
Homology Modeling and Molecular Dynamics-Driven Search for Natural Inhibitors That Universally Target Receptor-Binding Domain of Spike Glycoprotein in SARS-CoV-2 Variants

 , ,
, ,
Abstract

1. Introduction
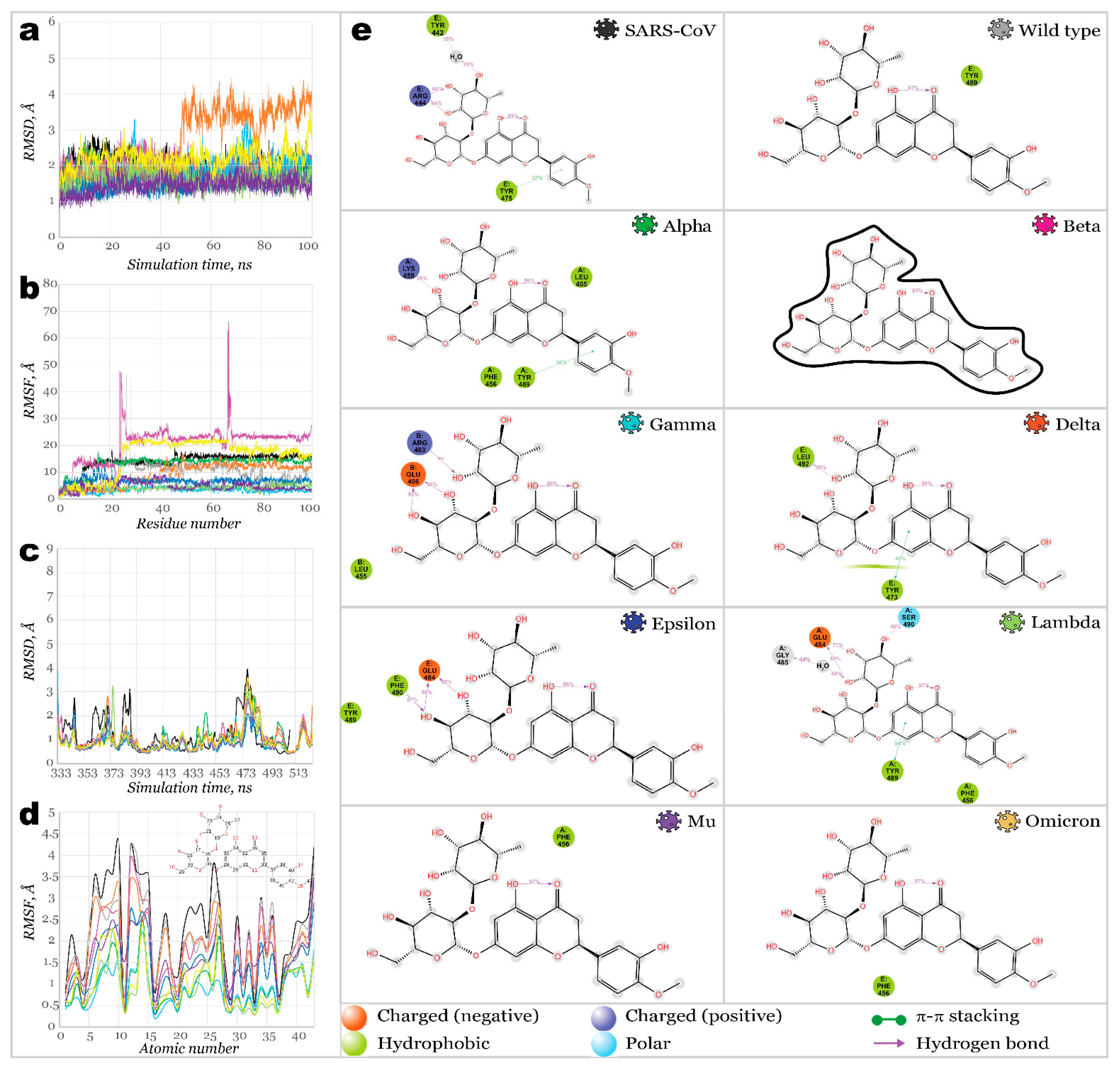
2. Results and Discussion
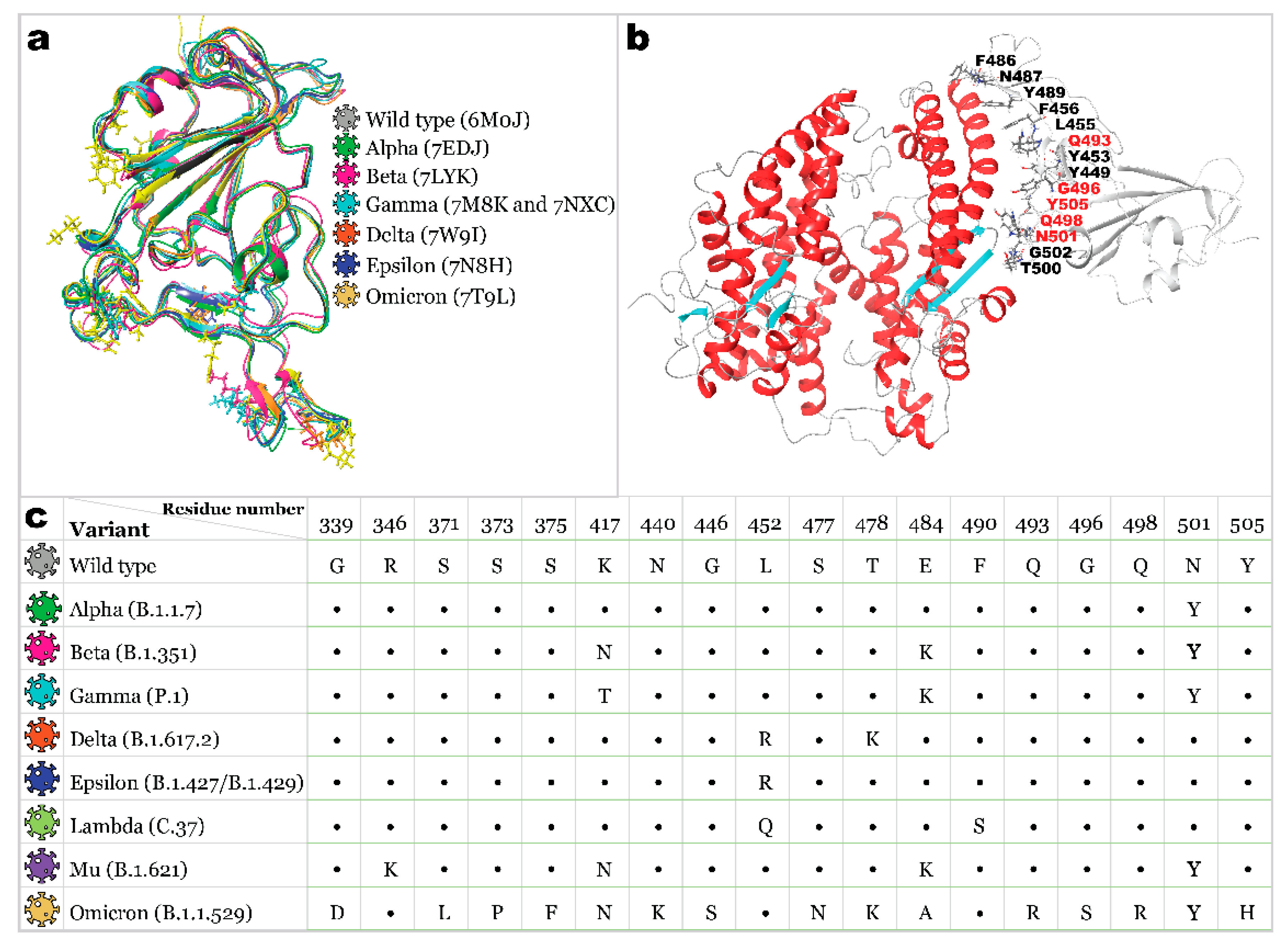
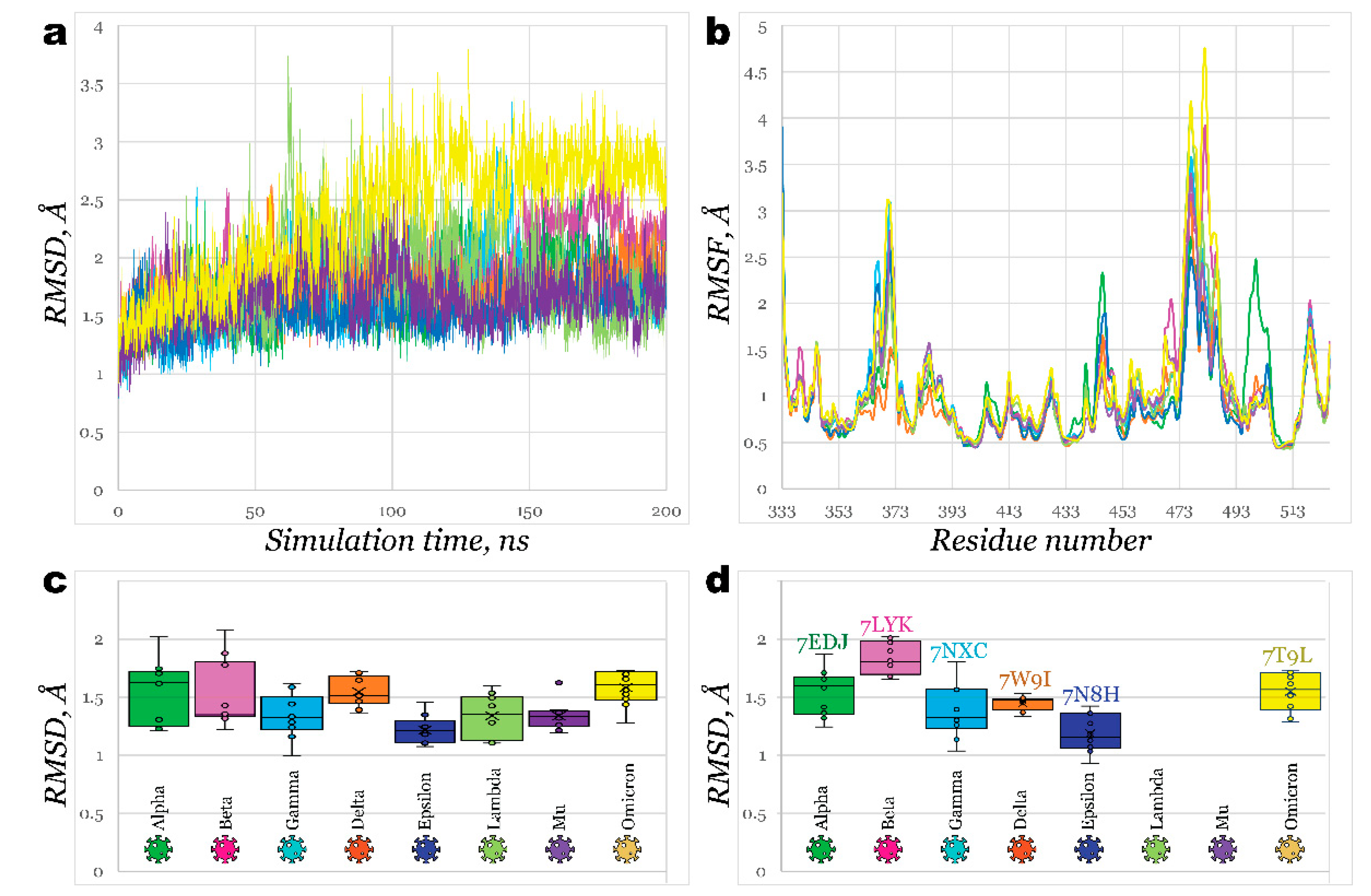
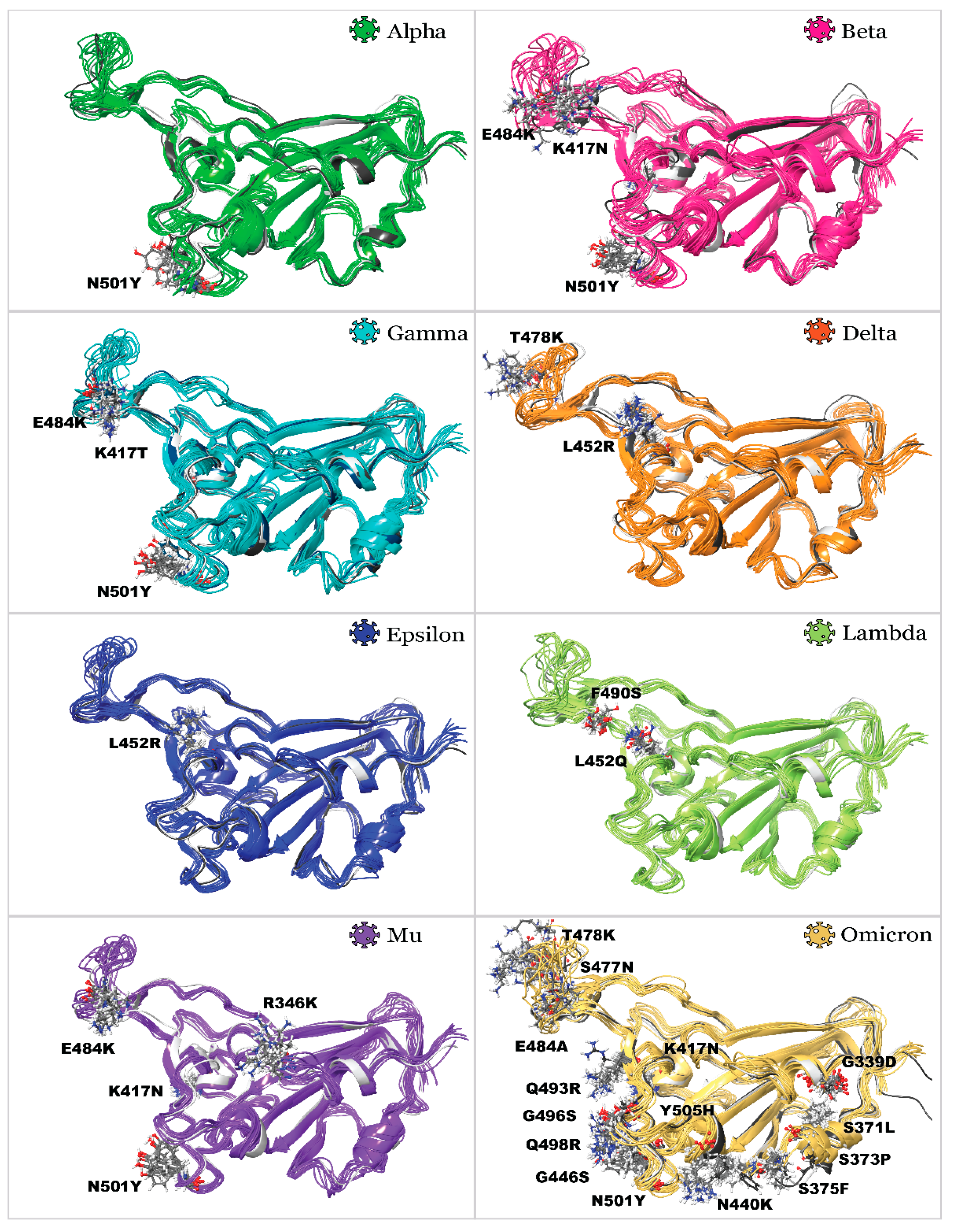
2.1. Receptor Binding Domain Mutations and Homology Models of SARS-CoV-2 Variants
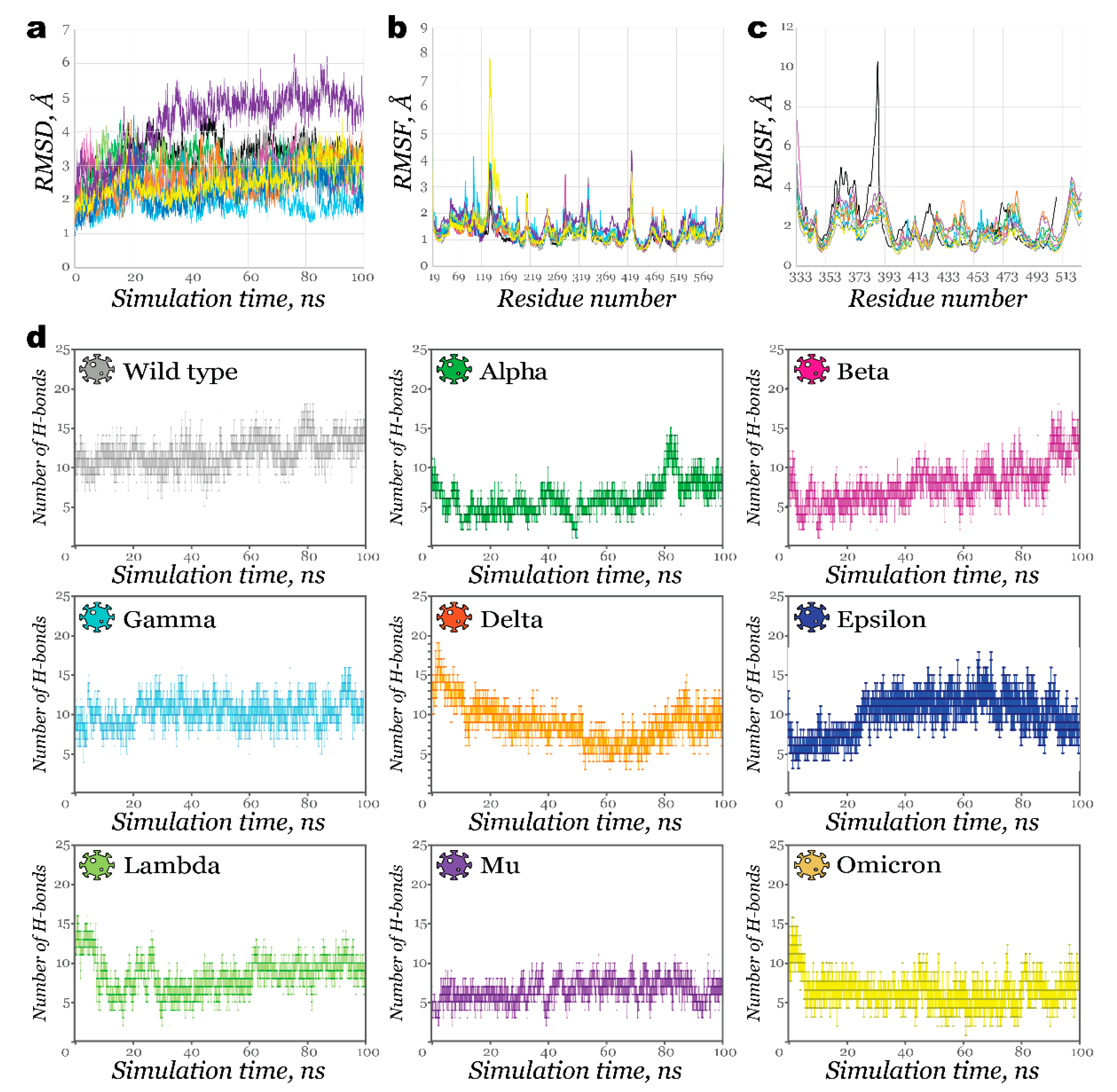
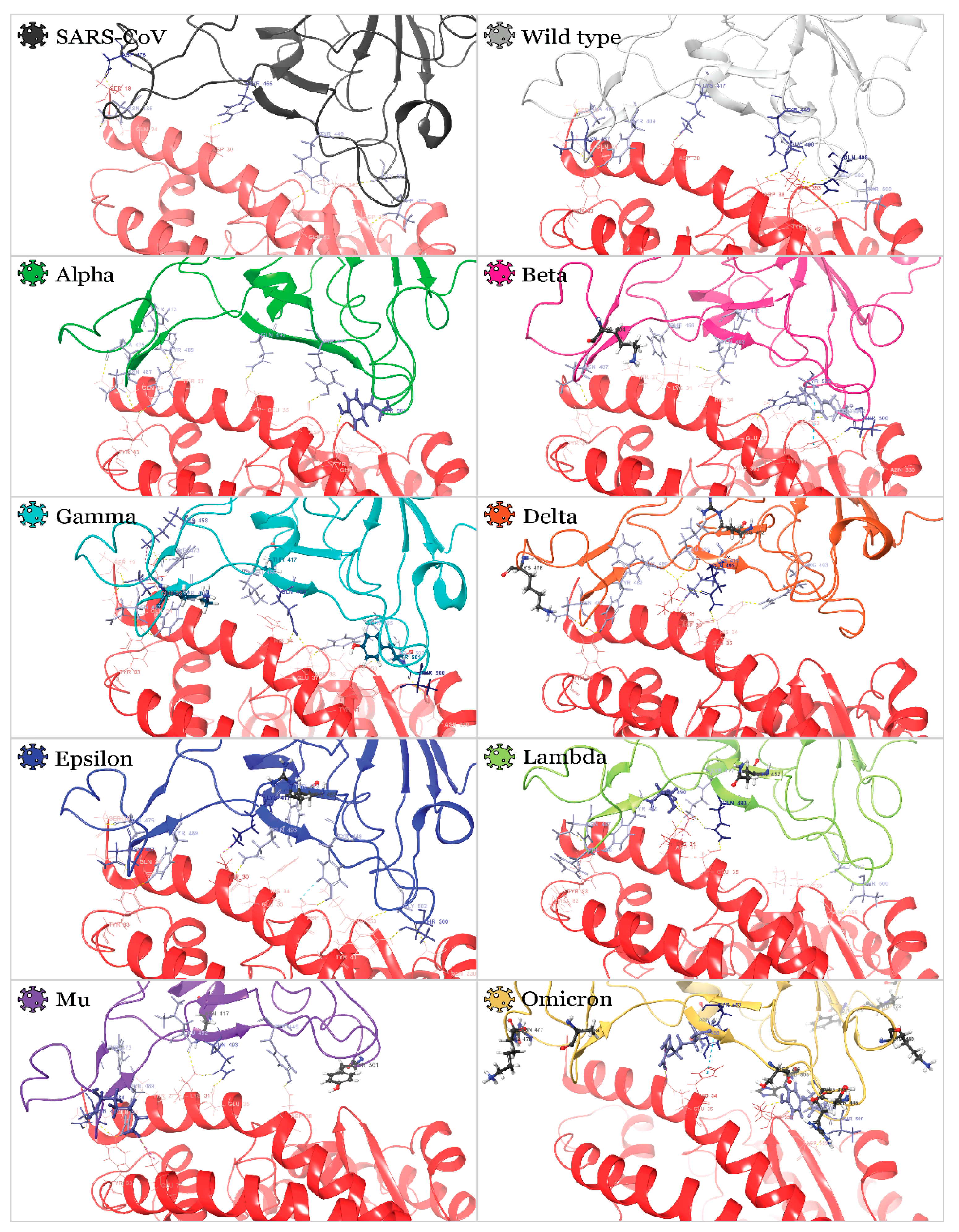
2.2. Interactions of RBDs with ACE2
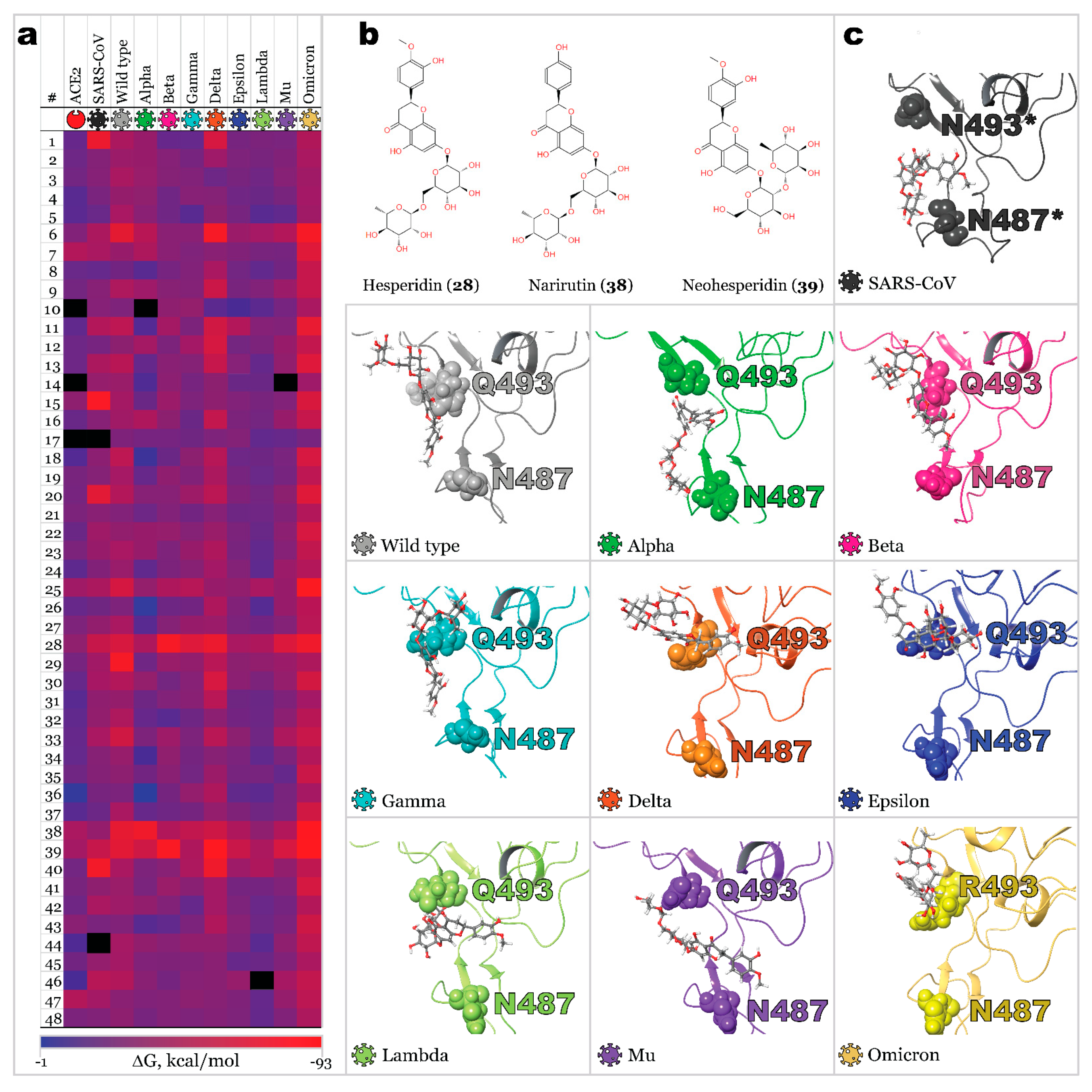
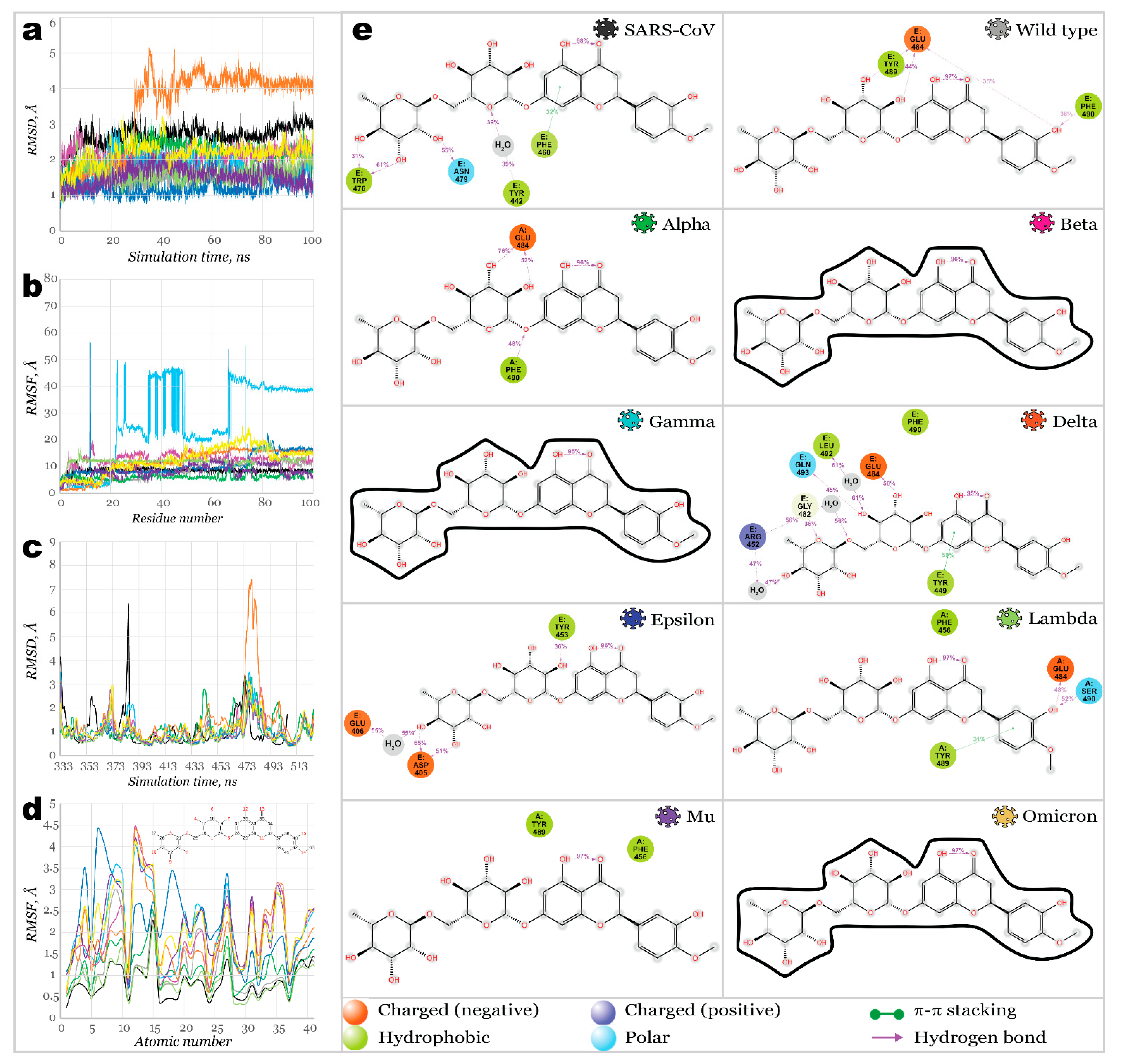
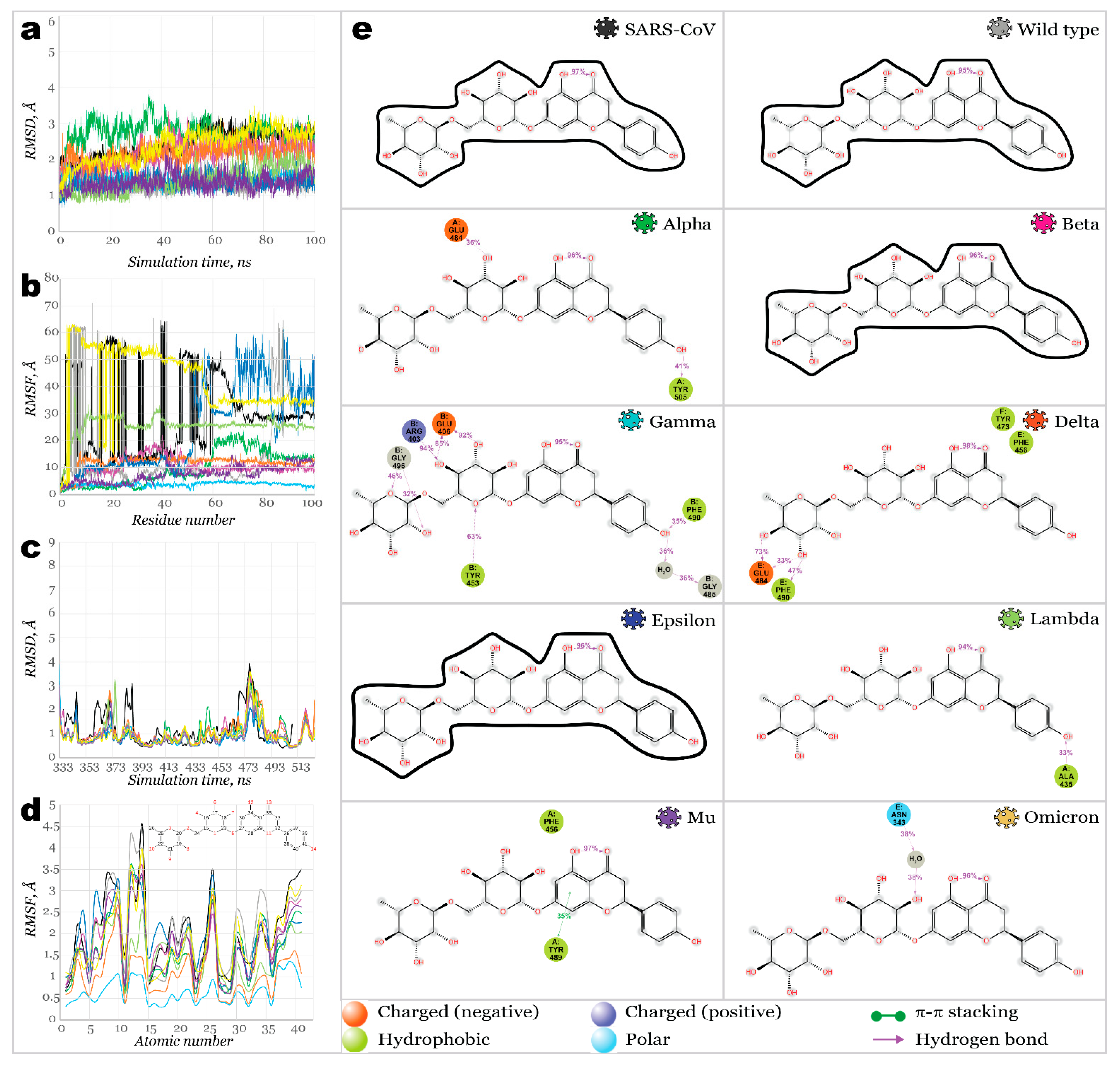
2.3. Benchmark Models for Drug Binding
3. Materials and Methods
3.1. Protein Preparation
3.2. Homology Modeling
3.3. Ligand Preparation
3.4. Molecular Docking and Molecular Mechanics
3.5. Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.; di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19); StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Gao, J.; Tian, Z.; Yang, X. Breakthrough: Chloroquine Phosphate Has Shown Apparent Efficacy in Treatment of COVID-19 Associated Pneumonia in Clinical Studies. Biosci. Trends 2020, 14, 72–73. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.L.; Vaca, C.E.; Paredes, R.; Mera, J.; Webb, B.J.; Perez, G.; Oguchi, G.; Ryan, P.; Nielsen, B.U.; Brown, M.; et al. Early Remdesivir to Prevent Progression to Severe COVID-19 in Outpatients. New Engl. J. Med. 2022, 386, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Coronavirus (COVID-19) Update: FDA Authorizes First Oral Antiviral for Treatment of COVID-19. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-COVID-19-update-fda-authorizes-first-oral-antiviral-treatment-COVID-19 (accessed on 1 January 2022).
- Kyriakidis, N.C.; López-Cortés, A.; González, E.V.; Grimaldos, A.B.; Prado, E.O. SARS-CoV-2 Vaccines Strategies: A Comprehensive Review of Phase 3 Candidates. NPJ. Vaccines 2021, 6, 28. [Google Scholar] [CrossRef]
