
Reagent-Controlled Highly Stereoselective Difluoromethylation: Efficient Access to Chiral α-Difluoromethylamines from Ketimines


Abstract
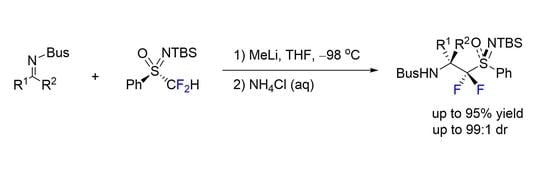
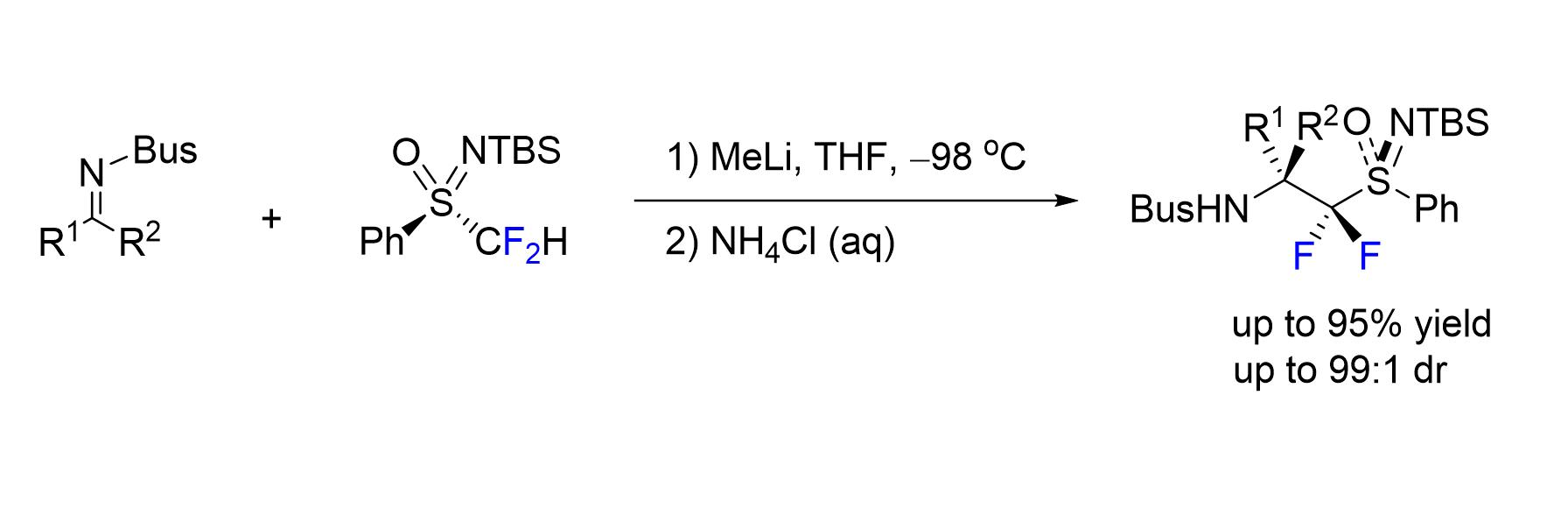
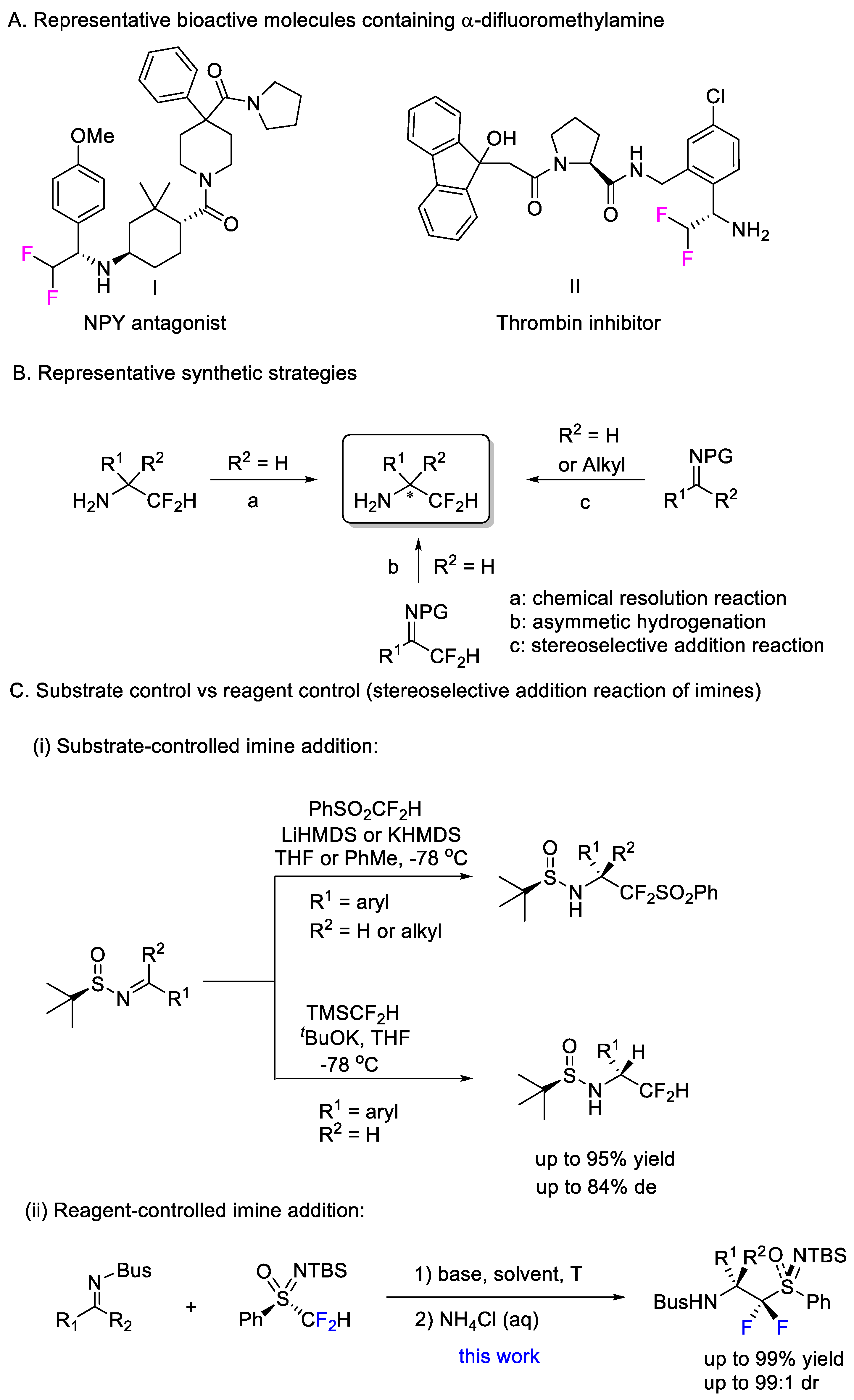
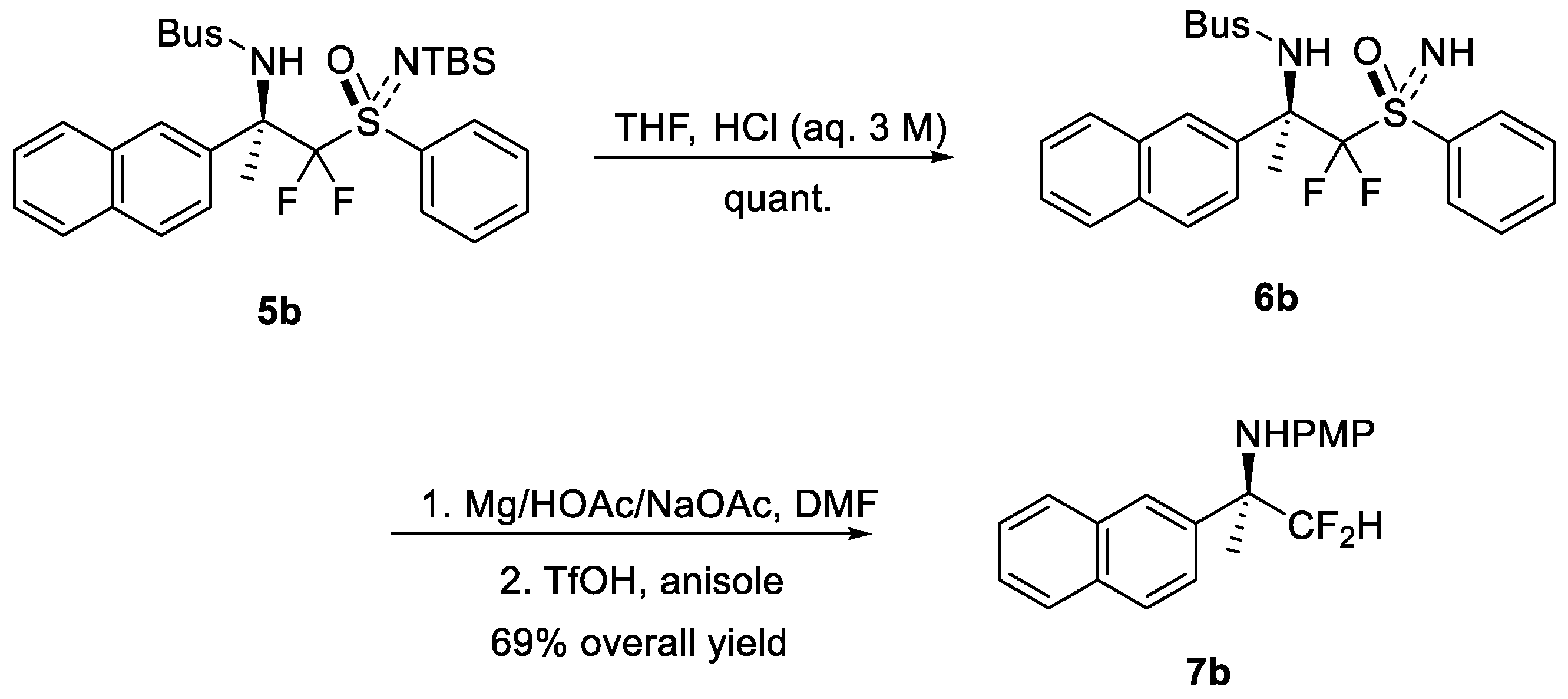
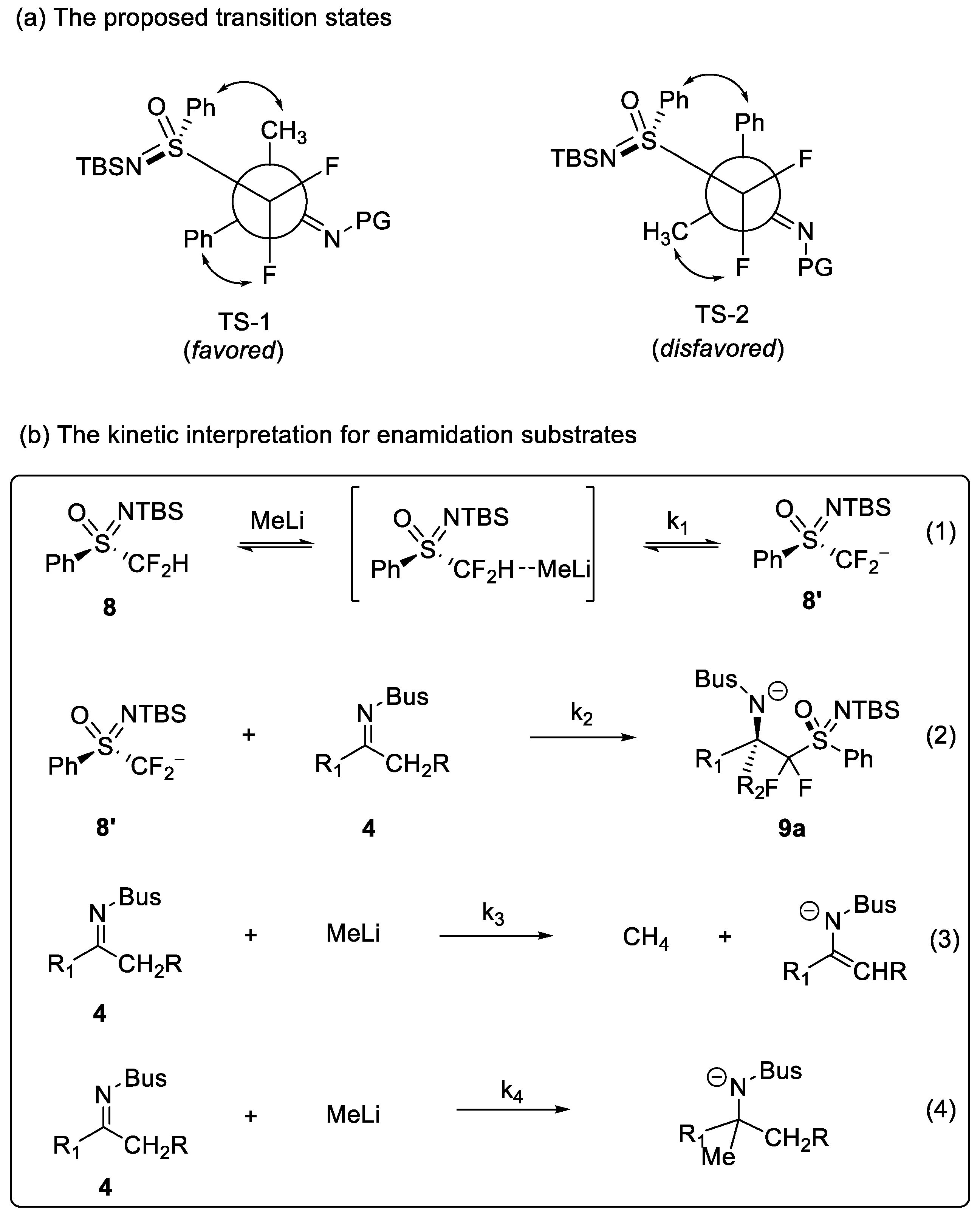
1. Introduction
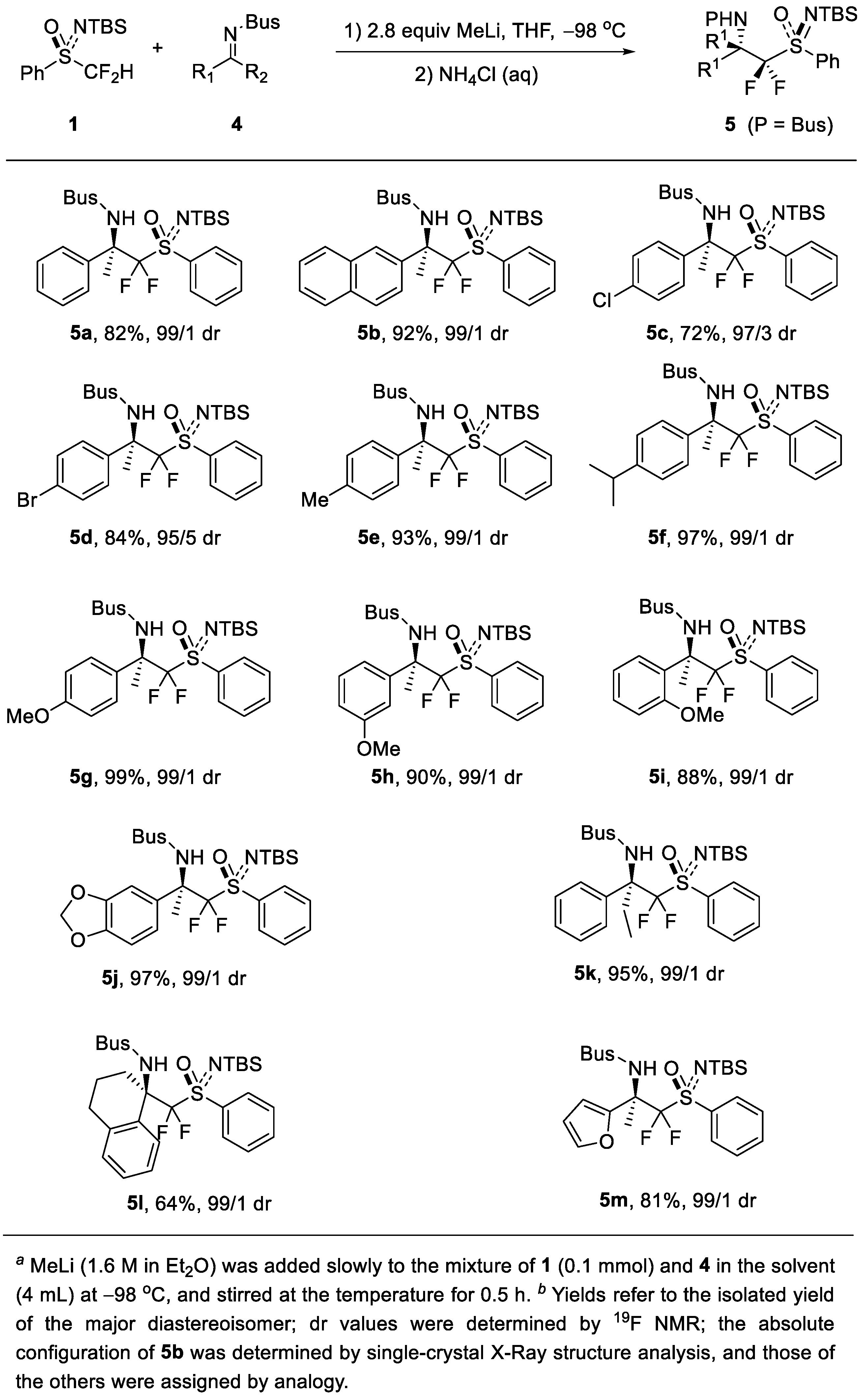
2. Results
3. Materials and Methods
3.1. General Information
3.2. General Procedure
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Meanwell, N.A. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.A.; McLoughlin, J.I. Hydrogen Bond Donor Properties of the Difluoromethyl Group. J. Org. Chem. 1995, 60, 1626–1631. [Google Scholar] [CrossRef]
- Narjes, F.; Koehler, K.F.; Koch, U.; Gerlach, B.; Colarusso, S.; Steinkühler, C.; Brunetti, M.; Altamura, S.; De Francesco, R.; Matassa, V.G. A designed P1 cysteine mimetic for covalent and non-covalent inhibitors of HCV NS3 protease. Bioorg. Med. Chem. Lett. 2002, 12, 701–704. [Google Scholar] [CrossRef]
