1. Introduction
Antisense oligonucleotides (ASOs), composed of approximately 20-bp DNA-like nucleotides, are classified as a kind of mRNA-targeted oligonucleotide therapeutics [
1]. Since the first ASO therapeutic, fomivirsen, was approved by the U.S. Food and Drug Administration (FDA) in 1998, nine ASO therapeutics have been approved [
2,
3,
4,
5,
6,
7,
8,
9,
10]. There is no doubt that further research on ASOs will be developed. ASOs can be divided into the following two types depending on the antisense mechanism: the ribonuclease H (RNase H)-dependent type and the splice-switching type [
11,
12,
13]. Both types of ASOs need to be taken up into cytoplasm or nucleoplasm for binding with the targeted mRNAs or pre-mRNAs, which would raise several challenges for the clinical application of ASOs. For example, nucleotides experience difficulty passing through cellular or nuclear membranes because of the negative charges on their phosphodiester (PO) linkages. Moreover, natural DNA strands are quickly degraded via nuclease-mediated hydrolysis inside the plasm, resulting in low pharmacological effects. To overcome these challenges, chemically modified nucleosides have been developed and utilized in approved ASOs.
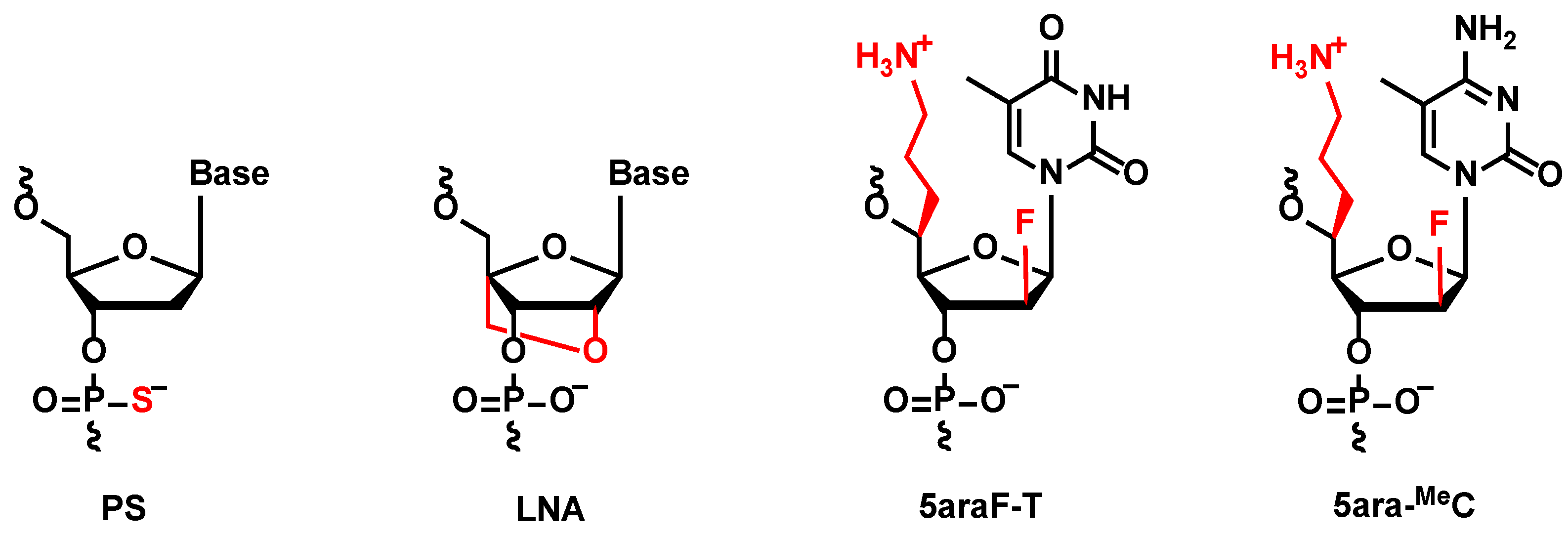
Phosphorothioate (PS) linkages, which contain sulphur substitutions of non-bridging oxygens at each PO linkage, are the most popular chemical modification to improve the properties of approved ASOs [
3,
4,
5,
6,
7,
8,
9,
10]. Due to the high degree of similarity between sulfur and oxygen atoms, PS linkages would not obstruct the recognition of ASO/RNA duplexes by RNase H and might even promote the RNase H-mediated cleavage mechanism [
14]. PS linkages also help to stabilize ASOs from nucleolytic degradation. In addition, the anionic sulfur in PS linkages could tightly interact with multiple proteins by electrostatic and hydrophobic interactions [
15]. Therefore, the application of PS linkages could enhance the membrane permeability of ASOs via binding to membrane proteins. However, the presence of apoptosis of off-target cells, relating to cytotoxicity, was reported in some cases, possibly due to unfavorable interactions between PS linkages and paraspeckle proteins [
15]. As general strategies, coordination with other chemical modifications and reducing the content of PS linkages might be beneficial for developing more effective PS-ASOs with less side effects [
16,
17,
18,
19,
20].