- Nasreen, S.; Chung, H.; He, S.; Brown, K.A.; Gubbay, J.B.; Buchan, S.A.; Fell, D.B.; Austin, P.C.; Schwartz, K.L.; Sundaram, M.E.; et al. Effectiveness of COVID-19 vaccines against symptomatic SARS-CoV-2 infection and severe outcomes with variants of concern in Ontario. Nat. Microbiol. 2022, 7, 379–385. [Google Scholar] [CrossRef]
- Baraniuk, C. COVID-19: How Effective Are Vaccines against the Delta Variant? BMJ 2021, 374, n1960. [Google Scholar] [CrossRef] [PubMed]
- Perlman, S.; Netland, J. Coronaviruses Post-SARS: Update on Replication and Pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, A.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013919. [Google Scholar] [CrossRef]
- Jackson, C.B.; Zhang, L.; Farzan, M.; Choe, H. Functional Importance of the D614G Mutation in the SARS-CoV-2 Spike Protein. Biochem. Biophys. Res. Commun. 2021, 538, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. eLife. 2021, 10, e69091. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Mannar, D.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.-P.; Saville, J.W.; Leopold, K.; Li, W.; Dimitrov, D.S.; Tuttle, K.S. Cryo-Electron Microscopy Structures of the N501Y SARS-CoV-2 Spike Protein in Complex with ACE2 and 2 Potent Neutralizing Antibodies. PLoS Biol. 2021, 19, e3001237. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Huang, D.; Lee, C.-C.D.; Wu, N.C.; Jackson, A.M.; Zhu, X.; Liu, H.; Peng, L.; van Gils, M.J.; Sanders, R.W. Structural and Functional Ramifications of Antigenic Drift in Recent SARS-CoV-2 Variants. Science 2021, 6556, 818–823. [Google Scholar] [CrossRef]
- Gobeil, S.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Saunders, K.O.; Edwards, R.J. Effect of Natural Mutations of SARS-CoV-2 on Spike Structure, Conformation and Antigenicity. Science 2021, 373, 6555. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host. Microbe. 2021, 29, 44–57. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N.; Azzaz, F.; Chahinian, H. Structural Dynamics of SARS-CoV-2 Variants: A Health Monitoring Strategy for Anticipating COVID-19 Outbreaks. J. Infect. 2021, 83, 197–206. [Google Scholar] [CrossRef]
