
A Computational Study on the Mechanism of Catalytic Cyclopropanation Reaction with Cobalt N-Confused Porphyrin: The Effects of Inner Carbon and Intramolecular Axial Ligand


Abstract
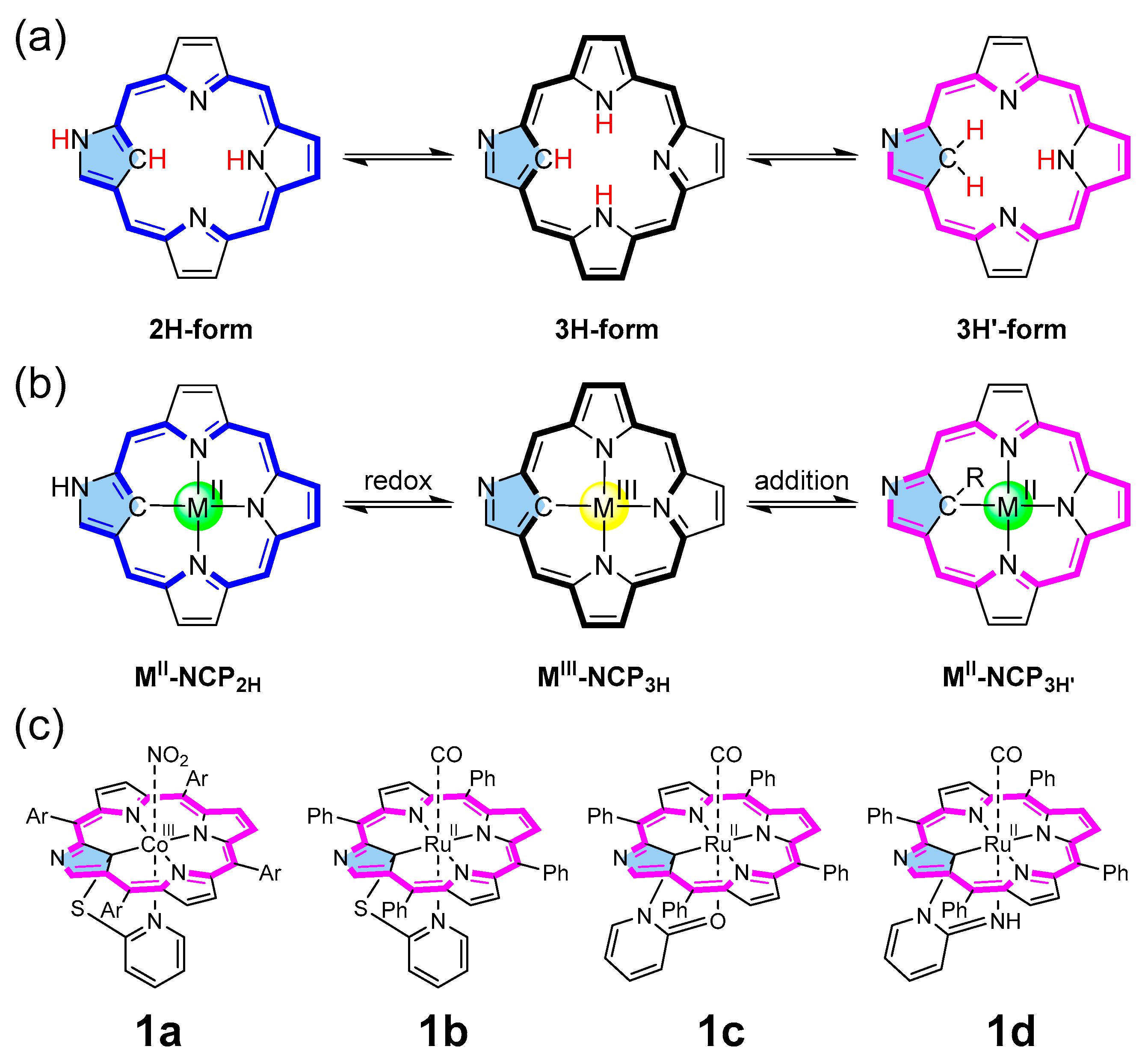
1. Introduction
2. Results and Discussion
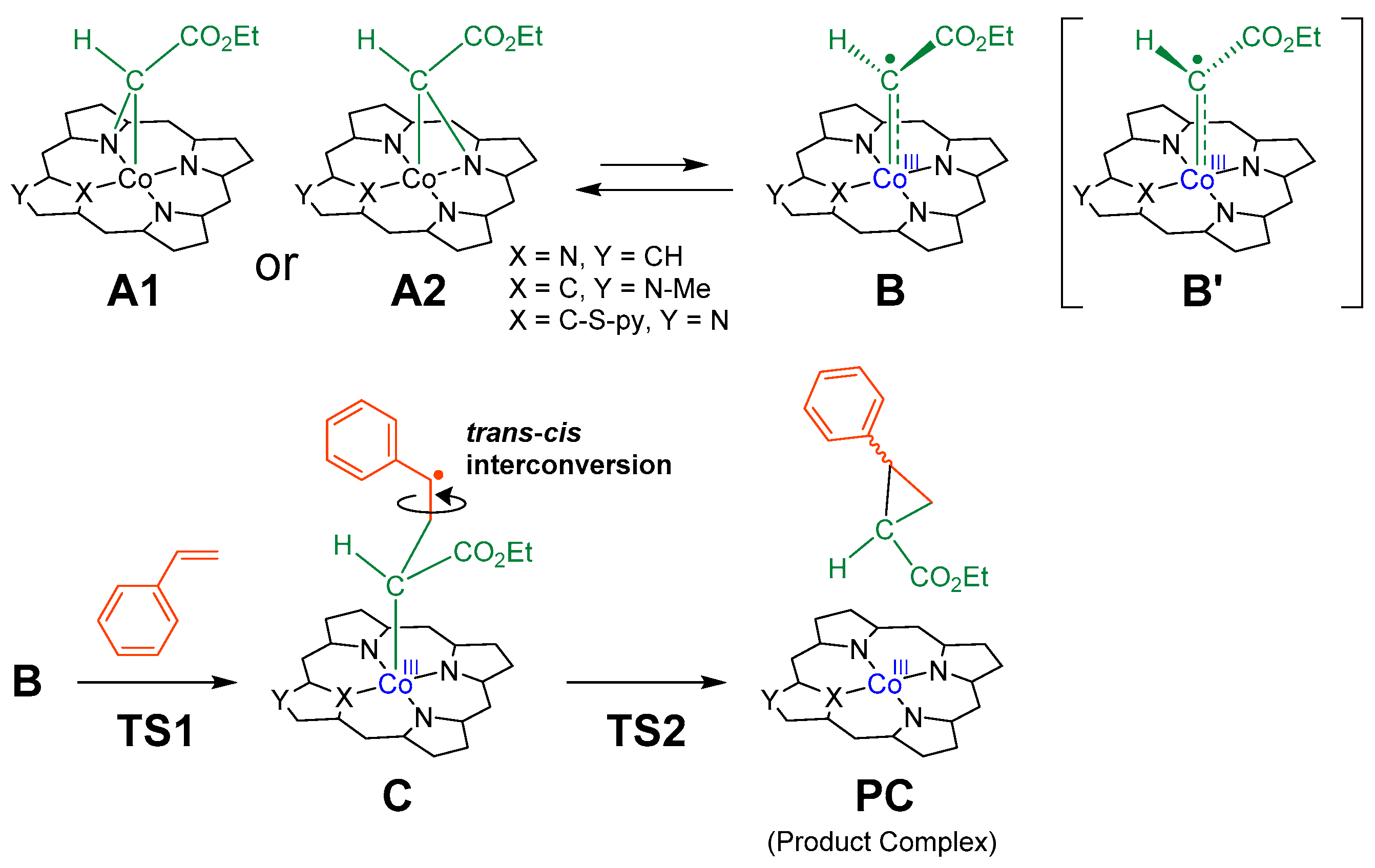
2.1. Reaction Mechanism
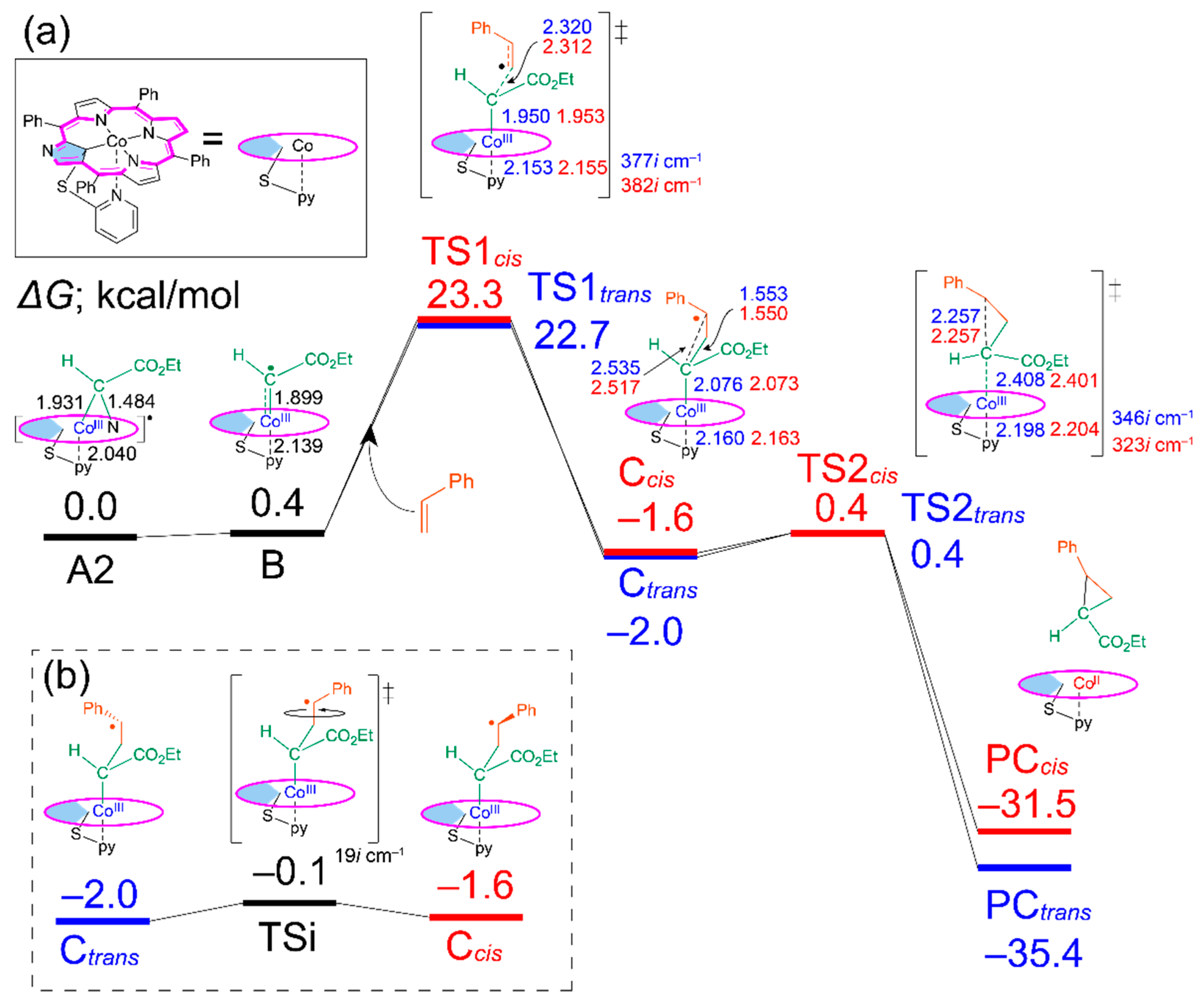
2.2. Energy Diagram for the Reaction with Co(NCTPPSpy)
2.3. Acceleration Effect
2.4. Structures of A and B
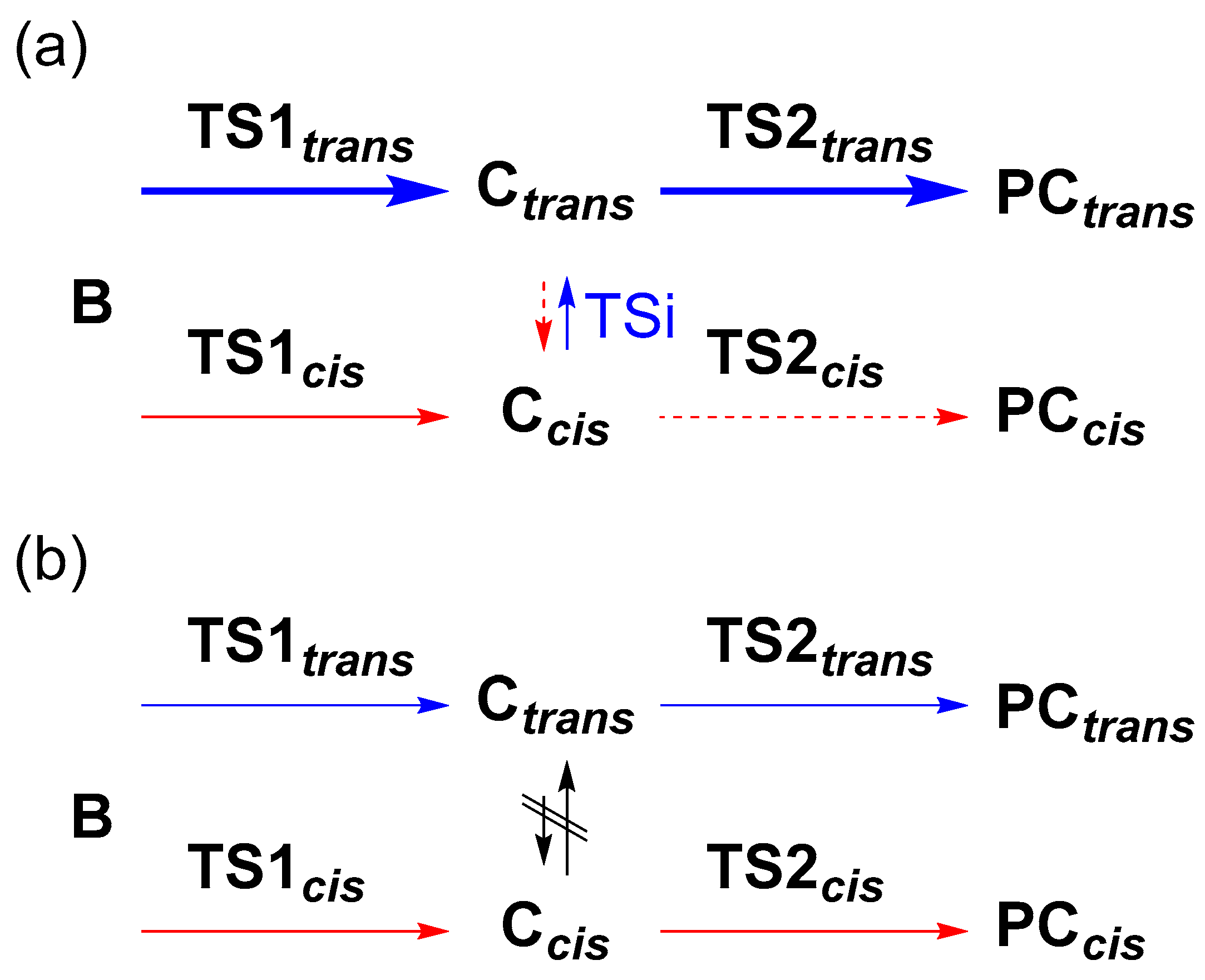
2.5. Trans/Cis Stereoselectivity
2.6. Structures of C
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, X.; Groves, J.T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef] [PubMed]
- Barona-Castaño, J.C.; Carmona-Vargas, C.C.; Brocksom, T.J.; De Oliveira, K.T. Porphyrins as Catalysts in Scalable Organic Reactions. Molecules 2016, 21, 310. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.M.Q.; Gonzaga, D.T.G.; Cardoso, M.F.C.; Forezi, L.D.S.M.; Gomes, A.T.P.C.; Da Silva, F.D.C.; Ferreira, V.F.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Carbene Transfer Reactions Catalysed by Dyes of the Metalloporphyrin Group. Molecules 2018, 23, 792. [Google Scholar] [CrossRef]
- Singh, R.; Mukherjee, A. Metalloporphyrin Catalyzed C–H Amination. ACS Catal. 2019, 9, 3604–3617. [Google Scholar] [CrossRef]
- Pereira, M.M.; Dias, L.D.; Calvete, M.J.F. Metalloporphyrins: Bioinspired Oxidation Catalysts. ACS Catal. 2018, 8, 10784–10808. [Google Scholar] [CrossRef]