In previous studies, our group designed and synthesized various chemically modified nucleoside analogs containing an aminoalkyl sidechain, which is expected to strengthen nuclease resistance and improve the intracellular internalization of oligonucleotides [
21,
22,
23,
24,
25,
26,
27]. As a result of evaluating (
S)-5ʹ-
C-aminopropyl- (
5deo) and (
S)-5ʹ-
C-aminopropyl-2ʹ-arabinofluoro- (
5ara) modified ASOs, it was revealed that ASOs with several
5ara modifications could significantly enhance the nuclease resistance and show a superior ability to activate the RNase H-dependent antisense mechanism compared with the
5deo-modified ones, although the cleavage of the complementary RNA strands would be obviously impeded compared with the natural DNAs [
27].
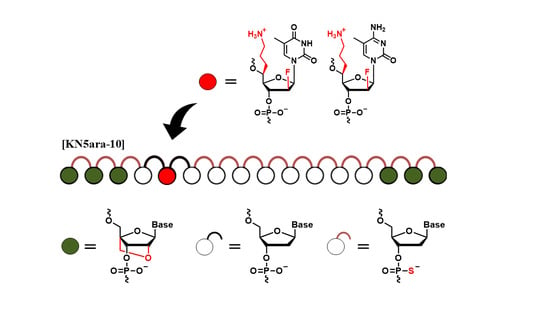
In this study, we focus on a special formation, named “gapmer”, composed of chemically modified wing regions for high RNA-binding affinity and robust nuclease resistance, and a DNA-based gap region for efficient RNase H-dependent antisense mechanism [
3,
6,
7].
We also focus on locked nucleic acids (LNAs), a class of chemically modified nucleosides, which contain a methylene bridge connecting the 2′ oxygen and 4′ carbon of nucleoside [
28,
29]. LNA demonstrated high RNA-binding affinity because it locks the ribose conformation into a C3′-endo type, which is beneficial for the A-type formation of ASO/RNA duplex, while the incorporation of LNAs inside oligonucleotides might suppress the recognition of RNase H. Therefore, LNA is often used in the wing regions of gapmers but not in the gap regions. However, even superior properties have been proved in the laboratory level, due to a lack of clinical knowledge, as there are not any cases yet approving LNA-containing ASOs. In this study, an LNA-wing antisense gapmer was selected as an excellent positive control, and the further improvements resulting from the collaboration with
5ara modification were evaluated, as well as the expedition of LNA into clinical usage.
The
KNTC2 (Kinetochore associated 2) gene, referred as to
KNTC2, was selected to be the target gene in this study, which is known to be highly expressed in various cancer cells. Selective knockdown of
KNTC2 was reported as a promising treatment for cancers [
30,
31,
32]. Based on previous findings, we designed a series of
KNTC2-targeted antisense gapmers with LNA-wings and
5ara modifications (described as “KN5ara gapmers”) in this study. Each structure of these chemical modifications is shown in
Figure 1. The following sections outline the synthesis of KN5ara gapmers by incorporating the reported (
S)-5ʹ-
C-aminopropyl-2ʹ-arabinofluoro-thymidine (
5ara-T) and newly synthesized (
S)-5ʹ-
C-aminopropyl-2ʹ-arabinofluoro-5-methyl-cytidine (
5ara-MeC) analogs, followed by the in vitro evaluations of their functional properties.