- Chowdhury, M.A.; Abdellatif, K.R.A.; Dong, Y.; Das, D.; Suresh, M.R.; Knaus, E.E. Synthesis of Celecoxib Analogues Possessing a N-Difluoromethyl-1,2-dihydropyrid-2-one 5-Lipoxygenase Pharmacophore: Biological Evaluation as Dual Inhibitors of Cyclooxygenases and 5-Lipoxygenase with Anti-Inflammatory Activity. J. Med. Chem. 2009, 52, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Graneto, M.J.; Philips, W.J. 3-Difluoromethylpyrazolecarboxamide Fungicides, Compositions and Use. U.S. Patent 5,093,347, 3 March 1992. [Google Scholar]
- Gauthier, J.Y.; Belley, M.; Deschênes, D.; Fournier, J.-F.; Gagné, S.; Gareau, Y.; Hamel, M.; Hénault, M.; Hyjazie, H.; Kargman, S.; et al. The identification of 4,7-disubstituted naphthoic acid derivatives as UDP-competitive antagonists of P2Y14. Bioorg. Med. Chem. Lett. 2011, 21, 2836–2839. [Google Scholar] [CrossRef]
- Cho, K.; Ando, M.; Kobayashi, K.; Miyazoe, H.; Tsujino, T.; Ito, S.; Suzuki, T.; Tanaka, T.; Tokita, S.; Sato, N. Design, synthesis and evaluation of a novel cyclohexanamine class of neuropeptide Y Y1 receptor antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 4781–4785. [Google Scholar] [CrossRef]
