
Tempol Inhibits the Growth of Lung Cancer and Normal Cells through Apoptosis Accompanied by Increased O2•− Levels and Glutathione Depletion
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Cell Growth Inhibition Assay
2.4. Sub-G1 Cell Analysis
2.5. Detection of Apoptosis
2.6. Measurement of MMP (ΔΨm)
2.7. Quantification of Caspase-3 Activity
2.8. Determination of Intracellular ROS and O2•− Levels
2.9. Detection of Intracellular GSH Levels
2.10. Statistical Analysis
3. Results
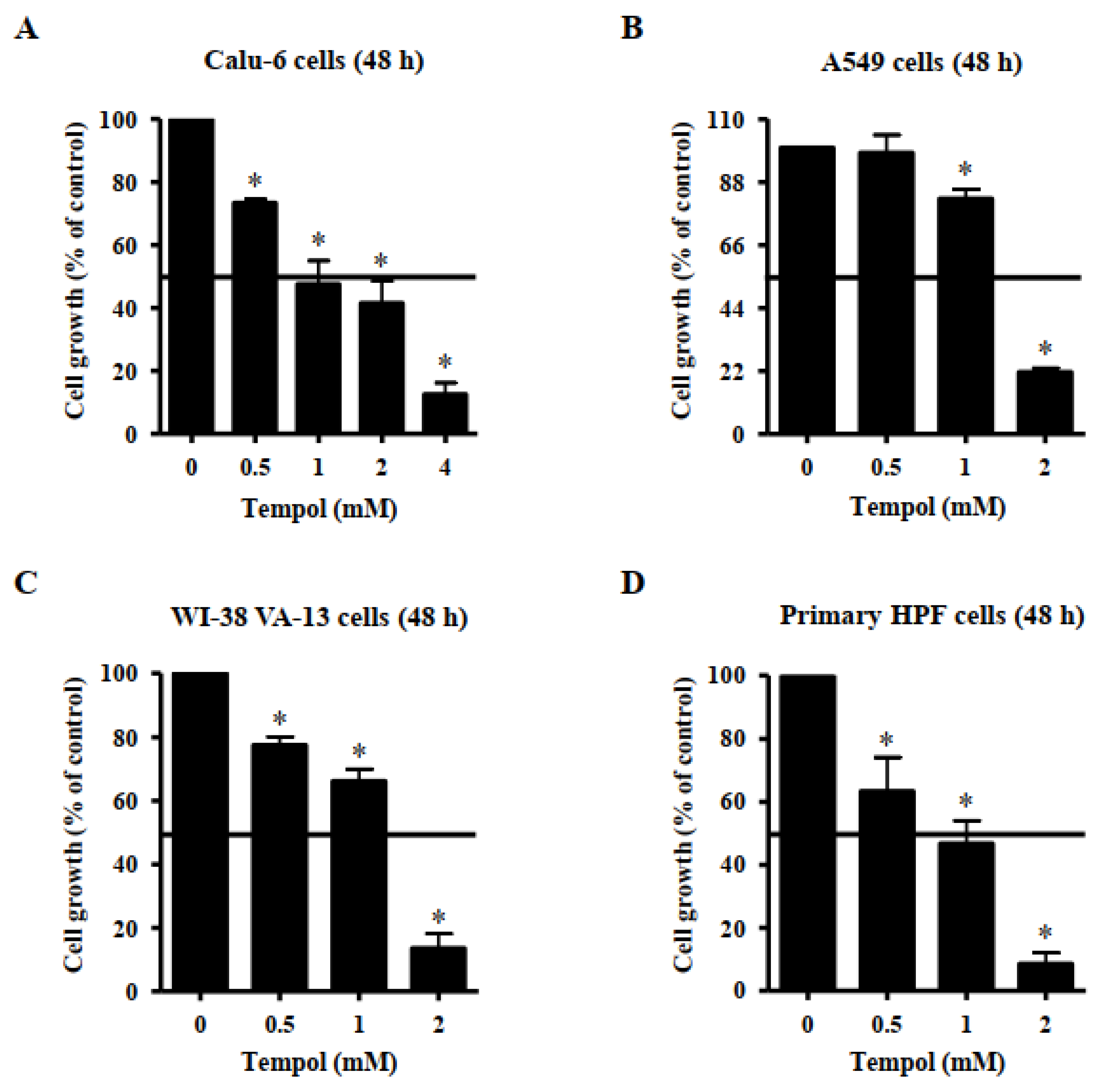
3.1. Effects of Tempol on Lung Cancer and Normal Cell Growth
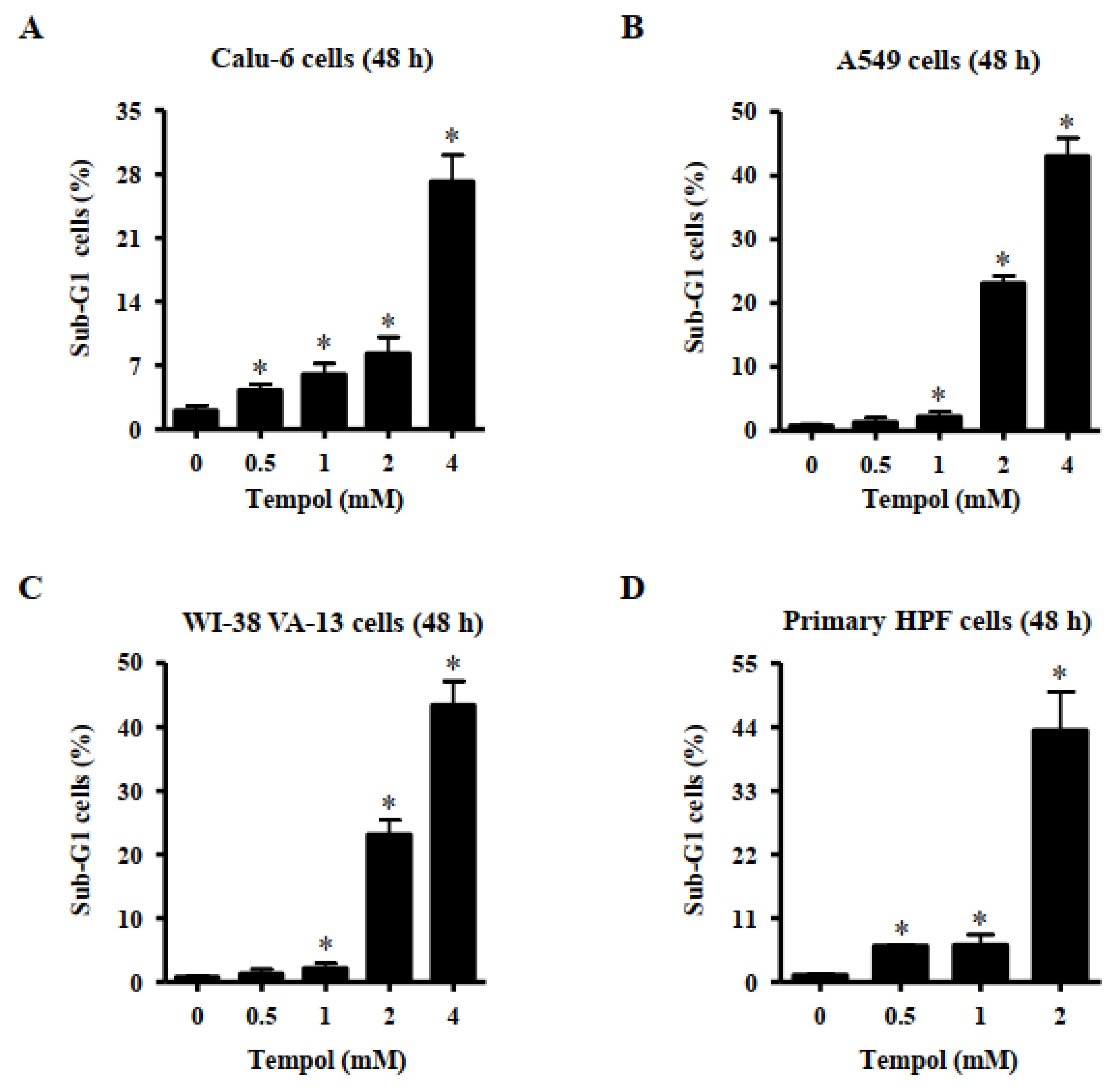
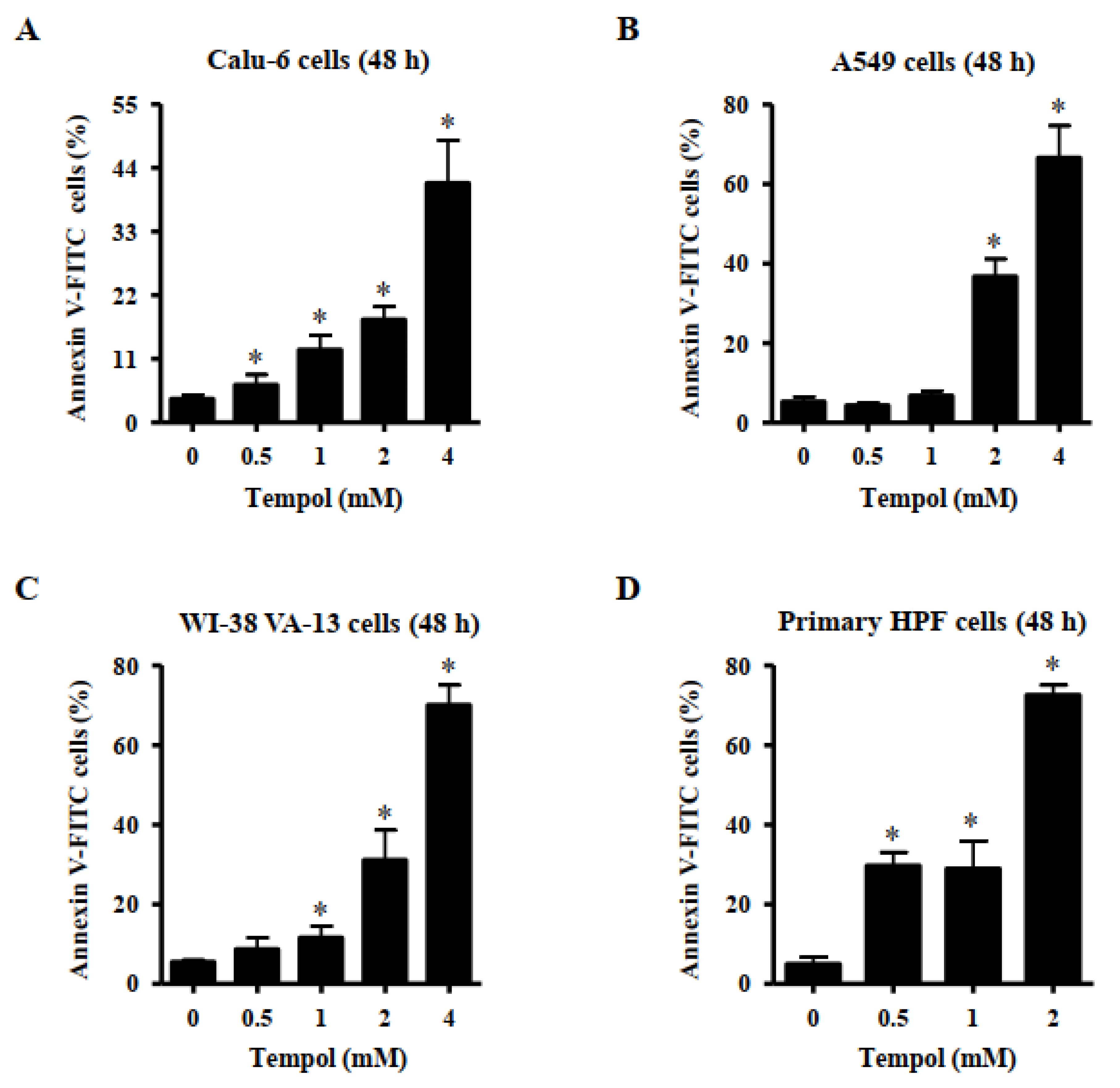
3.2. Effects of Tempol on Cell Death of Lung Cancer and Normal Cell Cells
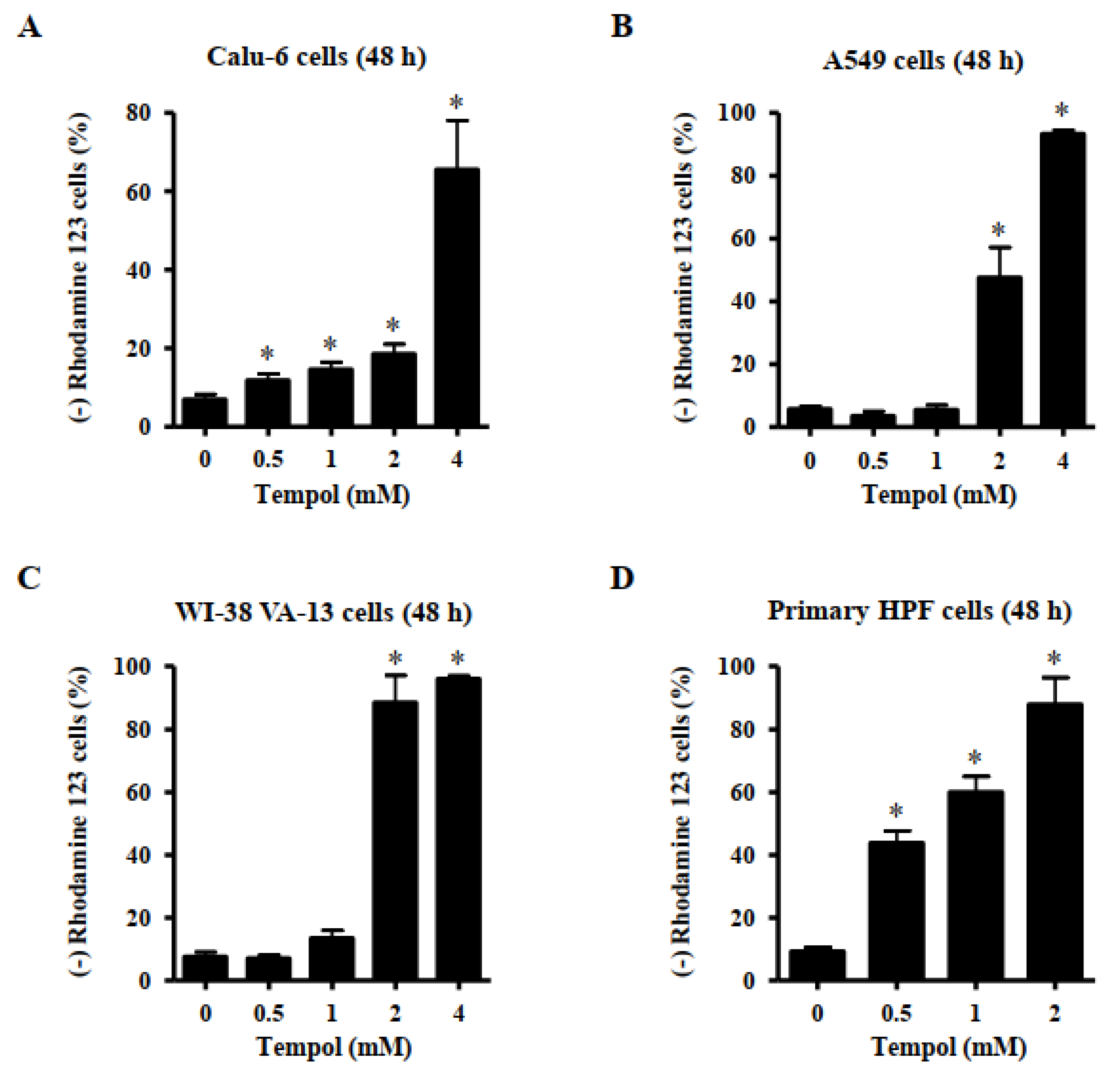
3.3. Effects of Tempol on Mitochondrial Membrane Potential (MMP; ∆Ψm) in Lung Cancer and Normal Cells
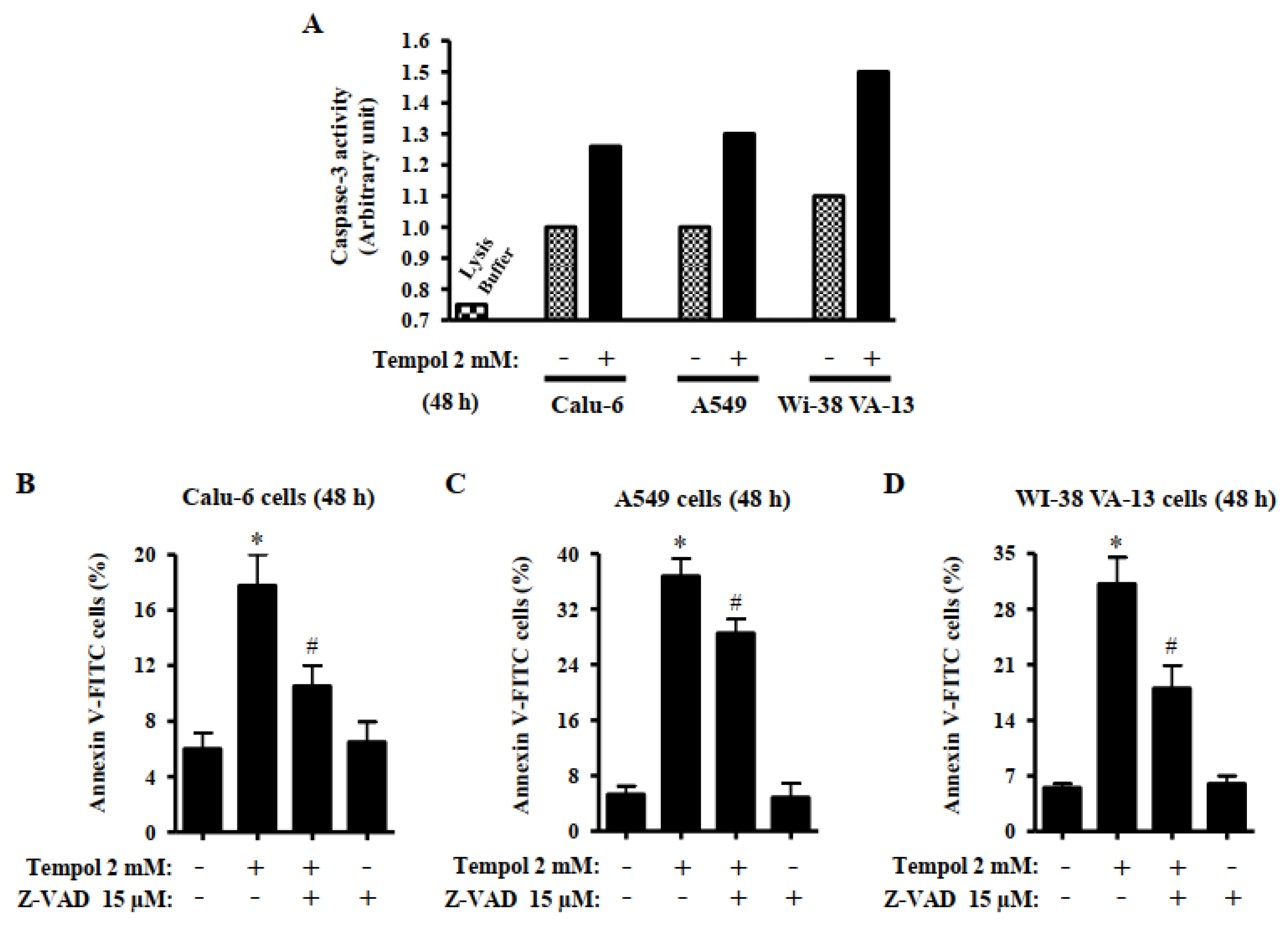
3.4. Effects of Tempol and/or Z-VAD on Caspase-3 Activity and Cell Death in Lung Cancer and Normal Cells
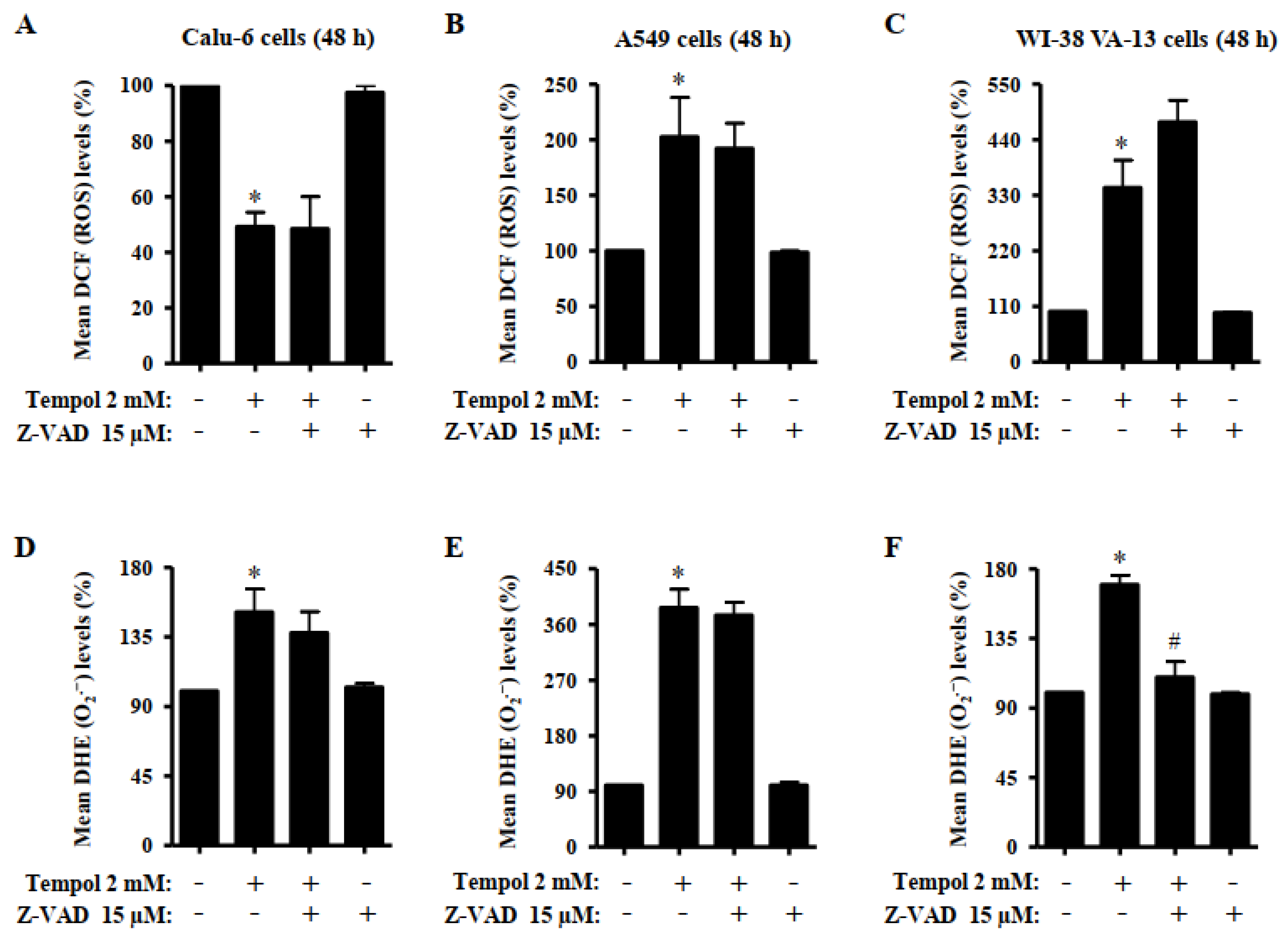
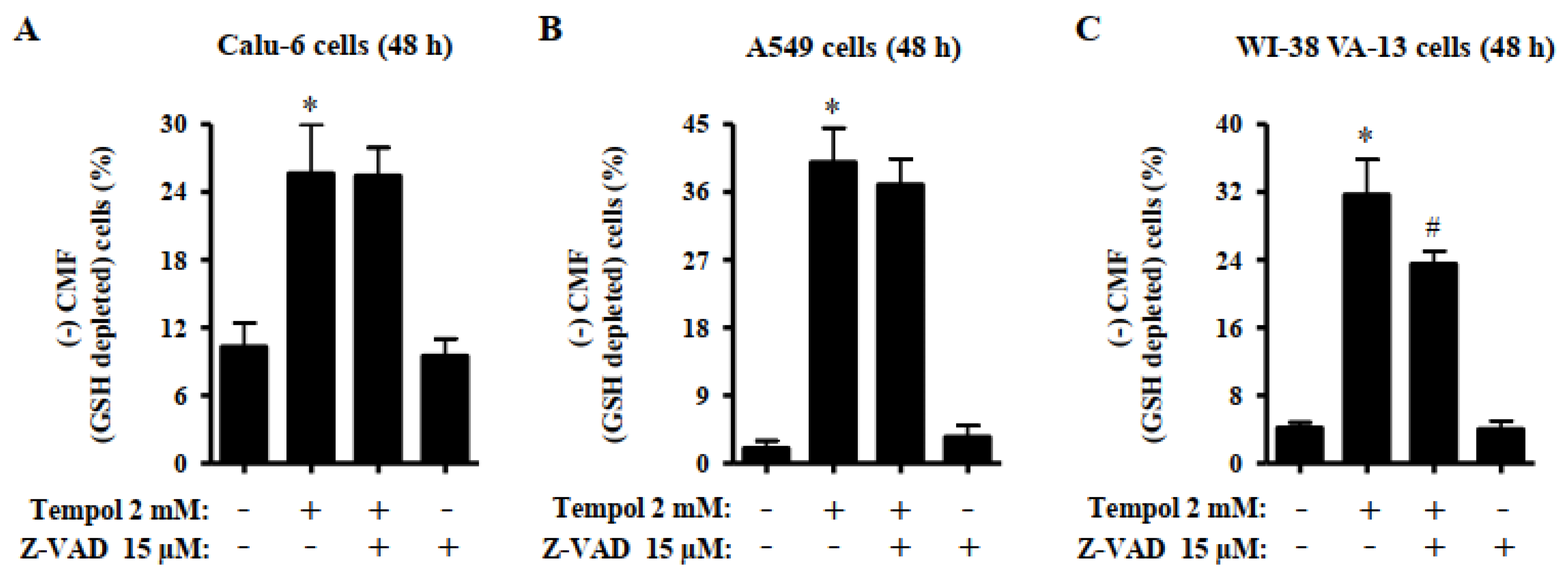
3.5. Effects of Tempol and/or Z-VAD on ROS and GSH Levels in Lung Cancer and Normal Cells
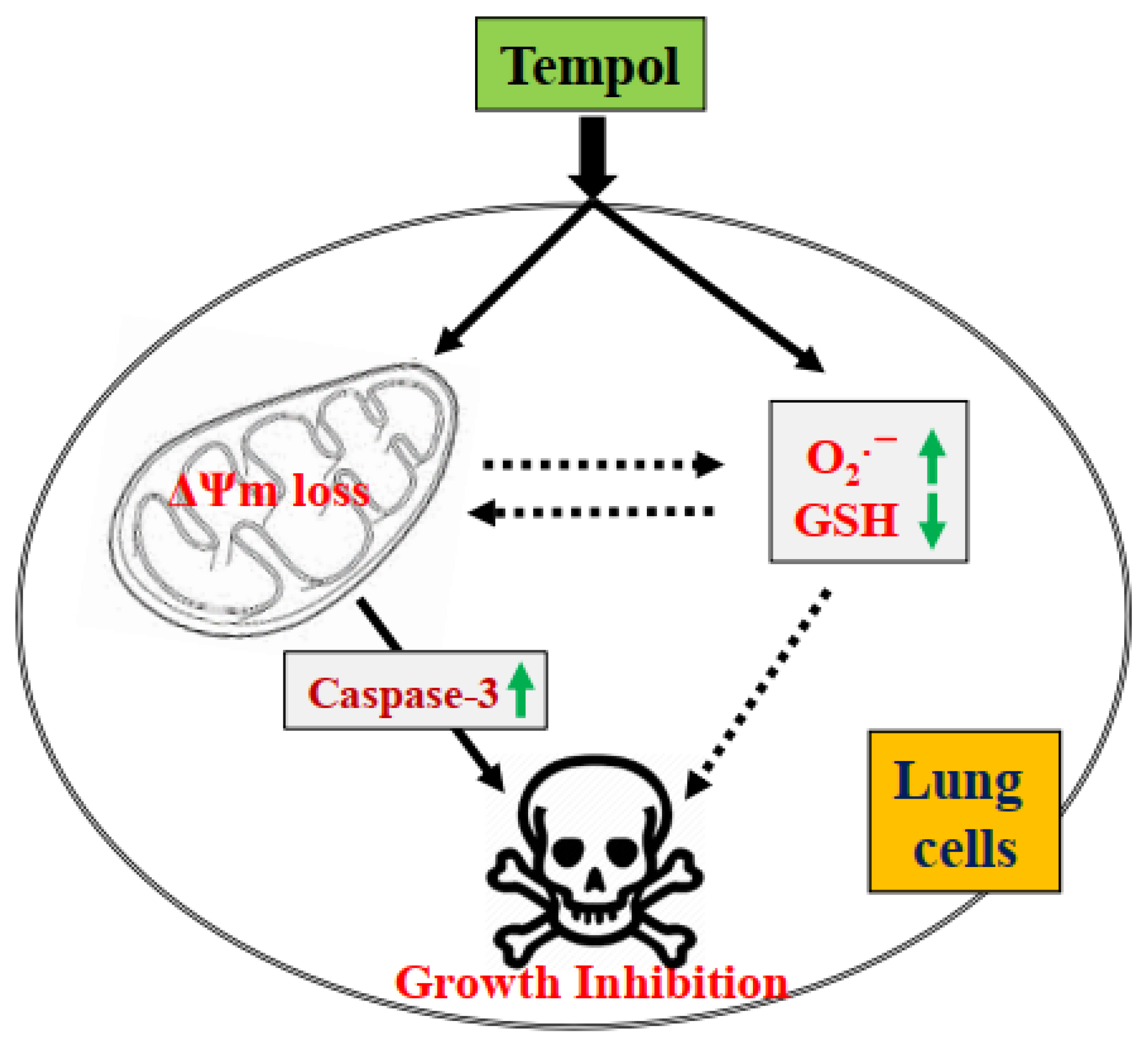
4. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmouliere, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal. Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Li, M.; Chen, Z.; Zhan, C.; Lin, Z.; Wang, Q. Advances in clinical trials of targeted therapy and immunotherapy of lung cancer in 2018. Transl. Lung Cancer Res. 2019, 8, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Rad, R.; Li, R.; Paul, M.K.; Dubinett, S.M.; Liu, B. The Biology of Lung Cancer: Development of More Effective Methods for Prevention, Diagnosis, and Treatment. Clin. Chest Med. 2020, 41, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Huska, J.D.; Lamb, H.M.; Hardwick, J.M. Overview of BCL-2 Family Proteins and Therapeutic Potentials. Methods Mol. Biol. 2019, 1877, 1–21. [Google Scholar]
- Chung, C. Restoring the switch for cancer cell death: Targeting the apoptosis signaling pathway. Am. J. Health Syst. Pharm. 2018, 75, 945–952. [Google Scholar] [CrossRef]
- Wurstle, M.L.; Laussmann, M.A.; Rehm, M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp. Cell Res. 2012, 318, 1213–1220. [Google Scholar] [CrossRef]
- Liu, X.; Yue, P.; Zhou, Z.; Khuri, F.R.; Sun, S.Y. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J. Natl. Cancer Instit. 2004, 96, 1769–1780. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Wilcox, C.S. Reactive oxygen species: Roles in blood pressure and kidney function. Curr. Hypertens. Rep. 2002, 4, 160–166. [Google Scholar] [CrossRef]
- Lauterburg, B.H. Analgesics and glutathione. Am. J. Ther. 2002, 9, 225–233. [Google Scholar] [CrossRef]
- Vassalle, C.; Maltinti, M.; Sabatino, L. Targeting Oxidative Stress for Disease Prevention and Therapy: Where Do We Stand, and Where Do We Go from Here. Molecules 2020, 25, 2653. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Wilcox, C.S. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 2010, 126, 119–145. [Google Scholar] [CrossRef]
- Samuni, A.; Krishna, C.M.; Mitchell, J.B.; Collins, C.R.; Russo, A. Superoxide reaction with nitroxides. Free Radic. Res. Commun. 1990, 9, 241–249. [Google Scholar] [CrossRef]
- Augusto, O.; Trindade, D.F.; Linares, E.; Vaz, S.M. Cyclic nitroxides inhibit the toxicity of nitric oxide-derived oxidants: Mechanisms and implications. An. Acad. Bras. Cienc. 2008, 80, 179–189. [Google Scholar] [CrossRef]