- Ebner, C.; Carreira, E.M. Cyclopropanation Strategies in Recent Total Syntheses. Chem. Rev. 2017, 117, 11651–11679. [Google Scholar] [CrossRef] [PubMed]
- Talele, T.T. The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 2016, 59, 8712–8756. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.L.; Brown, K.C.; Bartley, D.W.; Kodadek, T. Mechanism of the Rhodium Porphyrin-Catalyzed Cyclopropanation of Alkenes. Science 1992, 256, 1544–1547. [Google Scholar]
- Ciammaichella, A.; Cardoni, V.; Leoni, A.; Tagliatesta, P. Rhodium Porphyrin Bound to a Merrifield Resin as Heterogeneous Catalyst for the Cyclopropanation Reaction of Olefins. Molecules 2016, 21, 278. [Google Scholar] [CrossRef] [PubMed]
- Che, C.-M.; Huang, J.-S. Ruthenium and osmium porphyrin carbene complexes: Synthesis, structure, and connection to the metal-mediated cyclopropanation of alkenes. Coord. Chem. Rev. 2002, 231, 151–164. [Google Scholar] [CrossRef]
- Intrieri, D.; Gac, S.L.; Caselli, A.; Rose, E.; Boitrel, B.; Gallo, E. Highly diastereoselective cyclopropanation of α-methylstyrene catalysed by a C2-symmetrical chiral iron porphyrin complex. Chem. Commun. 2014, 50, 1811–1813. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Tinoco, A.; Steck, V.; Fasan, R.; Zhang, Y. Cyclopropanations via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution. J. Am. Chem. Soc. 2018, 140, 1649–1662. [Google Scholar] [CrossRef] [PubMed]
- Simonneaux, G.; Maux, P.L. Optically active ruthenium porphyrins: Chiral recognition and asymmetric catalysis. Coord. Chem. Rev. 2002, 228, 43–60. [Google Scholar] [CrossRef]