- Selnick, H.G.; Barrow, J.C.; Nantermet, P.G.; Williams, P.D.; Stauffer, K.J.; Sanderson, P.E.; Rittle, K.E.; Morrissette, M.M.; Wiscount, C.M.; Tran, L.O.; et al. Benzylamine Derivatives and Their Use as Thrombin Inhibitors. WO Patent 2002050056A1, 27 June 2002. [Google Scholar]
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The Significance of Chirality in Drug Design and Development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef]
- Kanai, M.; Ueda, K.; Yasumoto, M.; Kuriyama, Y.; Inomiya, K.; Ootsuka, T.; Katsuhara, Y.; Higashiyama, K.; Ishii, A. Asymmetric synthesis of α-monofluoromethyl- and α-difluoromethylbenzylamines through regioselective hydrogenolysis. J. Fluorine Chem. 2005, 126, 377–383. [Google Scholar] [CrossRef]
- Abe, H.; Amii, H.; Uneyama, K. Pd-Catalyzed Asymmetric Hydrogenation of α-Fluorinated Iminoesters in Fluorinated Alcohol: A New and Catalytic Enantioselective Synthesis of Fluoro α-Amino Acid Derivatives. Org. Lett. 2001, 3, 313–315. [Google Scholar] [CrossRef]
- Chen, M.-W.; Duan, Y.; Chen, Q.-A.; Wang, D.-S.; Yu, C.-B.; Zhou, Y.-G. Enantioselective Pd-Catalyzed Hydrogenation of Fluorinated Imines: Facile Access to Chiral Fluorinated Amines. Org. Lett. 2010, 12, 5075–5077. [Google Scholar] [CrossRef]