2. Results and Discussion
2.1. Oligonucleotide Synthesis
The synthesis of phosphoramidite corresponding to
5ara-MeC is shown in
Scheme S1 in the Supporting Information. The modified nucleoside analogs
5ara-T and
5ara-MeC were incorporated into a series of LNA-wing antisense gapmers utilizing a DNA/RNA synthesizer via the solid-phase phosphoramidite method. After synthesis, to prevent the additional reaction of acrylonitrile with 5ʹ-
C-aminopropyl groups, the controlled-pore glass (CPG) beads were treated with 10% dimethylamine in MeCN at room temperature for 5 min, followed by rinsing with MeCN to selectively remove cyanoethyl groups. The gapmers were then cleaved from CPG beads and deprotected by treatment with a concentrated NH
3 solution for 12 h at 55 °C. The corresponding RNA oligomers used in this study were prepared with a DNA/RNA synthesizer. After synthesis, in contrast to antisense gapmers, the RNA oligomers were cleaved from CPG beads and deprotected by treatment with concentrated NH
3 solution/40% methylamine (1:1,
v/
v) for 10 min at 65 °C. Then, 2ʹ-
O-TBDMS groups in RNA oligomers were removed using Et
3N∙3HF (125 µL) in DMSO (100 µL) for 1.5 h at 65 °C. The reaction was quenched with a 0.1 M TEAA buffer (pH 7.0) and the mixture was desalted using a Sep-Pak C18 cartridge. The modified antisense gapmers and RNA oligomers were finally purified by 20% denaturing polyacrylamide gel electrophoresis (PAGE) containing 7 M urea. The sequences of the oligonucleotides used in this study are shown in
Table 1 and
Table S1 (Supplementary Material).
2.2. RNA-Binding Affinity
The accurate binding of ASOs to target mRNA is necessary for RNase H recognition and the following antisense mechanism. As reported before, although PS linkages negatively affected thermal stability, LNAs could significantly increase RNA binding affinity and a single (
S)-5ʹ-
C-Aminopropyl-2ʹ-arabinofluoro modification caused few changes to the 50% melting temperature (
Tm) values of ASO/RNA duplexes [
27,
28,
29]. Therefore, we hypothesized that the series of KN5ara gapmers would have sufficient RNA binding affinity for therapeutic application. In this study, each KN5ara gapmer was mixed in the same volume of cRNA-1, and then annealed to form ASO/RNA duplexes. Temperature-induced melting was measured by ultraviolet (UV) spectroscopy in a 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl, and
Tm values were obtained from melting curves using the standard method. Each Δ
Tm was calculated from [
Tm (duplex containing each KN5ara gapmers) −
Tm (duplex containing KN-pos)].
As shown in
Table 1, all KN5ara gapmers maintained a stable duplex structure with complementary RNA strands at 37 °C. The incorporation of
5ara-modified analogs in the gap region showed moderate effects on
Tm values, while the replacement of LNAs in the wing region with
5ara-T or
5ara-MeC resulted in thermal destabilization. These results are consistent with previous studies showing that the thermal stability of
5ara modification is comparable to that of natural DNA but lower than that of LNA [
27]. Furthermore, the
Tm value of KN5ara-9 was similar to the addition of KN5ara-7 and KN5ara-8, indicating that the continuous introduction of modified analogs might not have an additional impact on thermal stability. The gapmers with fewer PS linkages (KN5ara-10 and KN5ara-11) were more stable than the corresponding full PS gapmers (KN5ara-3 and KN5ara-6).