- Mitchell, J.B.; Samuni, A.; Krishna, M.C.; DeGraff, W.G.; Ahn, M.S.; Samuni, U.; Russo, A. Biologically active metal-independent superoxide dismutase mimics. Biochemistry 1990, 29, 2802–2807. [Google Scholar] [CrossRef]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Apoptosis in pyrogallol-treated Calu-6 cells is correlated with the changes of intracellular GSH levels rather than ROS levels. Lung Cancer 2008, 59, 301–314. [Google Scholar] [CrossRef]
- Park, W.H.; Han, Y.W.; Kim, S.H.; Kim, S.Z. A superoxide anion generator, pyrogallol induces apoptosis in As4.1 cells through the depletion of intracellular GSH content. Mutat. Res. 2007, 619, 81–92. [Google Scholar] [CrossRef]
- Goralska, M.; Holley, B.; McGahan, M.C. The effects of Tempol on ferritin synthesis and Fe metabolism in lens epithelial cells. Biochim. Biophys. Acta 2000, 1497, 51–60. [Google Scholar] [CrossRef]
- Sies, H.; Mehlhorn, R. Mutagenicity of nitroxide-free radicals. Arch. Biochem. Biophys. 1986, 251, 393–396. [Google Scholar] [CrossRef]
- Gariboldi, M.B.; Lucchi, S.; Caserini, C.; Supino, R.; Oliva, C.; Monti, E. Antiproliferative effect of the piperidine nitroxide TEMPOL on neoplastic and nonneoplastic mammalian cell lines. Free Radic. Biol. Med. 1998, 24, 913–923. [Google Scholar] [CrossRef]