- Wang, P.; Casner, R.G.; Nair, M.S.; Wang, M.; Yu, J.; Cerutti, G.; Liu, L.; Kwong, P.D.; Huang, Y.; Shapiro, L. Increased Resistance of SARS-CoV-2 Variant P. 1 to Antibody Neutralization. Cell Host. Microbe. 2021, 29, 747–751. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Zhou, D.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R. Antibody Evasion by the P. 1 Strain of SARS-CoV-2. Cell 2021, 184, 2939–2954. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; Denis, K.S.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C. Multiple SARS-CoV-2 Variants Escape Neutralization by Vaccine-Induced Humoral Immunity. Cell 2021, 184, 2372–2383. [Google Scholar] [CrossRef]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of Common Mutations in the SARS-CoV-2 Spike RBD and Its Ligand, the Human ACE2 Receptor on Binding Affinity and Kinetics. Elife 2021, 10, e70658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, T.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; et al. Membrane Fusion and Immune Evasion by the Spike Protein of SARS-CoV-2 Delta Variant. Science 2021, 374, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cheng, G. Sequence Analysis of the Emerging SARS-CoV-2 Variant Omicron in South Africa. J. Med. Virol. 2021, 94, 1728–1733. [Google Scholar] [CrossRef]
- CDC COVID-19 Response Team. SARS-CoV-2 B. 1.1. 529 (Omicron) Variant—United States, 1–8 December 2021. Morb. Mortal. Wkly. Rep. 2021, 70, 1731–1734. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 Variant: A New Chapter in the COVID-19 Pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.; Kemp, S.A.; Goonawardane, N.; Papa, G.; Fatihi, S.; Charles, O.; Collier, D.; Choi, J. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, S.; Wu, B.; Yang, Q.; Chen, A.; Li, Y.; Zhang, Y.; Pan, T.; Zhang, H.; He, X. SARS-CoV-2 Omicron Strain Exhibits Potent Capabilities for Immune Evasion and Viral Entrance. Signal Transduct. Target Ther. 2021, 6, 1–3. [Google Scholar] [CrossRef]