2.3. The Ability for E. coli RNase H1 Activation
Before the in vitro cell experiments, we established a simple enzymatic reaction system using RNase H1 purified from
E. coli to determine whether RNase H-mediated cleavage could be activated by KN5ara gapmers [
33,
34]. The KN5ara gapmers used in this experiment were mixed with fluorescein-labeled cRNA-2 at 1:5 before annealing. These duplexes were dissolved in a buffer containing 50 mM Tris–HCl (pH 8.0), 75 mM KCl, 3 mM MgCl
2, and 10 mM dithiothreitol. Diluted
E. coli RNase H1 solution (60 unit/L in H
2O) was then added, and the mixture was incubated at 37 °C for the required time (0, 1, 5, 15, and 30 min and 1, 2, and 4 h). The aliquots were analyzed using 20% denaturing PAGE and then quantified using a luminescent image analyzer LAS-4000 (Fujifilm). As shown in
Figure 2, even though the initial reaction velocity showed a slight change, it was confirmed that the complete cRNA strand was almost cleaved in KN5ara gapmers as well as in the positive control KN-pos after a 5 min reaction. Despite differences in each cleavage pattern, a single
5ara modification moderately affected the cleavage mechanism of
E. coli RNase H1, which was consistent with previous reports, because of the remaining recognition portions [
27].
Notably, not all KN5ara gapmers were evaluated in this assay. Since the recognition of E. coli RNase H1 would begin with a few nucleotides from the 5ʹ-terminal of ASO, KN5ara-1 and -2, in which the 5ara modification was inserted into 5ʹ-gap region, it might activate E. coli RNase H1 similarly with KN-pos. Meanwhile, as there are no significant differences for KN5ara-6 and -7, KN5ara-4 and -5 were found to be similar to the others. The antisense activity of all KN5ara gapmers is directly evaluated in the following section.
ASOs containing multiple
5ara modifications showed extremely high enzyme tolerance in a 3% bovine serum (BS) experiment system [
27]. LNAs and PS linkages are also expected to improve the stability of KN5ara gapmers during enzyme-mediated hydrolysis. However, PS linkages may lead to undesirable apoptosis because of their high affinity with plasma proteins, such as paraspeckle proteins [
15]. We hypothesized that the application of the
5ara modification could reduce the number of PS linkages while maintaining sufficient nuclease resistance. In this research, a series of fluorescein-labeled KN5ara gapmers were synthesized (
Table S1), including positive control (KN-pos-F), and gapmers with full-PS (KN5ara-6-F) or with less PS linkages (KN5ara-10/11-F). For comparison, the gapmer without any
5ara modification corresponding to KN5ara-11-F was prepared as well. The gapmers described above were dissolved in OPTI-MEM and incubated with 50% BS at 37 °C. During incubation, aliquots from the reactions were taken for the required time (0, 1, 3, 6, 12, 24 and 48 h), then analyzed with 20% PAGE containing 7 M urea and quantified using the Luminescent Image analyzer LAS-4000 (Fujifilm). The results are shown in
Figure 3.
After 48 h incubation, approximately 44% of the KN-pos-F strands remained, while only 26% of KN5ara-12-F strands persisted, apparently owing to the decrease in the two PS linkages. A comparison of KN-pos-F and KN5ara-6-F showed that 5ara modification could further increase the nuclease resistance of gapmers, resulting in a half-life of >48 h with 50% BS treatment. Meanwhile, 44% and 42% of the complete strands of KN5ara-10-F and KN5ara-11-F remained, respectively, indicating the same nuclease resistance as KN-pos-F. It is presumed that incorporating one 5ara modification into the two PO linkages could reverse destabilization in enzymatic hydrolysis, which is caused by fewer PS linkages.