- Han, Y.H.; Park, W.H. Tempol inhibits growth of As4.1 juxtaglomerular cells via cell cycle arrest and apoptosis. Oncol. Rep. 2012, 27, 842–848. [Google Scholar] [PubMed]
- Ye, S.; Xu, P.; Huang, M.; Chen, X.; Zeng, S.; Wang, Q.; Chen, J.; Li, K.; Gao, W.; Liu, R.; et al. The heterocyclic compound Tempol inhibits the growth of cancer cells by interfering with glutamine metabolism. Cell Death Dis. 2020, 11, 312. [Google Scholar] [CrossRef]
- Wang, M.; Li, K.; Zou, Z.; Li, L.; Zhu, L.; Wang, Q.; Gao, W.; Wang, Y.; Huang, W.; Liu, R.; et al. Piperidine nitroxide Tempol enhances cisplatin-induced apoptosis in ovarian cancer cells. Oncol. Lett. 2018, 16, 4847–4854. [Google Scholar] [CrossRef]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a G2 arrest of the cell cycle and apoptosis accompanied with the depletion of GSH. Cancer Lett. 2008, 270, 40–55. [Google Scholar] [CrossRef]
- Park, W.H. Hydrogen peroxide inhibits the growth of lung cancer cells via the induction of cell death and G1phase arrest. Oncol. Rep. 2018, 40, 1787–1794. [Google Scholar]
- Han, Y.H.; Park, W.H. Propyl gallate inhibits the growth of HeLa cells via regulating intracellular GSH level. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2009, 47, 2531–2538. [Google Scholar] [CrossRef]