- Funabiki, K.; Nagamori, M.; Goushi, S.; Matsui, M. First catalytic asymmetric synthesis of β-amino-β-polyfluoroalkyl ketones via proline-catalysed direct asymmetric carbon–carbon bond formation reaction of polyfluoroalkylated aldimines. Chem. Commun. 2004, 40, 1928–1929. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, J. Highly Diastereoselective Synthesis of α-Difluoromethyl Amines from N-tert-Butylsulfinyl Ketimines and Difluoromethyl Phenyl Sulfone. Chem. Eur. J. 2010, 16, 11443–11454. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, W.; Zheng, J.; Hu, J. Efficient and Direct Nucleophilic Difluoromethylation of Carbonyl Compounds and Imines with Me3SiCF2H at Ambient or Low Temperature. Org. Lett. 2011, 13, 5342–5345. [Google Scholar] [CrossRef]
- Shen, X.; Hu, J. Fluorinated Sulfoximines: Preparation, Reactions and Applications. Eur. J. Org. Chem. 2014, 2014, 4437–4451. [Google Scholar] [CrossRef]
- Liu, Q.; Ni, C.; Hu, J. China’s flourishing synthetic organofluorine chemistry: Innovations in the new millennium. Natl. Sci. Rev. 2017, 4, 303–325. [Google Scholar] [CrossRef]
- Liu, Q.; Shen, X.; Ni, C.; Hu, J. Stereoselective Carbonyl Olefination with Fluorosulfoximines: Facile Access to Z or E Terminal Monofluoroalkenes. Angew. Chem. Int. Ed. 2017, 56, 619–623. [Google Scholar] [CrossRef]