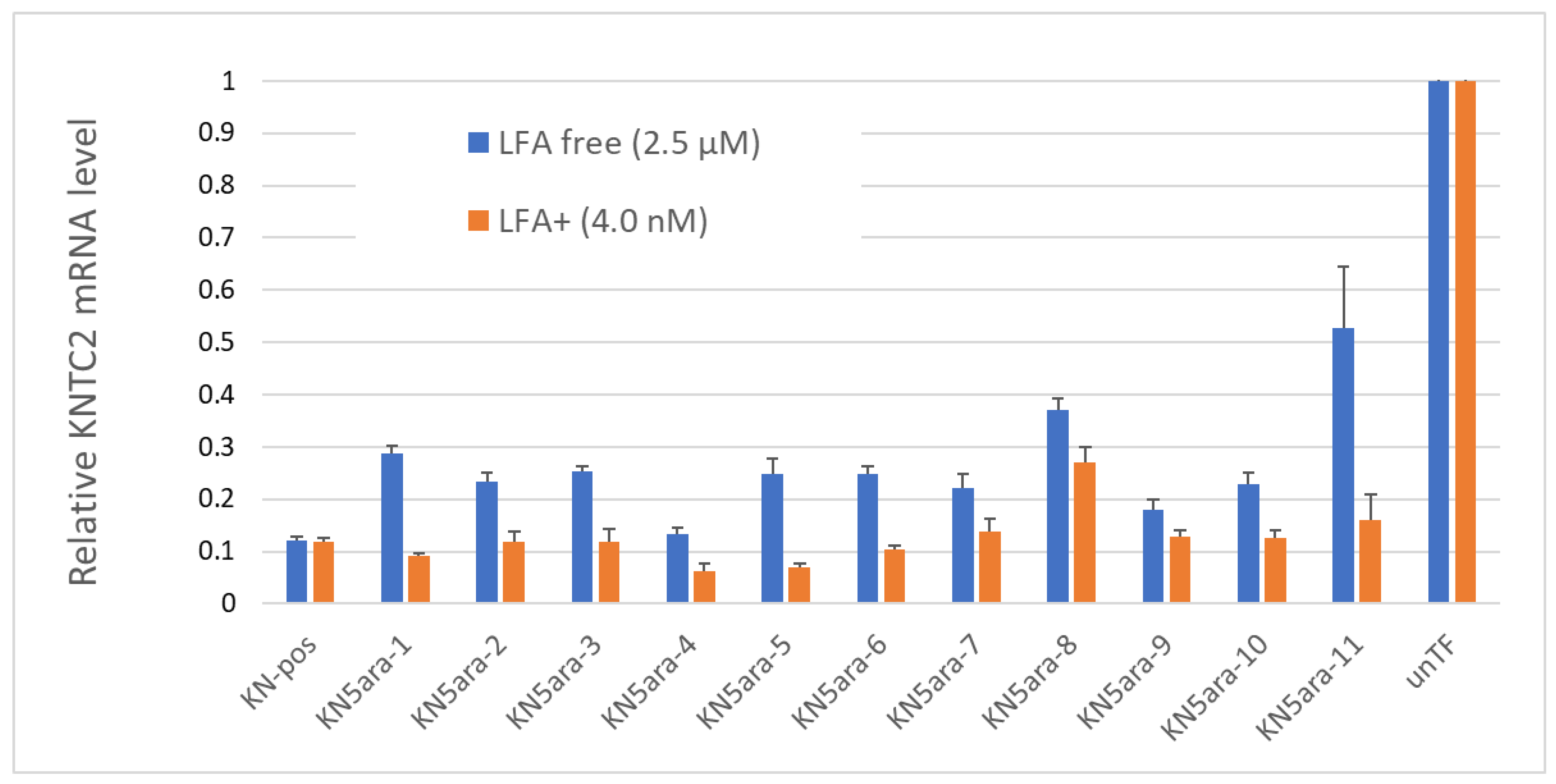
2.4. Antisense Activity
In addition to the physical experiments and simple in vitro enzymatic assays, in vitro cellular experiments were performed to knock down
KNTC2 in A549tGFP cells to evaluate the antisense activity of each KN5ara gapmer. The pre-cultured A549tGFP cells were treated with KN5ara gapmers at a final concentration of 4.0 nM or 2.5 µM, and were transfected with 0.3% Lipofectamine
® 2000 (LFA) or not, respectively. After incubation, total mRNA inside the cells was extracted, followed by reverse transcription of the targeted
KNTC2 mRNA. A quantitative real-time polymerase chain reaction (qRT-PCR) was performed in duplicate, and relative
KNTC2 mRNA levels were calculated, as shown in
Figure 4.