- Wolf, M.W.; Vargas, D.A.; Lehnert, N. Engineering of RuMb: Toward a Green Catalyst for Carbene Insertion Reactions. Inorg. Chem. 2017, 56, 5623–5635. [Google Scholar] [CrossRef] [PubMed]
- Anding, B.J.; Ellern, A.; Woo, L.K. Olefin Cyclopropanation Catalyzed by Iridium(III) Porphyrin Complexes. Organometallics 2012, 31, 3628–3635. [Google Scholar] [CrossRef]
- Huang, L.; Chen, Y.; Gao, G.-Y.; Zhang, X.P. Diastereoselective and Enantioselective Cyclopropanation of Alkenes Catalyzed by Cobalt Porphyrins. J. Org. Chem. 2003, 68, 8179–8184. [Google Scholar] [CrossRef]
- Chen, Y.; Fields, K.B.; Zhang, X.P. Bromoporphyrins as Versatile Synthons for Modular Construction of Chiral Porphyrins: Cobalt-Catalyzed Highly Enantioselective and Diastereoselective Cyclopropanation. J. Am. Chem. Soc. 2004, 126, 14718–14719. [Google Scholar] [CrossRef] [PubMed]
- De Montellano, P.R.O. Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Yamamoto, T.; Mashita, S.; Deguchi, Y.; Fukuyama, K.; Ishida, M.; Mori, S.; Furuta, H. N-Confused Porphyrin Metal Complexes with an Axial Pyridine Directly Tethered from an Inner Carbon: A Bioinspired Ligand as a Versatile Platform for Catalysis. Eur. J. Inorg. Chem. 2018, 2018, 203–207. [Google Scholar] [CrossRef]
- Miyazaki, T.; Fukuyama, K.; Mashita, S.; Deguchi, Y.; Yamamoto, T.; Ishida, M.; Mori, S.; Furuta, H. Ruthenium N-Confused Porphyrins: Selective Reactivity for Ambident 2-Heteroatom-Substituted Pyridines Serving as Axial Ligands. ChemPlusChem 2019, 84, 603–607. [Google Scholar] [CrossRef]