- Liu, Q.; Ni, C.; Wang, Q.; Meng, D.; Hu, J. Difluoromethyl 2-Pyridyl Sulfoximine: A Stereoselective Nucleophilic Reagent for Difluoro(aminosulfinyl)methylation and Difluoro(aminosulfonyl)methylation. CCS Chem. 2022, 1–12. [Google Scholar] [CrossRef]
- Liu, Q.; Kong, T.; Ni, C.; Hu, J. Dynamic Kinetic Resolution-Enabled Highly Stereoselective Nucleophilic Fluoroalkylation to Access Chiral β-Fluoro Amines. Org. Lett. 2022, 24, 5982–5987. [Google Scholar] [CrossRef]
- Zhu, G.; Negishi, E.-I. Fully Reagent-Controlled Asymmetric Synthesis of (−)-Spongidepsin via the Zr-Catalyzed Asymmetric Carboalumination of Alkenes (ZACA Reaction). Org. Lett. 2007, 9, 2771–2774. [Google Scholar] [CrossRef]
- You, Y.; Zhang, L.; Luo, S. Reagent-controlled enantioselectivity switch for the asymmetric fluorination of β-ketocarbonyls by chiral primary amine catalysis. Chem. Sci. 2017, 8, 621–626. [Google Scholar] [CrossRef]
- Cui, L.; You, Y.; Mi, X.; Luo, S. Asymmetric Fluorination of α-Branched Aldehydes by Chiral Primary Amine Catalysis: Reagent-Controlled Enantioselectivity Switch. J. Org. Chem. 2018, 83, 4250–4256. [Google Scholar] [CrossRef]
