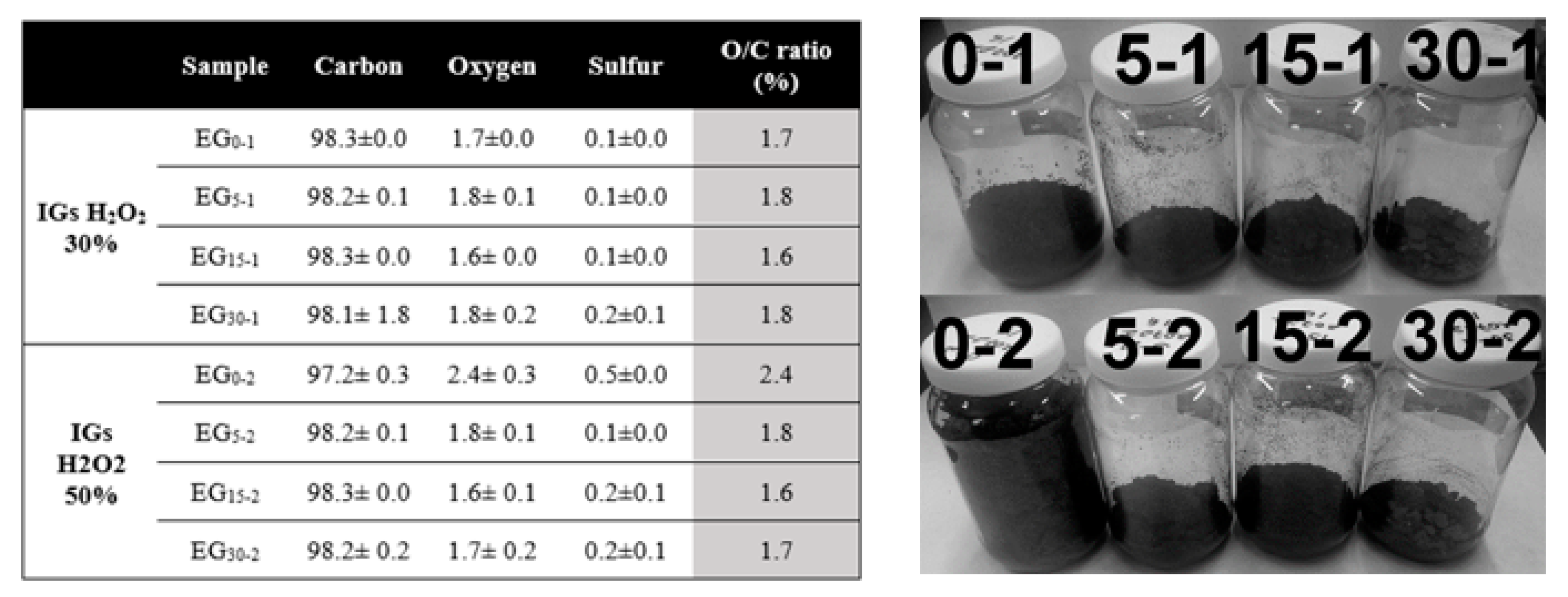
2.1. Powders Characterization
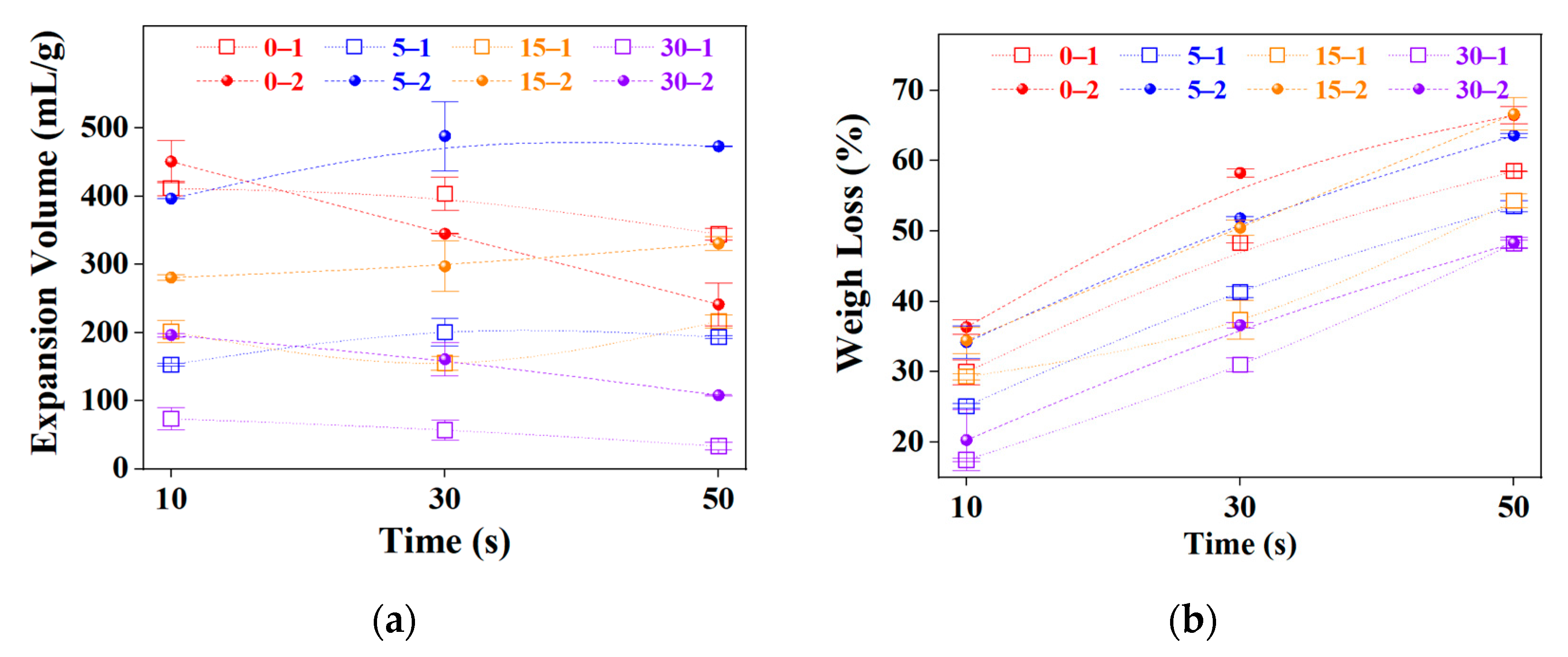
2.2. Graphite Pre-Expansion
2.3. Microwave Time Optimization
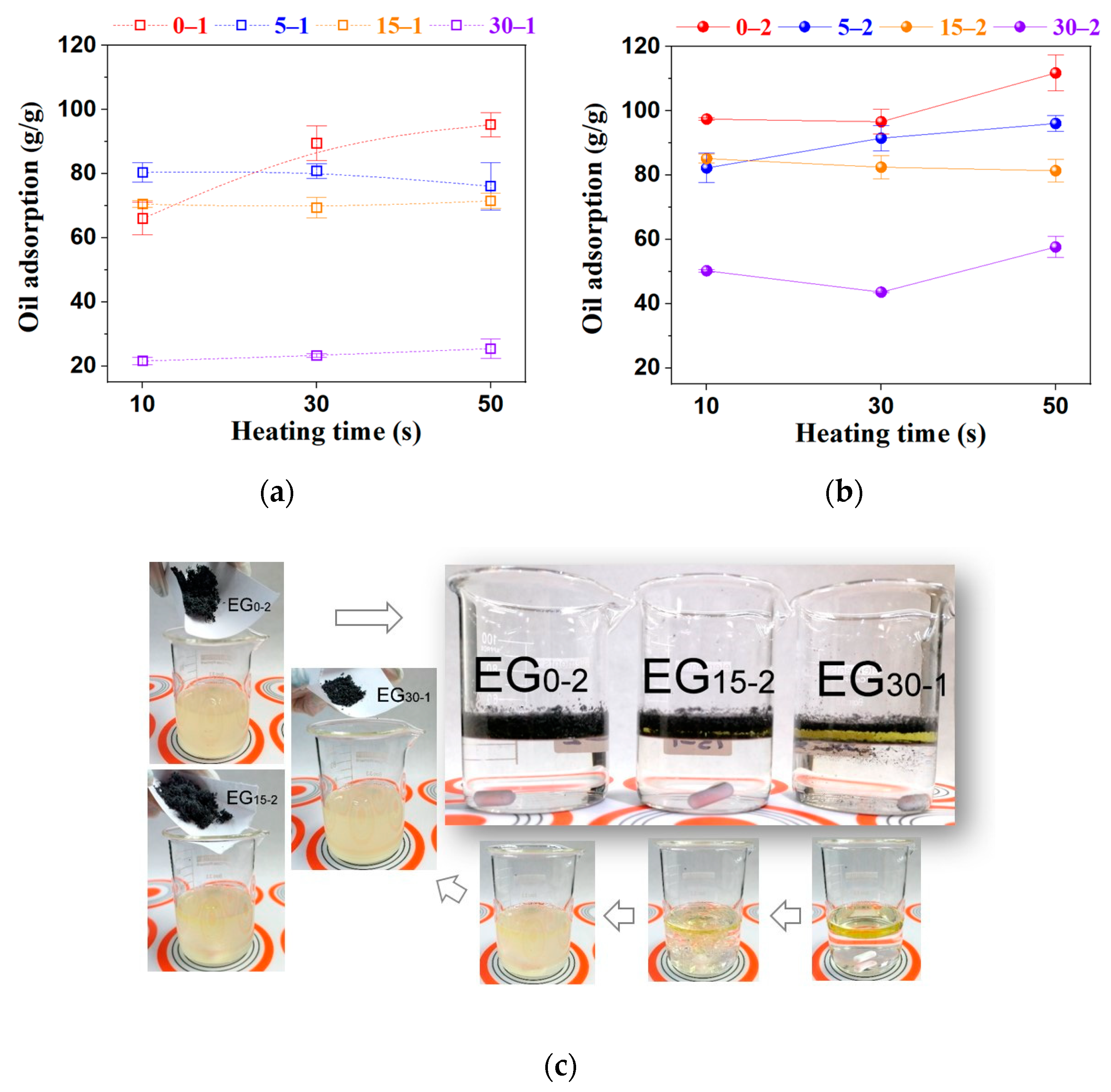
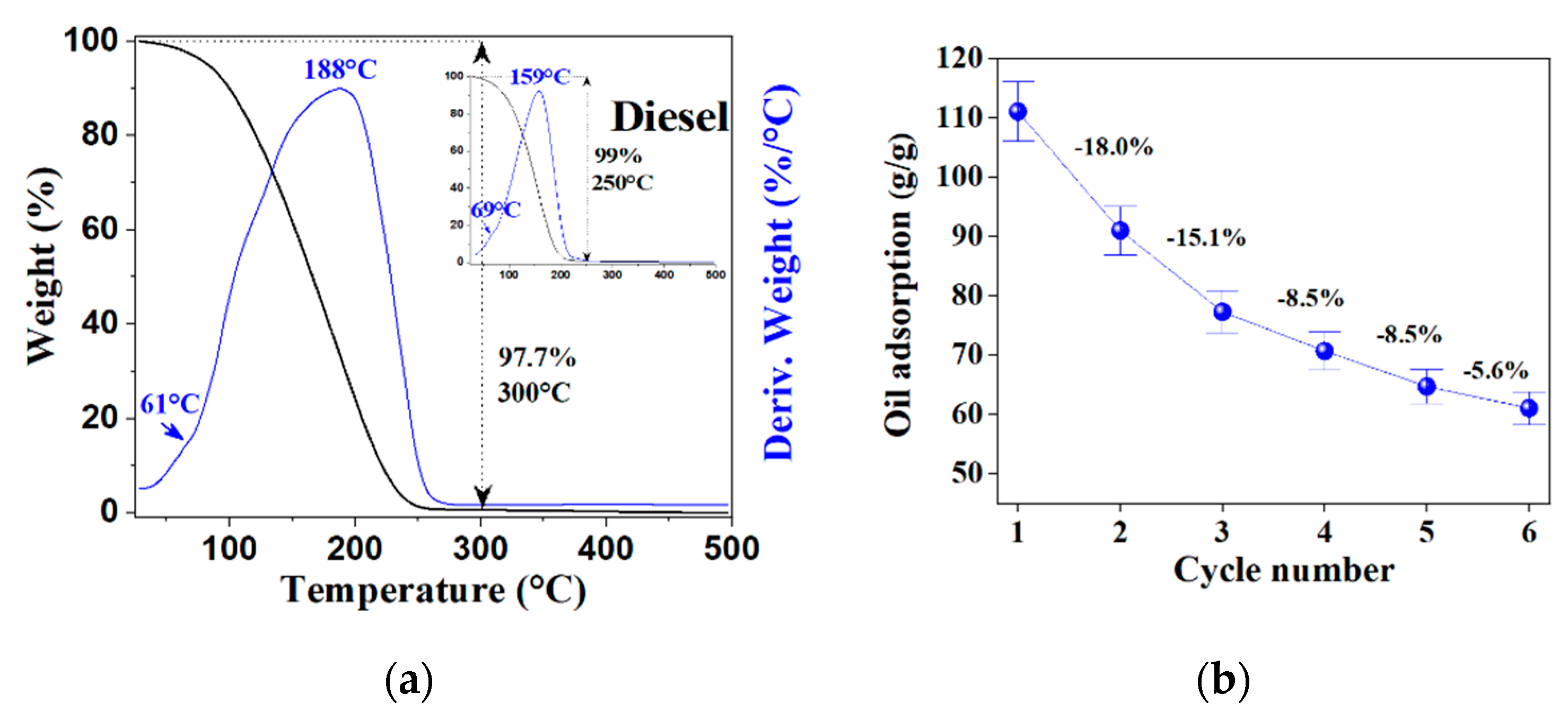
2.4. Oil Adsorption Capacity (OAC)
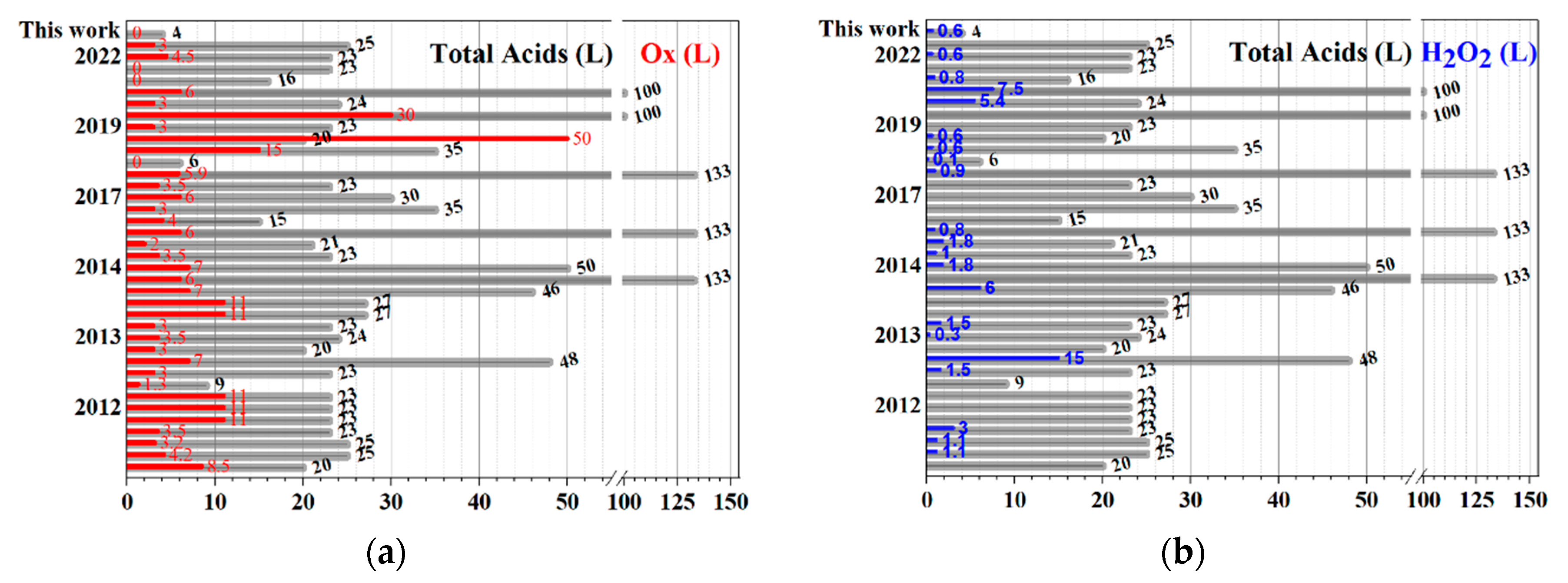
3. Discussion
4. Experimental Procedure
4.1. Materials
4.2. Intercalated Graphite (IG) Preparation
4.3. Graphite Expansion
4.4. Expansion Rate
4.5. Diesel Sorption Studies
4.6. Materials Characterization
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vocciante, M.; D’auris, A.D.F.; Reverberi, A.P. A Novel Graphite-Based Sorbent for Oil Spill Cleanup. Materials 2022, 15, 609. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Zhu, T.; Shen, W.; Kang, F.; Inagaki, M.; Huang, Z.H. Exfoliated Graphite Blocks with Resilience Prepared by Room Temperature Exfoliation and Their Application for Oil-Water Separation. J. Hazard. Mater. 2022, 424, 127724. [Google Scholar] [CrossRef] [PubMed]
- Mitter, E.K.; Germida, J.J.; de Freitas, J.R. Impact of Diesel and Biodiesel Contamination on Soil Microbial Community Activity and Structure. Sci. Rep. 2021, 11, 10856. [Google Scholar] [CrossRef] [PubMed]
- Gul Zaman, H.; Baloo, L.; Pendyala, R.; Singa, P.K.; Ilyas, S.U.; Kutty, S.R.M. Produced Water Treatment with Conventional Adsorbents and MOF as an Alternative: A Review. Materials 2021, 14, 7607. [Google Scholar] [CrossRef]
- Ewis, D.; Mahmud, N.; Benamor, A.; Ba-Abbad, M.M.; Nasser, M.; El-Naas, M. Enhanced Removal of Diesel Oil Using New Magnetic Bentonite-Based Adsorbents Combined with Different Carbon Sources. Water Air Soil Pollut. 2022, 233, 195. [Google Scholar] [CrossRef]
- Ambika, S.; Srilekha, V. Eco-Safe Chemicothermal Conversion of Industrial Graphite Waste to Exfoliated Graphene and Evaluation as Engineered Adsorbent to Remove Toxic Textile Dyes. Environ. Adv. 2021, 4, 100072. [Google Scholar] [CrossRef]
- Hou, S.; Li, J.; Huang, X.; Wang, X.; Ma, L.; Shen, W.; Kang, F.; Huang, Z.H. Silver Nanoparticles-Loaded Exfoliated Graphite and Its Anti-Bacterial Performance. Appl. Sci. 2017, 7, 852. [Google Scholar] [CrossRef]
- Bradder, P.; Ling, S.K.; Wang, S.; Liu, S. Dye Adsorption on Layered Graphite Oxide. J. Chem. Eng. Data 2011, 56, 138–141. [Google Scholar] [CrossRef]
- Xu, C.; Jiao, C.; Yao, R.; Lin, A.; Jiao, W. Adsorption and Regeneration of Expanded Graphite Modified by CTAB-KBr/H3PO4 for Marine Oil Pollution. Environ. Pollut. 2018, 233, 194–200. [Google Scholar] [CrossRef]
- Saikam, L.; Dhanpal Jayram, N.; Sykam, N. Rapid Removal of Organic Micropollutants under Aqueous Solutions Using Mesoporous Exfoliated Graphite. Available online: https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4111974 (accessed on 20 October 2022).