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.W.; Wang, M.; Liu, L.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S. Striking Antibody Evasion Manifested by the Omicron Variant of SARS-CoV-2. Nature 2021, 602, 676–681. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Sharma, K.; Chand, H.S.; Byrareddy, S.N.; Singh, K. Omicron SARS-CoV-2 Variant: Unique Features and Their Impact on Pre-Existing Antibodies. J. Autoimmun. 2022, 126, 102779. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R. Omicron Escapes the Majority of Existing SARS-CoV-2 Neutralizing Antibodies. Nature 2021, 602, 657–663. [Google Scholar] [CrossRef]
- Ali, A.; Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV Spike Protein Interface Reveal Unique Mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef]
- Rath, S.L.; Kumar, K. Investigation of the Effect of Temperature on the Structure of SARS-CoV-2 Spike Protein by Molecular Dynamics Simulations. Front. Mol. Biosci. 2020, 7, 583523. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zhu, Z.; Shi, Y.; Wang, X.; Mu, K.; Yang, Y.; Zhang, X.; Xu, Z.; Zhu, W. Computational Insights into the Conformational Accessibility and Binding Strength of SARS-CoV-2 Spike Protein to Human Angiotensin-Converting Enzyme 2. J. Phys. Chem. Lett. 2020, 11, 10482–10488. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, M.T.; Aki-Yalcin, E. Homology Modeling in Drug Discovery: Overview, Current Applications, and Future Perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor Binding and Complex Structures of Human ACE2 to Spike RBD from Omicron and Delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Han, P.; Su, C.; Zhang, Y.; Bai, C.; Zheng, A.; Qiao, C.; Wang, Q.; Niu, S.; Chen, Q.; Zhang, Y.; et al. Molecular Insights into Receptor Binding of Recent Emerging SARS-CoV-2 Variants. Nat. Commun. 2021, 12, 6103. [Google Scholar] [CrossRef]