- Schreiber, B.; Martinek, H.; Wolschann, P.; Schuster, P. Kinetic studies on the nucleophilic addition to double bonds. 1. Addition of amines to electrophilic carbon-carbon double bonds. J. Am. Chem. Soc. 1979, 101, 4708–4713. [Google Scholar] [CrossRef]
- Henderson, W.M.; Shelver, W.H. Kinetic Study of the Nucleophilic Addition of Diethyl Phosphonate to Benzylideneanilines. J. Pharm. Sci. 1969, 58, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Miao, W.; Ni, C.; Hu, J. Stereoselective Nucleophilic Fluoromethylation of Aryl Ketones: Dynamic Kinetic Resolution of Chiral α-Fluoro Carbanions. Angew. Chem. Int. Ed. 2014, 53, 775–779. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Variation from Standard Conditions | Yield (%) b | dr b |
| 1 | none | 38 | 99/1 |
| 2 | nBuLi instead of MeLi | 35 | 99/1 |
| 3 | NaHMDS instead of MeLi | 3 | n.d. |
| 4 | CH2Cl2 instead of THF | 0 | n.d. |
| 5 | Toluene instead of THF | 8 | n.d. |
| 6 | Et2O instead of THF | <5 | n.d. |
| 7 | THF/HMPA (10/1, v/v) instead of THF | 9 | 99/1 |
| 8 | 2.0 equiv. 4a and 2.8 equiv. MeLi were used | 77 | 99/1 |
| 9 c | N-Ts-4a was used | 40 | n.d. |