- Zhao, M.; Liu, P. Adsorption of Methylene Blue from Aqueous Solutions by Modified Expanded Graphite Powder. Desalination 2009, 249, 331–336. [Google Scholar] [CrossRef]
- Bannov, A.G.; Ukhina, A.V.; Maksimovskii, E.A.; Prosanov, I.Y.; Shestakov, A.A.; Lapekin, N.I.; Lazarenko, N.S.; Kurmashov, P.B.; Popov, M.V. Highly Porous Expanded Graphite: Thermal Shock vs. Programmable Heating. Materials 2021, 14, 7687. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, M.; Inagaki, M. Heavy Oil Sorption Using Exfoliated Graphite New Application of Exfoliated Graphite to Protect Heavy Oil Pollution. Carbon 2000, 38, 199–210. [Google Scholar] [CrossRef]
- EPA United States Environmental Protection Agency Basics of Green Chemistry. Available online: https://www.epa.gov/greenchemistry/basics-green-chemistry (accessed on 9 October 2022).
- Hirschvogel, A.; Wagner, F. Method for the Production of Graphite-Hydrogensulfate. United States Patent 4,091,083, 23 May 1978. [Google Scholar]
- Yoo, M.J.; Park, H.B. Effect of Hydrogen Peroxide on Properties of Graphene Oxide in Hummers Method. Carbon N. Y. 2019, 141, 515–522. [Google Scholar] [CrossRef]
- Hou, S.; He, S.; Zhu, T.; Li, J.; Ma, L.; Du, H.; Shen, W.; Kang, F.; Huang, Z.H. Environment-Friendly Preparation of Exfoliated Graphite and Functional Graphite Sheets. J. Mater. 2021, 7, 136–145. [Google Scholar] [CrossRef]
- Mahmoud, A.E.D.; Stolle, A.; Stelter, M. Sustainable Synthesis of High-Surface-Area Graphite Oxide via Dry Ball Milling. ACS Sustain. Chem. Eng. 2018, 6, 6358–6369. [Google Scholar] [CrossRef]
- Hidayah, N.M.S.; Liu, W.W.; Lai, C.W.; Noriman, N.Z.; Khe, C.S.; Hashim, U.; Lee, H.C. Comparison on Graphite, Graphene Oxide and Reduced Graphene Oxide: Synthesis and Characterization. In AIP Conference Proceedings; American Institute of Physics Inc.: College Park, MD, USA, 2017; Volume 1892. [Google Scholar]
- Muzyka, R.; Kwoka, M.; Smȩdowski, Ł.; Díez, N.; Gryglewicz, G. Oxidation of Graphite by Different Modified Hummers Methods. New Carbon Mater. 2017, 32, 15–20. [Google Scholar] [CrossRef]
- MOOSA, A.A.; ABED, M.S. Graphene Preparation and Graphite Exfoliation. Turk. J. Chem. 2021, 45, 493–519. [Google Scholar] [CrossRef]
- Tavakoli, F.; Salavati-Niasari, M.; Badiei, A.; Mohandes, F. Green Synthesis and Characterization of Graphene Nanosheets. Mater. Res. Bull. 2015, 63, 51–57. [Google Scholar] [CrossRef]
- Salvatore, M.; Carotenuto, G.; de Nicola, S.; Camerlingo, C.; Ambrogi, V.; Carfagna, C. Synthesis and Characterization of Highly Intercalated Graphite Bisulfate. Nanoscale Res. Lett. 2017, 12, 167. [Google Scholar] [CrossRef]
- Cao, J.; He, P.; Mohammed, M.A.; Zhao, X.; Young, R.J.; Derby, B.; Kinloch, I.A.; Dryfe, R.A.W. Two-Step Electrochemical Intercalation and Oxidation of Graphite for the Mass Production of Graphene Oxide. J. Am. Chem. Soc. 2017, 139, 17446–17456. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Al-Gaashani, R.; Najjar, A.; Zakaria, Y.; Mansour, S.; Atieh, M.A. XPS and Structural Studies of High Quality Graphene Oxide and Reduced Graphene Oxide Prepared by Different Chemical Oxidation Methods. Ceram. Int. 2019, 45, 14439–14448. [Google Scholar] [CrossRef]