- An, X.; Zhang, Y.; Duan, L.; Jin, D.; Zhao, S.; Zhou, R.; Duan, Y.; Lian, F.; Tong, X. The Direct Evidence and Mechanism of Traditional Chinese Medicine Treatment of COVID-19. Biomed. Pharmacother. 2021, 137, 111267. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, W.; Sun, J.; Ou, G.; Zhong, N.-S.; Liu, Z. Exploring the Potential Pharmacological Mechanism of Hesperidin and Glucosyl Hesperidin against COVID-19 Based on Bioinformatics Analyses and Antiviral Assays. Am. J. Chin. Med. 2022, 50, 351–369. [Google Scholar] [CrossRef]
- Cheng, F.-J.; Huynh, T.-K.; Yang, C.-S.; Hu, D.-W.; Shen, Y.-C.; Tu, C.-Y.; Wu, Y.-C.; Tang, C.-H.; Huang, W.-C.; Chen, Y.; et al. Hesperidin Is a Potential Inhibitor against SARS-CoV-2 Infection. Nutrients 2021, 13, 2800. [Google Scholar] [CrossRef]
- Behloul, N.; Baha, S.; Guo, Y.; Yang, Z.; Shi, R.; Meng, J. In Silico Identification of Strong Binders of the SARS-CoV-2 Receptor-Binding Domain. Eur. J. Pharmacol. 2021, 890, 173701. [Google Scholar] [CrossRef] [PubMed]
- Pojskic, L. Screening of Preferential Binding Affinity of Selected Natural Compounds to SARS-CoV-2 Proteins Using in Silico Methods. Eurasian J. Med. Oncol. 2020, 4, 311. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of Therapeutic Targets for SARS-CoV-2 and Discovery of Potential Drugs by Computational Methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-3; Schrödinger LLC: New York, NY, USA, 2021.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for PK a Prediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L. OPLS3e: Extending Force Field Coverage for Drug-like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson Toby, J.; Higgins, D.G. Multiple Sequence Alignment Using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2002, 2, 2–3. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein− Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins: Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Olafuyi, O.; Kapusta, K.; Reed, A.; Kolodziejczyk, W.; Saloni, J.; Hill, G.A. Investigation of cannabidiol’s potential targets in limbic seizures. In-silico approach. J. Biomol. Struct. Dyn. 2022, 1–13. [Google Scholar] [CrossRef]