- Furuta, H.; Asano, T.; Ogawa, T. “N-Confused Porphyrin”: A New Isomer of Tetraphenylporphyrin. J. Am. Chem. Soc. 1994, 116, 767–768. [Google Scholar] [CrossRef]
- Chmielewski, P.J.; Latos-Grażyński, L.; Rachlewicz, K.; Głowiak, T. Tetra-p-tolylporphyrin with an Inverted Pyrrole Ring: A Novel Isomer of Porphyrin. Angew. Chem. Int. Ed. Engl. 1994, 33, 779–781. [Google Scholar] [CrossRef]
- Toganoh, M.; Furuta, H. Creation from Confusion and Fusion in Porphyrin World—The Last Three Decades of N-Confused Porphyrinoid Chemistry. Chem. Rev. 2022, 122, 8313–8437. [Google Scholar] [CrossRef] [PubMed]
- Furuta, H.; Ishizuka, T.; Osuka, A.; Dejima, H.; Nakagawa, H.; Ishikawa, Y. NH Tautomerism of N-Confused Porphyrin. J. Am. Chem. Soc. 2001, 123, 6207–6208. [Google Scholar] [CrossRef] [PubMed]
- Furuta, H.; Ogawa, T.; Uwatoko, Y.; Araki, K. N-Confused Tetraphenylporphyrin–Silver(III) Complex. Inorg. Chem. 1999, 38, 2676–2682. [Google Scholar] [CrossRef]
- Maeda, H.; Ishikawa, Y.; Matsuda, T.; Osuka, A.; Furuta, H. Control of Cu(II) and Cu(III) States in N-Confused Porphyrin by Protonation/Deprotonation at the Peripheral Nitrogen. J. Am. Chem. Soc. 2003, 125, 11822–11823. [Google Scholar] [CrossRef] [PubMed]
- Niino, T.; Toganoh, M.; Andrioletti, B.; Furuta, H. Rhodium N-confused porphyrin-catalyzed alkene cyclopropanation. Chem. Commun. 2006, 41, 4335–4337. [Google Scholar] [CrossRef]
- Fields, K.B.; Engle, J.T.; Sripothongnak, S.; Kim, C.; Zhang, X.P.; Ziegler, C.J. Cobalt carbaporphyrin-catalyzed cyclopropanation. Chem. Commun. 2011, 47, 749–751. [Google Scholar] [CrossRef]