- Park, W.H. Upregulation of thioredoxin and its reductase attenuates arsenic trioxideinduced growth suppression in human pulmonary artery smooth muscle cells by reducing oxidative stress. Oncol. Rep. 2020, 43, 358–367. [Google Scholar]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef]
- Han, Y.H.; Park, W.H. Proteasome inhibitor MG132 reduces growth of As4.1 juxtaglomerular cells via caspase-independent apoptosis. Arch. Toxicol. 2010, 84, 689–698. [Google Scholar] [CrossRef]
- Suy, S.; Mitchell, J.B.; Samuni, A.; Mueller, S.; Kasid, U. Nitroxide tempo, a small molecule, induces apoptosis in prostate carcinoma cells and suppresses tumor growth in athymic mice. Cancer 2005, 103, 1302–1313. [Google Scholar] [CrossRef]
- Monti, E.; Supino, R.; Colleoni, M.; Costa, B.; Ravizza, R.; Gariboldi, M.B. Nitroxide TEMPOL impairs mitochondrial function and induces apoptosis in HL60 cells. J. Cell Biochem. 2001, 82, 271–276. [Google Scholar] [CrossRef]
- Martinez-Alonso, D.; Malumbres, M. Mammalian cell cycle cyclins. Semin. Cell Dev. Biol. 2020, 107, 28–35. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.I.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef]