- Kapusta, K.; Kar, S.; Collins, J.T.; Franklin, L.M.; Kolodziejczyk, W.; Leszczynski, J.; Hill, G.A. Protein reliability analysis and virtual screening of natural inhibitors for SARS-CoV-2 main protease (Mpro) through docking, molecular mechanic & dynamic, and ADMET profiling. J. Biomol. Struct. Dyn. 2021, 39, 6810–6827. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; IEEE: Piscataway, NJ, USA, 2006; Volume 43. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant |  |  |  |  |  |  |  |  |  |
|---|---|---|---|---|---|---|---|---|---|
| Wild Type | Alpha | Beta | Gamma | Delta | Epsilon | Lambda | Mu | Omicron | |
| R403 | H34 | ||||||||
| HB 1 | |||||||||
| K417 | D30 | D30 | D30 | D30 | |||||
| HB + SB | HB + SB | HB + SB + vdW | HB + SB | ||||||
| Y449 | D38 | D38 | D38 | D38 | |||||
| HB + vdW | HB | HB | HB | ||||||
| Y453 | H34 | H34 | H34 | ||||||
| HB | vdW | π + vdW | |||||||
| L455 | H34 | ||||||||
| vdW | |||||||||
| F456 | T27 | ||||||||
| vdW | |||||||||
| K458 | E23 | ||||||||
| HB + SB | |||||||||
| Y473 | T27 | E23 | T27 | ||||||
| HB | HB | vdW | |||||||
| A475 | S19 | Q24 | S19 + Q24 | S19 | |||||
| HB | HB | 2 HB | HB | ||||||
| E484 | E75 | ||||||||
| HB + SB | |||||||||
| F486 | Q24 | ||||||||
| HB | |||||||||
| N487 | Q24 + Y83 | Y83 | Y83 | Y83 | Y83 | Q24 + Y83 | Y83 | Q24 + Y83 | |
| 2 HB | HB | HB | HB | HB | 2 HB | HB | 2 HB | ||
| Y489 | Y83 | Y83 | Y83 | Y83 | Y83 | Y83 | |||
| HB | HB | HB | HB | HB | HB | ||||
| L492 | K31 | K31 | |||||||
| HB | HB | ||||||||
| Q493 | E35 | K31 | H34 + D38 | K31 + E35 | E35 | E35 | K31 + E35 | E35 | |
| HB | HB | 2 HB | 2 HB + vdW | HB | HB | 2 HB | HB | ||
| G496 | K353 | ||||||||
| HB + vdW | |||||||||
| Q498 | Q42 + K353 | ||||||||
| 3 HB | |||||||||
| T500 | Y41 | Y41 + N330 | Y41 + N330 | Y41 + N330 | D355 | D355 | |||
| HB | 2 HB | 2vdW + HB | 2 HB | HB | HB | ||||
| N501 | Y41 + Q42 | Y41 | K353 | ||||||
| HB + π | π | HB | |||||||
| G502 | K353 | K353 | K353 | K353 | K353 | K353 | |||
| HB | HB | HB | HB | HB | HB | ||||