- Yamamoto, T.; Toganoh, M.; Furuta, H. Cooperation between metal and ligand in oxygen atom transport by N-confused porphyrin oxorhenium(V) complexes. Dalton Trans. 2012, 41, 9154–9157. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.-H.; Mahmood, M.H.; Zou, H.-B.; Yang, S.-B.; Liu, H.-Y. The first manganese N-confused porphyrins catalyzed oxidation of alkene. J. Mol. Catal. A-Chem. 2014, 395, 180–185. [Google Scholar] [CrossRef]
- Dela Cruz, J.B.; Ruamps, M.; Arco, S.; Hung, C.-H. Ni and Pd N-confused porphyrin complexes as catalysts for the synthesis of cyclic carbonates from epoxides and CO2. Dalton Trans. 2019, 48, 7527–7531. [Google Scholar] [CrossRef]
- Ge, Y.; Cheng, G.; Xu, N.; Wang, W.; Ke, H. Zinc 2-N-methyl N-confused porphyrin: An efficient catalyst for the conversion of CO2 into cyclic carbonates. Catal. Sci. Technol. 2019, 9, 4255–4261. [Google Scholar] [CrossRef]
- Dzik, W.I.; Xu, X.; Zhang, X.P.; Reek, J.N.H.; de Bruin, B. ‘Carbene Radicals’ in CoII(por)-Catalyzed Olefin Cyclopropanation. J. Am. Chem. Soc. 2010, 132, 10891–10902. [Google Scholar] [CrossRef]
- Lu, H.; Dzik, W.I.; Xu, X.; Wojtas, L.; de Bruin, B.; Zhang, X.P. Experimental Evidence for Cobalt(III)-Carbene Radicals: Key Intermediates in Cobalt(II)-Based Metalloradical Cyclopropanation. J. Am. Chem. Soc. 2011, 133, 8518–8521. [Google Scholar] [CrossRef] [PubMed]