- Kusrini, E.; Ramadhani, I.; Alhamid, M.I.; Voo, N.Y.; Usman, A. Synthesis and Adsorption Performance of Graphene Oxide-Polyurethane Sponge for Oil-Water Separation. Eng. J. 2022, 26, 1–9. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, X.; Wu, W.; Liu, T.; Liu, Y.; Guo, B.; Zhang, R. One-Step Chemical Exfoliation of Graphite to ∼100% Few-Layer Graphene with High Quality and Large Size at Ambient Temperature. Chem. Eng. J. 2019, 355, 181–185. [Google Scholar] [CrossRef]
- Krishnamoorthy, K.; Veerapandian, M.; Yun, K.; Kim, S.J. The Chemical and Structural Analysis of Graphene Oxide with Different Degrees of Oxidation. Carbon N. Y. 2013, 53, 38–49. [Google Scholar] [CrossRef]
- Chen, J.; Yao, B.; Li, C.; Shi, G. An Improved Hummers Method for Eco-Friendly Synthesis of Graphene Oxide. Carbon N. Y. 2013, 64, 225–229. [Google Scholar] [CrossRef]
- Poh, H.L.; Šaněk, F.; Ambrosi, A.; Zhao, G.; Sofer, Z.; Pumera, M. Graphenes Prepared by Staudenmaier, Hofmann and Hummers Methods with Consequent Thermal Exfoliation Exhibit Very Different Electrochemical Properties. Nanoscale 2012, 4, 3515–3522. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, B.; Bulin, C.; Li, R.; Xing, R. High-Efficient Synthesis of Graphene Oxide Based on Improved Hummers Method. Sci. Rep. 2016, 6, 36143. [Google Scholar] [CrossRef]
- Zhao, G.; Li, J.; Ren, X.; Chen, C.; Wang, X. Few-Layered Graphene Oxide Nanosheets as Superior Sorbents for Heavy Metal Ion Pollution Management. Environ. Sci. Technol. 2011, 45, 10454–10462. [Google Scholar] [CrossRef]
- Yue, X.; Wang, H.; Wang, S.; Zhang, F.; Zhang, R. In-Plane Defects Produced by Ball-Milling of Expanded Graphite. J. Alloys Compd. 2010, 505, 286–290. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Zhang, L. Green and Simple Production of Graphite Intercalation Compound Used Sodium Bicarbonate as Intercalation Agent. BMC Chem. 2022, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Zaaba, N.I.; Foo, K.L.; Hashim, U.; Tan, S.J.; Liu, W.W.; Voon, C.H. Synthesis of Graphene Oxide Using Modified Hummers Method: Solvent Influence. Procedia Eng. 2017, 184, 469–477. [Google Scholar] [CrossRef]
- Jeong, H.K.; Lee, Y.P.; Jin, M.H.; Kim, E.S.; Bae, J.J.; Lee, Y.H. Thermal Stability of Graphite Oxide. Chem. Phys. Lett. 2009, 470, 255–258. [Google Scholar] [CrossRef]
- Chua, C.K.; Sofer, Z.; Pumera, M. Graphite Oxides: Effects of Permanganate and Chlorate Oxidants on the Oxygen Composition. Chem.—A Eur. J. 2012, 18, 13453–13459. [Google Scholar] [CrossRef]
- Guerrero-Contreras, J.; Caballero-Briones, F. Graphene Oxide Powders with Different Oxidation Degree, Prepared by Synthesis Variations of the Hummers Method. Mater. Chem. Phys. 2015, 153, 209–220. [Google Scholar] [CrossRef]
- Shao, G.; Lu, Y.; Wu, F.; Yang, C.; Zeng, F.; Wu, Q. Graphene Oxide: The Mechanisms of Oxidation and Exfoliation. J. Mater. Sci. 2012, 47, 4400–4409. [Google Scholar] [CrossRef]
- Akhavan, O.; Bijanzad, K.; Mirsepah, A. Synthesis of Graphene from Natural and Industrial Carbonaceous Wastes. RSC Adv. 2014, 4, 20441–20448. [Google Scholar] [CrossRef]
- Botas, C.; Álvarez, P.; Blanco, P.; Granda, M.; Blanco, C.; Santamaría, R.; Romasanta, L.J.; Verdejo, R.; López-Manchado, M.A.; Menéndez, R. Graphene Materials with Different Structures Prepared from the Same Graphite by the Hummers and Brodie Methods. Carbon N. Y. 2013, 65, 156–164. [Google Scholar] [CrossRef]
- Zhao, J.; Dumont, J.H.; Martinez, U.; Macossay, J.; Artyushkova, K.; Atanassov, P.; Gupta, G. Graphite Intercalation Compounds Derived by Green Chemistry as Oxygen Reduction Reaction Catalysts. ACS Appl. Mater. Interfaces 2020, 12, 42678–42685. [Google Scholar] [CrossRef]