| 10 c | N-SPh-4a was used | <5 | n.d. |
| 11 c | 0.025 M instead of 0.05 M | 84 | 99/1 |
| 12 d | −98 °C instead of −78 °C | 90 | 99/1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Kong, T.; Ni, C.; Hu, J. Reagent-Controlled Highly Stereoselective Difluoromethylation: Efficient Access to Chiral α-Difluoromethylamines from Ketimines. Molecules 2022, 27, 7076. https://doi.org/10.3390/molecules27207076
Liu Q, Kong T, Ni C, Hu J. Reagent-Controlled Highly Stereoselective Difluoromethylation: Efficient Access to Chiral α-Difluoromethylamines from Ketimines. Molecules. 2022; 27(20):7076. https://doi.org/10.3390/molecules27207076
Chicago/Turabian StyleLiu, Qinghe, Taige Kong, Chuanfa Ni, and Jinbo Hu. 2022. "Reagent-Controlled Highly Stereoselective Difluoromethylation: Efficient Access to Chiral α-Difluoromethylamines from Ketimines" Molecules 27, no. 20: 7076. https://doi.org/10.3390/molecules27207076
APA StyleLiu, Q., Kong, T., Ni, C., & Hu, J. (2022). Reagent-Controlled Highly Stereoselective Difluoromethylation: Efficient Access to Chiral α-Difluoromethylamines from Ketimines. Molecules, 27(20), 7076. https://doi.org/10.3390/molecules27207076