- Park, H.K.; Han, B.R.; Park, W.H. Combination of Arsenic Trioxide and Valproic Acid Efficiently Inhibits Growth of Lung Cancer Cells via G2/M-Phase Arrest and Apoptotic Cell Death. Int. J. Mol. Sci. 2020, 21, 2649. [Google Scholar] [CrossRef]
- Wilcox, C.S.; Pearlman, A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol. Rev. 2008, 60, 418–469. [Google Scholar] [CrossRef]
- Kroemer, G.; Dallaporta, B.; Resche-Rigon, M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998, 60, 619–642. [Google Scholar] [CrossRef]
- Moon, H.J.; Park, W.H. Butylated hydroxyanisole inhibits the growth of HeLa cervical cancer cells via caspase-dependent apoptosis and GSH depletion. Mol. Cell. Biochem. 2011, 349, 179–186. [Google Scholar] [CrossRef]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Induction of apoptosis in arsenic trioxide-treated lung cancer A549 cells by buthionine sulfoximine. Mol. Cells 2008, 26, 158–164. [Google Scholar] [PubMed]
- Park, W.H.; Han, B.R.; Park, H.K.; Kim, S.Z. Arsenic trioxide induces growth inhibition and death in human pulmonary artery smooth muscle cells accompanied by mitochondrial O2•− increase and GSH depletion. Environ. Toxicol. 2018. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Park, W.H. Auranofin induces mesothelioma cell death through oxidative stress and GSH depletion. Oncol. Rep. 2016, 35, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Suppression of arsenic trioxide-induced apoptosis in HeLa cells by N-acetylcysteine. Mol. Cells 2008, 26, 18–25. [Google Scholar] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, W.H. Tempol Inhibits the Growth of Lung Cancer and Normal Cells through Apoptosis Accompanied by Increased O2•− Levels and Glutathione Depletion. Molecules 2022, 27, 7341. https://doi.org/10.3390/molecules27217341
Park WH. Tempol Inhibits the Growth of Lung Cancer and Normal Cells through Apoptosis Accompanied by Increased O2•− Levels and Glutathione Depletion. Molecules. 2022; 27(21):7341. https://doi.org/10.3390/molecules27217341
Chicago/Turabian StylePark, Woo Hyun. 2022. "Tempol Inhibits the Growth of Lung Cancer and Normal Cells through Apoptosis Accompanied by Increased O2•− Levels and Glutathione Depletion" Molecules 27, no. 21: 7341. https://doi.org/10.3390/molecules27217341
APA StylePark, W. H. (2022). Tempol Inhibits the Growth of Lung Cancer and Normal Cells through Apoptosis Accompanied by Increased O2•− Levels and Glutathione Depletion. Molecules, 27(21), 7341. https://doi.org/10.3390/molecules27217341