| Y505 | E37 + R393 | E37 | |||||||
| 2 HB | HB |
| # | Ligand | TCM Type 1 | PubChem CID | Molecular Formula | # | Ligand | TCM Type | PubChem CID | Molecular Formula |
|---|---|---|---|---|---|---|---|---|---|
| 1 | (−)-taxifolin | QPD | 712316 | C15H12O7 | 25 | Glycyrrhizic acid | QPD, MSD, CP, GC | 14982 | C42H62O16 |
| 2 | (+)-Epicatechin | QPD | 182232 | C15H14O6 | 26 | Hederagenin | HPD | 73299 | C30H48O4 |
| 3 | (2S)-dihydrobaicalein | QPD | 14135323 | C15H12O5 | 27 | Herbacetin | MSD | 5280544 | C15H10O7 |
| 4 | 3-O-Methylviolanone | QPD | 10019512 | C18H18O6 | 28 | Hesperidin | QPD, MSD | 10621 | C28H34O15 |
| 5 | 7-Methoxy-2-methyl isoflavone | DYY | 354368 | C17H14O3 | 29 | Inflacoumarin A | MSD | 5318437 | C20H18O4 |
| 6 | Amygdalin | QPD, MSD | 656516 | C20H27NO11 | 30 | Isolicoflavonol | RDS | 5318585 | C20H18O6 |
| 7 | Arenobufagin | LC | 12305198 | C24H32O6 | 31 | Isotrifoliol | MSD | 5318679 | C16H10O6 |
| 8 | Astragalus polysaccharide | HPD | 2782115 | C10H7ClN2O2S | 32 | Kaempferol | MSD, CP, DYY | 5280863 | C15H10O6 |
| 9 | Baicalin | QPD, MSD | 64982 | C21H18O11 | 33 | Kanzonol F | MSD | 101666840 | C26H28O5 |
| 10 | Bufotalin | LC | 12302120 | C26H36O6 | 34 | Licoisoflavone B | RDS | 5481234 | C20H16O6 |
| 11 | Cianidanol | QPD | 9064 | C15H14O6 | 35 | Luteolin | RDS | 5280445 | C15H10O6 |
| 12 | Cinobufotalin | LC | 259776 | C26H34O7 | 36 | Mairin | HPD | 64971 | C30H48O3 |
| 13 | Cyclo(L-Tyr-l-Phe) | QPD | 11438306 | C18H18N2O3 | 37 | naringenin | DYY, QPD | 932 | C15H12O5 |
| 14 | Dammaradienyl acetate | HPD | 179610 | C32H52O2 | 38 | Narirutin | QPD, MSD | 442431 | C27H32O14 |
| 15 | Delphinidin | MSD | 68245 | C15H11ClO7 | 39 | Neohesperidin | QPD, MSD | 442439 | C28H34O15 |
| 16 | Desacetylcinobufotalin | LC | 15513544 | C24H32O6 | 40 | Oxysanguinarine | HPD and TP | 443716 | C20H13NO5 |
| 17 | Ephedrine | QPD, MSD | 9294 | C10H15NO | 41 | Quercetin | MSD, RDS, CP, DYY | 5280343 | C15H10O7 |
| 18 | Eriodyctiol (flavanone) | QPD | 373261 | C15H12O6 | 42 | Resivit | MSD | 71629 | C15H14O7 |