- Alsulami, I.K.; Saeed, A.; Abdullahi, S.; Alshahrie, A.; Salah, N. Nanodiamonds as a Knife for Cutting Graphene Multilayers into Ultrafine Pieces under Microwave Irradiation. FlatChem 2022, 36, 100432. [Google Scholar] [CrossRef]
- Kang, J.H.; Kim, T.; Choi, J.; Park, J.; Kim, Y.S.; Chang, M.S.; Jung, H.; Park, K.T.; Yang, S.J.; Park, C.R. Hidden Second Oxidation Step of Hummers Method. Chem. Mater. 2016, 28, 756–764. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, R.; Zhang, X.; Liu, K.; Liu, Y.; Yan, P. One-Step Room-Temperature Preparation of Expanded Graphite. Carbon N. Y. 2017, 119, 544–547. [Google Scholar] [CrossRef]
- Sun, L.; Fugetsu, B. Mass Production of Graphene Oxide from Expanded Graphite. Mater. Lett. 2013, 109, 207–210. [Google Scholar] [CrossRef]
- Sridhar, V.; Jeon, J.H.; Oh, I.K. Synthesis of Graphene Nano-Sheets Using Eco-Friendly Chemicals and Microwave Radiation. Carbon N. Y. 2010, 48, 2953–2957. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, C.; Huang, H.; Guo, W.; Yu, J.; Qiu, J. Carbon-Enabled Microwave Chemistry: From Interaction Mechanisms to Nanomaterial Manufacturing. Nano Energy 2021, 85, 106027. [Google Scholar] [CrossRef]
- You, S.; Luzan, S.M.; Szabó, T.; Talyzin, A.V. Effect of Synthesis Method on Solvation and Exfoliation of Graphite Oxide. Carbon N. Y. 2013, 52, 171–180. [Google Scholar] [CrossRef]
- Sykam, N.; Jayram, N.D.; Rao, G.M. Highly Efficient Removal of Toxic Organic Dyes, Chemical Solvents and Oils by Mesoporous Exfoliated Graphite: Synthesis and Mechanism. J. Water Process Eng. 2018, 25, 128–137. [Google Scholar] [CrossRef]
- Diraki, A.; Mackey, H.R.; Mckay, G.; Abdala, A. Removal of Emulsified and Dissolved Diesel Oil from High Salinity Wastewater by Adsorption onto Graphene Oxide. J. Environ. Chem. Eng. 2019, 7, 103106. [Google Scholar] [CrossRef]
- Inagaki, M.; Toyoda, M.; Iwashita, N.; Nishi, Y.; Konno, H. Exfoliated Graphite for Spilled Heavy Oil Recovery. Carbon Lett. 2001, 2, 1–8. [Google Scholar]
- Jaafar, M.; Alatabe, A. Sustainable Oil Adsorption from Produced Water Using Cane Papyrus as Natural Biosorbent. Preprints 2021, 2021100157. [Google Scholar] [CrossRef]
- Wilson, N.R.; Pandey, P.A.; Beanland, R.; Young, R.J.; Kinloch, I.A.; Gong, L.; Liu, Z.; Suenaga, K.; Rourke, J.P.; York, S.J.; et al. Graphene Oxide: Structural Analysis and Application as a Highly Transparent Support for Electron Microscopy. ACS Nano 2009, 3, 2547–2556. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Li, B.; Qu, Z.G.; Zhang, J.F.; Jin, Z.G. Effects of Graphite Microstructure Evolution on the Anisotropic Thermal Conductivity of Expanded Graphite/Paraffin Phase Change Materials and Their Thermal Energy Storage Performance. Int. J. Heat Mass Transf. 2020, 155, 119853. [Google Scholar] [CrossRef]
- Lavin-Lopez, M.P.; Valverde, J.L.; Sanchez-Silva, L.; Romero, A. Solvent-Based Exfoliation via Sonication of Graphitic Materials for Graphene Manufacture. Ind. Eng. Chem. Res. 2016, 55, 845–855. [Google Scholar] [CrossRef]
- Shribman, S.; Davis, A.; Vella, N.; Giovannoni, G. Heavy Metal Toxicity: Lessons from a Case of Simultaneous Occupational Exposure to Manganese and Mercury. J. Neurol. Neurosurg. Psychiatry 2013, 84, e2. [Google Scholar] [CrossRef]