The results suggested that there was no obvious change in the antisense activity of KN5ara gapmers with a single 5ara modification when LFA was used. KN5ara-4/5 showed much higher KNTC2 knockdown efficiency than the positive control KN-pos, although KN5ara-8 showed a 2-fold decrease. Interestingly, compared to KN5ara-8, KN5ara-9, which has another 5ara modification at the same site as KN5ara-7, maintained antisense activity similar to that of KN5ara-7, suggesting that continuous 5ara modifications may induce further improvement. Contrastingly, KN5ara-3 and KN5ara-10 exhibited similar antisense activity, whereas KN5ara-11 had slightly lower activity than KN5ara-6.
In the LFA-free group, KN-pos showed antisense activity comparable to that in the LFA+ condition. All KN5ara gapmers were clearly less active, indicating that cellular uptake might be reduced, although the extent of the reduction varied greatly depending on the site of the 5ara modification. Meanwhile, KN5ara-4 efficiently knocked down KNTC2 mRNA, and the decrease in antisense activity of KN5ara-8 was also reversed by continuous 5ara modifications (KN5ara-9). Notably, significant property degradation was observed in KN5ara-11, as compared to KN5ara-6, whereas KN5ara-10 showed similar or slightly better antisense activity than KN5ara-3.
In more detail, it is obvious that KN5ara-4 showed the most effective antisense activity in both methods, when used with lipofection or not. The relative
KNTC2 mRNA level tended to increase according to the shift of
5ara modification to the 3ʹ-terminal, which suggests
5ara modification might be accepted well in the center of the gap region. Further investigation is need, however, of the synthesis of
5ara modified adenosine and guanosine analogs. Moreover, according to previous studies, the incorporation of a single 2ʹ-OMe modification at gap position 2 could reduce the PS-derived toxicity while maintaining enough antisense activity [
16]. In this study, KN5ara-3 and -10 obtained a
5ara modification at gap position 2; additionally, KN5ara-10 retained even less antisense activity for PS linkages. Therefore, KN5ara-10 might be the most promising candidate for reducing PS-derived cytotoxicity. Further experiments studying the cytotoxicity are planned.
3. Conclusions
In summary, the novel synthesis of (S)-5ʹ-C-Aminopropyl-2ʹ-arabinofluoro-5-methyl-cytidine (5ara-MeC) was accomplished in this study. The properties of a series of LNA-wing antisense gapmers containing (S)-5ʹ-C-Aminopropyl-2ʹ-arabinofluoro (5ara) modification, named KN5ara gapmers, were evaluated. It was revealed that each KN5ara gapmer could bind to the complementary RNA strand with high affinity and could be thermally stable under biological condition. The ability for E. coli RNase H1 activation was moderately affected by the incorporation of a single 5ara modification, although the cleavage pattern was obviously changed, which is consistent with previous reports. To determine whether the 5ara modification could be an alternative to PS linkage, the nuclease resistance of several KN5ara gapmers was evaluated using a simple enzymatic reaction system. As a result, incorporating one 5ara modification inside the two PO linkages could reverse the destabilization in enzymatic hydrolysis caused by fewer PS linkages. In addition, in vitro cellular experiments were performed to knockdown KNTC2 in A549tGFP cells. The LFA+ group showed a relatively higher antisense activity than the LFA-free group, while KN5ara-4 showed superior antisense activity in both methods. Compared to KN5ara-7, KN5ara-8, and KN5ara-9, continuous 5ara modifications at certain sites might induce further improvement. KN5ara-10, which contained fewer PS linkages, showed similar or slightly better antisense activity than the corresponding KN5ara-3. Hence, given the possibility of lower PS-derived cytotoxicity, KN5ara-10 might be the best candidate for KNTC2-targeted ASO for cancer therapy in this study. However, the further cytotoxicity assay of these KN5ara gapmers should be performed in future studies, and gapmers with fewer PS linkages than KN5ara-10 should also be synthesized and evaluated as well.