| 19 | Estrone | MSD | 5870 | C18H22O2 | 43 | Semilicoisoflavone-B | RDS | 5481948 | C20H16O6 |
| 20 | Fisetin | RDS | 5281614 | C15H10O6 | 44 | Sitosterol | MSD | 222284 | C29H50O |
| 21 | Formononetin | MSD, DYY | 5280378 | C16H12O4 | 45 | SR-01000767148 | QPD | 676152 | C16H14O6 |
| 22 | Gamabufotalin | LC | 259803 | C24H34O5 | 46 | Stigmasterol | MSD, HPD | 5280794 | C29H48O |
| 23 | Glyasperin F | RDS | 392442 | C20H18O6 | 47 | telocinobufagin | LC | 259991 | C24H34O5 |
| 24 | Glycyrrhetinic Acid | GC | 10114 | C30H46O4 | 48 | ZINC13130930 | QPD | 25721350 | C16H14O5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ovchynnykova, O.; Kapusta, K.; Sizochenko, N.; Sukhyy, K.M.; Kolodziejczyk, W.; Hill, G.A.; Saloni, J. Homology Modeling and Molecular Dynamics-Driven Search for Natural Inhibitors That Universally Target Receptor-Binding Domain of Spike Glycoprotein in SARS-CoV-2 Variants. Molecules 2022, 27, 7336. https://doi.org/10.3390/molecules27217336
Ovchynnykova O, Kapusta K, Sizochenko N, Sukhyy KM, Kolodziejczyk W, Hill GA, Saloni J. Homology Modeling and Molecular Dynamics-Driven Search for Natural Inhibitors That Universally Target Receptor-Binding Domain of Spike Glycoprotein in SARS-CoV-2 Variants. Molecules. 2022; 27(21):7336. https://doi.org/10.3390/molecules27217336
Chicago/Turabian StyleOvchynnykova, Olha, Karina Kapusta, Natalia Sizochenko, Kostyantyn M. Sukhyy, Wojciech Kolodziejczyk, Glake A. Hill, and Julia Saloni. 2022. "Homology Modeling and Molecular Dynamics-Driven Search for Natural Inhibitors That Universally Target Receptor-Binding Domain of Spike Glycoprotein in SARS-CoV-2 Variants" Molecules 27, no. 21: 7336. https://doi.org/10.3390/molecules27217336
APA StyleOvchynnykova, O., Kapusta, K., Sizochenko, N., Sukhyy, K. M., Kolodziejczyk, W., Hill, G. A., & Saloni, J. (2022). Homology Modeling and Molecular Dynamics-Driven Search for Natural Inhibitors That Universally Target Receptor-Binding Domain of Spike Glycoprotein in SARS-CoV-2 Variants. Molecules, 27(21), 7336. https://doi.org/10.3390/molecules27217336