- Oohora, K.; Meichin, H.; Kihira, Y.; Sugimoto, H.; Shiro, Y.; Hayashi, T. Manganese(V) Porphycene Complex Responsible for Inert C–H Bond Hydroxylation in a Myoglobin Matrix. J. Am. Chem. Soc. 2017, 139, 18460–18463. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, J.P.T.; Yosca, T.H.; Siegler, M.A.; Moënne-Loccoz, P.; Green, M.T.; Goldberg, D.P. Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex. J. Am. Chem. Soc. 2017, 139, 13640–13643. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Wachters, A.J.H. Gaussian Basis Set for Molecular Wavefunctions Containing Third-Row Atoms. J. Chem. Phys. 1970, 52, 1033–1036. [Google Scholar] [CrossRef]
- Hay, P.J. Gaussian basis sets for molecular calculations. The representation of 3d orbitals in transition-metal atoms. J. Chem. Phys. 1977, 66, 4377–4384. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W. Highly correlated systems. Excitation energies of first-row transition metals Sc–Cu. J. Chem. Phys. 1989, 91, 1062–1065. [Google Scholar] [CrossRef]
- Dunning, T.H.; Hay, P.J. Modern Theoretical Chemistry; Schaefer, H.F., III, Ed.; Plenum: New York, NY, USA, 1976; Volume 3, pp. 1–27. [Google Scholar]






| Entry | Catalyst | Cat. (mol%) | Yield (%) | trans/cis | Ref |
|---|---|---|---|---|---|
| 1 | Co(MeNCTPP)(py) | 1 | 85 | 93/7 | [28] |
| 2 2 | Co(TPP) | 1 | 67 | 74/26 | [28] |
| 3 | reduced 1a [Co(NCTPPSpy)] | 0.5 | 78 | 92/8 | [19] |
| 4 | Co(MeNCTPP)(py) | 0.5 | 31 | 92/8 | [19] |
| 5 | Co(TPP) | 0.5 | 6 | 72/28 | [19] |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔG(B–A) | Ea(TS1trans) 1 | ΔG(Ctrans–Ccis) | Ea(TS2cis) 2 | Ea[TSi(Ccis→Ctrans)] 3 | |
|---|---|---|---|---|---|
| Co(TPP) | 4.8 | 18.7 | −1.8 | 0.2 | 2.9 |
| Co(MeNCTPP) | 1.8 | 19.4 | −0.5 | 1.3 | 2.4 |