4. Experimental Sections
4.1. Solid-Phase Oligonucleotide Synthesis
The synthesis was carried out with a DNA/RNA synthesizer by the phosphoramidite method. After the synthesis, the RNA oligomers were cleaved from CPG beads and deprotected by treatment with concentrated NH3 solution/40% methylamine (1:1, v/v) for 10 min at 65 °C, while the DNA-based oligomers were treated with concentrated NH3 solution for 12 h at 55 °C. Notably, before the treatment of the NH3 solution, the CPG beads were treated with 10% dimethylamine in MeCN for 5 min followed by rinsing with MeCN to selectively remove cyanoethyl groups, if there are analogs introduced in oligomers. Then, 2ʹ-O-TBDMS groups in RNA oligomers were removed by Et3N∙3HF (125 µL) in DMSO (100 µL) for 1.5 h at 65 °C. The reaction was quenched with 0.1 M TEAA buffer (pH 7.0), and the mixture was desalted using a Sep-Pak C18 cartridge. The oligomers were purified by 20% PAGE containing 7 M urea to give highly purified oligonucleotides.
4.2. MALDI-TOF/MS Analysis of ONs
The spectra were obtained with a time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). A solution of 3-hydroxypicolinic acid (3-HPA) and diammonium hydrogen citrate in H2O was used as a matrix. Data of synthetic ONs: KN-pos: m/z = 5346.49 (calcd for C165H202N58O88P15S15 [M-H]−, 5348.32); KNpos-F: m/z = 5884.62 (calcd for C192H226N59O87P16S15 [M-H]−, 5882.91); KN5ara-1: m/z = 5395.54 (calcd for C167H208N59O87FP15S15 [M-H]−, 5393.54); KN5ara-2: m/z = 5396.54 (calcd for C167H207N59O87FP15S15 [M-H]−, 5397.39); KN5ara-3: m/z = 5428.03 (calcd for C168H208N59O88FP15S15 [M-H]−, 5425.40); KN5ara-4: m/z = 5438.57 (calcd for C169H210N59O88FP15S15 [M-H]−, 5439.43); KN5ara-5: m/z = 5424.63 (calcd for C168H208N59O88FP15S15 [M-H]−, 5425.40); KN5ara-6: m/z = 5425.59 (calcd for C168H208N59O88FP15S15 [M-H]−, 5425.40); KN5ara-6-F: m/z = 5959.67 (calcd for C195H232N60O97FP16S15 [M-H]−, 5960.54); KN5ara-7: m/z = 5427.77 (calcd for C168H208N59O88FP15S15 [M-H]−, 5425.40); KN5ara-8: m/z = 5393.92 (calcd for C167H208N59O87FP15S15 [M-H]−, 5393.54); KN5ara-9: m/z = 5472.64 (calcd for C170H214N60O87F2P15S15 [M-H]−, 5468.59); KN5ara-10: m/z = 5392.62 (calcd for C168H208N59O90FP15S13 [M-H]−, 5389.59); KN5ara-10-F: m/z = 5927.72 (calcd for C195H232N60O99FP16S13 [M-H]−, 5928.59); KN5ara-11: m/z = 5391.73 (calcd for C168H208N59O90FP15S13 [M-H]−, 5389.59); KN5ara-11-F: m/z = 5927.72 (calcd for C195H232N60O99FP16S13 [M-H]−, 5928.94); KN5ara-12-F: m/z = 5852.67 (calcd for C192H226N59O99P16S13 [M-H]−, 5853.65); cRNA-1: m/z = 5046.38 (calcd for C152H190N61O107P15 [M-H]−, 5045.74); cRNA-2: m/z = 5585.96 (calcd for C179H215N62O116P16 [M-H]−, 5583.87).
4.3. Thermal Denaturation Study
The solution containing 3.0 µM ASO/RNA duplexes were prepared by mixing the ASOs (600 pmol) with complementary target cRNA-1 (600 pmol) in a buffer of 10 mM sodium phosphate (pH 7.0) containing 100 mM NaCl, and then heated at 90–100 °C, followed by being cooled gradually to room temperature. Thermally induced transitions were monitored at 260 nm with a UV/vis spectrometer fitted with a temperature controller in quartz cuvettes with a path length of 1.0 cm. The sample temperature was increased by 0.5 °C/min.