- Wang, K.; Ruan, J.; Song, H.; Zhang, J.; Wo, Y.; Guo, S.; Cui, D. Biocompatibility of Graphene Oxide. Nanoscale Res. Lett. 2011, 6, 8. [Google Scholar] [CrossRef]
- Eigler, S.; Dotzer, C.; Hof, F.; Bauer, W.; Hirsch, A. Sulfur Species in Graphene Oxide. Chem.—A Eur. J. 2013, 19, 9490–9496. [Google Scholar] [CrossRef]
- Chng, E.L.K.; Pumera, M. The Toxicity of Graphene Oxides: Dependence on the Oxidative Methods Used. Chem.—A Eur. J. 2013, 19, 8227–8235. [Google Scholar] [CrossRef]
- Alghyamah, A.A.; Haider, S.; Khalil, U.; Khan, R.; Haider, A.; Almasry, W.A.; Ihsan, R.; Perveen, T.; Wazeer, I.; Chafidz, A. Synthesis and Characterization of Graphene Oxide, Reduced Graphene Oxide and Their Nanocomposites with Polyethylene Oxide. Curr. Appl. Phys. 2022, 40, 1–11. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year, [Reference] | Total Acids = (L) | Total Oxidants (Kg) | H2O2 (100%, L) |
|---|---|---|---|
| 2009, [37] | 20.0 | 8.5 | |
| 2011, [59] | 25.0 | 4.2 | 1.05 |
| 2011, [8] | 25.0 | 3.2 | 1.05 |
| 2012, [40] | 23.0 | 3.5 | 3 |
| 2012, [31] | 22.9 8.5 | 11.0 1.3 | |
| 2013, [30] | 23.3 | 3.0 | 1.5 |
| 2013, [42] | 48.0 | 7.0 | 15 |
| 2013, [47] | 20.0 | 3.0 | |
| 2013, [60] | 24.0 | 3.5 | 0.3 |
| 2013, [29] | 22.5 | 3.0 | 1.5 |
| 2013, [61] | 26.5 46.0 133.3 | 11.0 7.0 6.0 | 6 |
| 2014, [41] | 50.0 | 7.0 | 1.8 |
| 2015, [39] | 23.0 | 3.5 | 1 |
| 2015, [22] | 21.3 | 2.0 | 1.8 |
| 2017, [19] | 133.3 | 6.0 | 0.81 |
| 2017, [20] | 15.0 35.0 30.0 22.5 | 4.0 3.0 6.0 3.5 | |
| 2017, [36] | 133.3 | 5.9 | 0.9 |
| 2017, [7] | 6.0 | 0.0 | 0.06 |
| 2017, [23] | 35.0 20 | 15.0 50 | 0.6 0.6 |
| 2019, [16] | 23.0 | 3.0 | |
| 2019, [28] | 100.0 | 30.0 | |
| 2019, [62] | 24.5 | 3.0 | 5.4 |
| 2019, [26] | 100.0 | 6.0 | 7.5 |
| 2021, [17] | 16.0 | 0.0 | 0.84 |
| 2021, [21] | 22.9 | 0.0 | |
| 2022, [5] | 23.0 | 4.5 | 0.6 |
| 2022, [2] | 25.0 | 3.0 | |
| Average | 37.8 ± 5.6 | 7.1 ± 1.5 | 2.6 ± 0.8 |
| This work | 4.0 | 0.0 | 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarango-Rivero, G.; Mendoza-Duarte, J.M.; Santos-Beltrán, A.; Estrada-Guel, I.; Garay-Reyes, C.G.; Pizá-Ruiz, P.; Gómez-Esparza, C.D.; Rocha-Rangel, E.; Martínez-Sánchez, R. Effect of Process Parameters on the Graphite Expansion Produced by a Green Modification of the Hummers Method. Molecules 2022, 27, 7399. https://doi.org/10.3390/molecules27217399
Tarango-Rivero G, Mendoza-Duarte JM, Santos-Beltrán A, Estrada-Guel I, Garay-Reyes CG, Pizá-Ruiz P, Gómez-Esparza CD, Rocha-Rangel E, Martínez-Sánchez R. Effect of Process Parameters on the Graphite Expansion Produced by a Green Modification of the Hummers Method. Molecules. 2022; 27(21):7399. https://doi.org/10.3390/molecules27217399
Chicago/Turabian StyleTarango-Rivero, Gabriela, José M. Mendoza-Duarte, Audel Santos-Beltrán, Ivanovich Estrada-Guel, Carlos G. Garay-Reyes, Pedro Pizá-Ruiz, Cynthia D. Gómez-Esparza, Enrique Rocha-Rangel, and Roberto Martínez-Sánchez. 2022. "Effect of Process Parameters on the Graphite Expansion Produced by a Green Modification of the Hummers Method" Molecules 27, no. 21: 7399. https://doi.org/10.3390/molecules27217399
APA StyleTarango-Rivero, G., Mendoza-Duarte, J. M., Santos-Beltrán, A., Estrada-Guel, I., Garay-Reyes, C. G., Pizá-Ruiz, P., Gómez-Esparza, C. D., Rocha-Rangel, E., & Martínez-Sánchez, R. (2022). Effect of Process Parameters on the Graphite Expansion Produced by a Green Modification of the Hummers Method. Molecules, 27(21), 7399. https://doi.org/10.3390/molecules27217399