| Co(MeNCTPP)(py) | 0.9 | 20.6 | −1.4 | 2.3 | 1.9 |
| Co(NCTPPSpy) | 0.4 | 22.3 | −0.4 | 2.0 | 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwanaga, O.; Miyanishi, M.; Tachibana, T.; Miyazaki, T.; Shiota, Y.; Yoshizawa, K.; Furuta, H. A Computational Study on the Mechanism of Catalytic Cyclopropanation Reaction with Cobalt N-Confused Porphyrin: The Effects of Inner Carbon and Intramolecular Axial Ligand. Molecules 2022, 27, 7266. https://doi.org/10.3390/molecules27217266
Iwanaga O, Miyanishi M, Tachibana T, Miyazaki T, Shiota Y, Yoshizawa K, Furuta H. A Computational Study on the Mechanism of Catalytic Cyclopropanation Reaction with Cobalt N-Confused Porphyrin: The Effects of Inner Carbon and Intramolecular Axial Ligand. Molecules. 2022; 27(21):7266. https://doi.org/10.3390/molecules27217266
Chicago/Turabian StyleIwanaga, Osamu, Mayuko Miyanishi, Toshihiro Tachibana, Takaaki Miyazaki, Yoshihito Shiota, Kazunari Yoshizawa, and Hiroyuki Furuta. 2022. "A Computational Study on the Mechanism of Catalytic Cyclopropanation Reaction with Cobalt N-Confused Porphyrin: The Effects of Inner Carbon and Intramolecular Axial Ligand" Molecules 27, no. 21: 7266. https://doi.org/10.3390/molecules27217266
APA StyleIwanaga, O., Miyanishi, M., Tachibana, T., Miyazaki, T., Shiota, Y., Yoshizawa, K., & Furuta, H. (2022). A Computational Study on the Mechanism of Catalytic Cyclopropanation Reaction with Cobalt N-Confused Porphyrin: The Effects of Inner Carbon and Intramolecular Axial Ligand. Molecules, 27(21), 7266. https://doi.org/10.3390/molecules27217266