4.4. RNase H Assay
The ASO/RNA duplexes used for RNase H assay were prepared by mixing the ASOs (600 pmol) with fluorescein labeled complementary target cRNA-2 (3000 pmol) in 75 µL of 50 mM Tris–HCl (pH 8.0) containing 75 mM KCl, 3 mM MgCl2 and 10 mM dithiothreitol, followed by heating at 90–100 °C for 5 min and cooling gradually to room temperature. Then, 70 µL diluted RNase H solution (60 unit/L in H2O) was added, and subsequently the mixture was incubated at 37 °C for the required time. Aliquots of 5 µL were diluted with 100% formamide (10 µL). Samples were subjected to electrophoresis in 20% PAGE containing 7M urea and quantified by Luminescent Image analyzer LAS-4000 (Fujifilm).
4.5. Nuclease Resistance of Single-Stranded ASO
Fluorescein labeled ASOs (300 pmol) were dissolved in OPTI-MEM (37 µL) and used for the serum stability test. 1.0 µL of the oligomer solution was diluted in 10 µL stop solution (10% formamide in 10 mM EDTA) as the control sample (0 min). Then, 36 µL bovine serum was added to achieve a final concentration of 50% (v/v), and subsequently the mixture was incubated at 37 °C for the required time. Aliquots of 2.0 µL were diluted with 10 µL stop solution. Samples were subjected to electrophoresis in 20% PAGE containing 7M urea and quantified by Luminescent Image analyzer LAS-4000 (Fujifilm).
4.6. ASOs In Vitro Activity Assay
A549tGFP cells were established by transfection with pGIPZ (Horizon Discovery) into a human lung cancer cell line A549 (ATCC). For LFA-free (LFA-) group, A549tGFP cells were plated into 96-well plates at 2 × 103 cells/well, followed by the cultivation in D-MEM (Nissui) containing 10% fetal bovine serum (FBS), streptomycin (100 µg/mL) and penicillin (100 U/mL). Cells were incubated at 37 °C with 5% carbon dioxide for 24 h prior to the ASO administration. Then, the medium was exchanged into OPTI-MEM containing 2% FBS, and the KNTC2-targeting antisense gapmers were added into specified wells at the final concentration of 2.5 µM. After a 48-h incubation in 2% FBS_OPTI-MEM and a further 24-h incubation in 10% FBS_D-MEM, cells were retrieved, and the mRNAs were extracted using CellAmp Direct RNA Prep Kit for RT-PCR (Takara Bio). For LFA+ group, A549tGFP cells (ATCC) were plated into 96-well plates at 4 × 103 cells/well and cultured in 10% FBS_D-MEM for 24 h prior to the ASO administration. After exchanging the medium into 10% FBS_OPTI-MEM, the KNTC2-targeting antisense gapmers, mixed with 0.3% Lipofectamine® 2000 (Invitrogen) in advance, were added into specified wells at the final concentration of 4.0 nM and incubated for 24 h. Then, as the same as LFA- group, the medium was replaced by 10% FBS_D-MEM and cells were incubated for further 24 h, followed by the extraction of mRNAs. Treatment without ASOs was used as a control, named “unTF”.
Quantitative real-time polymerase chain reaction (qRT-PCR) of the targeted KNTC2 mRNA was performed using One Step TB Green PrimeScript RT-PCR kit II (Takara), and Thermal Cycler Dice Real Time System (Takara). Each PCR reaction was performed in duplicate, and the relative KNTC2 mRNA levels were calculated with the ddCT method, using beta-actin (ACTB) as a reference. The sequences of primers used in the qRT-PCR for Human KNTC2 are 5ʹ-CCTCTCCATGCAGGAGTTAAGA-3ʹ for the forward primer, 5ʹ-GGTCTCGGGTCCTTGATTTTCT-3ʹ for the reverse primer. For Human ACTB, the sequences of forward primer and reverse primer are 5ʹ-GGAGCAATGATCTTGATCTT-3ʹ and 5ʹ-CCTTCCTGGGCATGGAGTCCT-3ʹ, respectively.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}