
Personalized Treatment for Infantile Ascending Hereditary Spastic Paralysis Based on In Silico Strategies

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
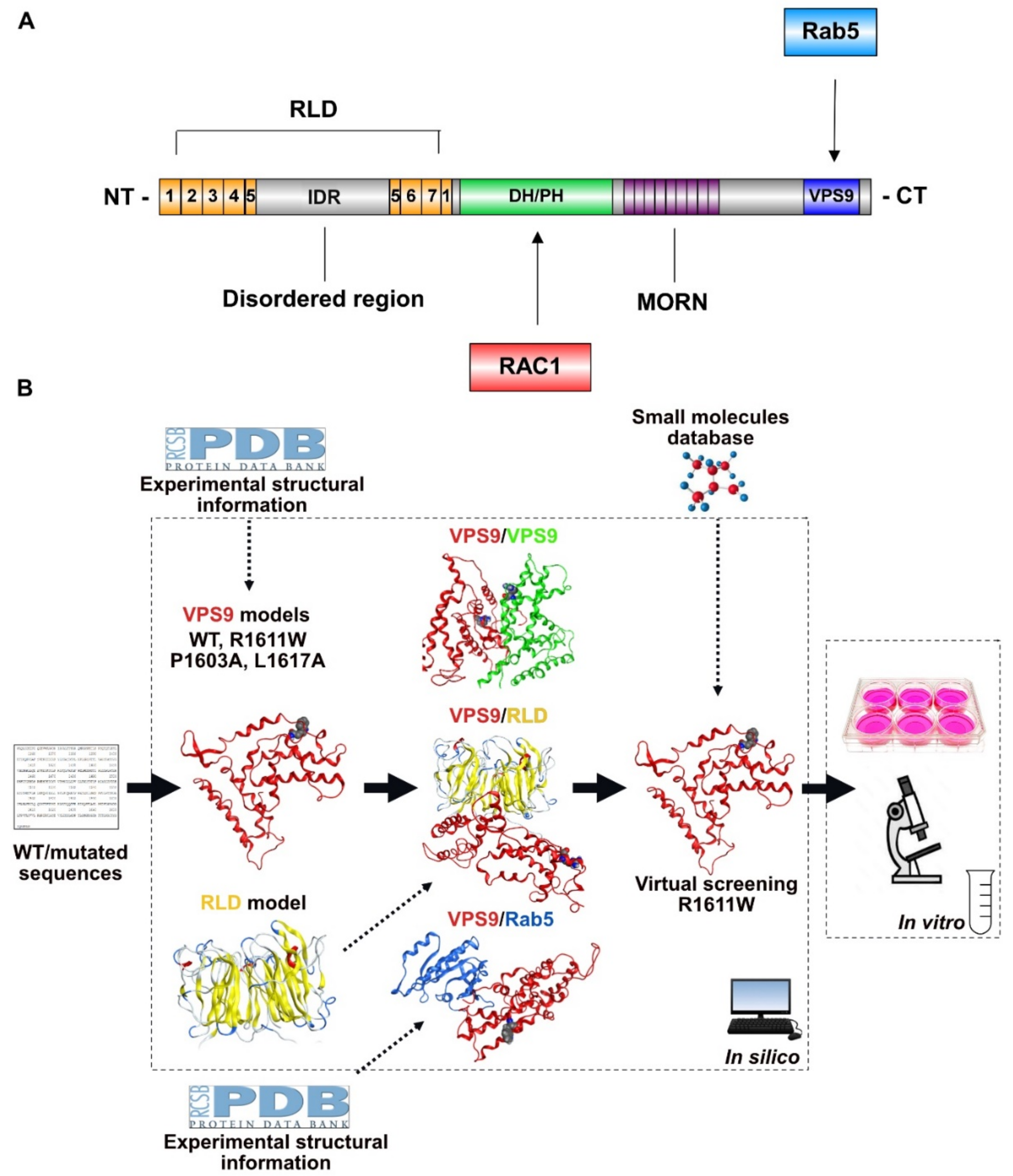
2.1. Setting-Up the Computational Strategy
2.2. 3D Models of VPS9 and RLD Domains and Related Mutants
2.3. VPS9 Models Stability
2.4. Homo and Heterodimers: Building and Structural/Energetic Analysis
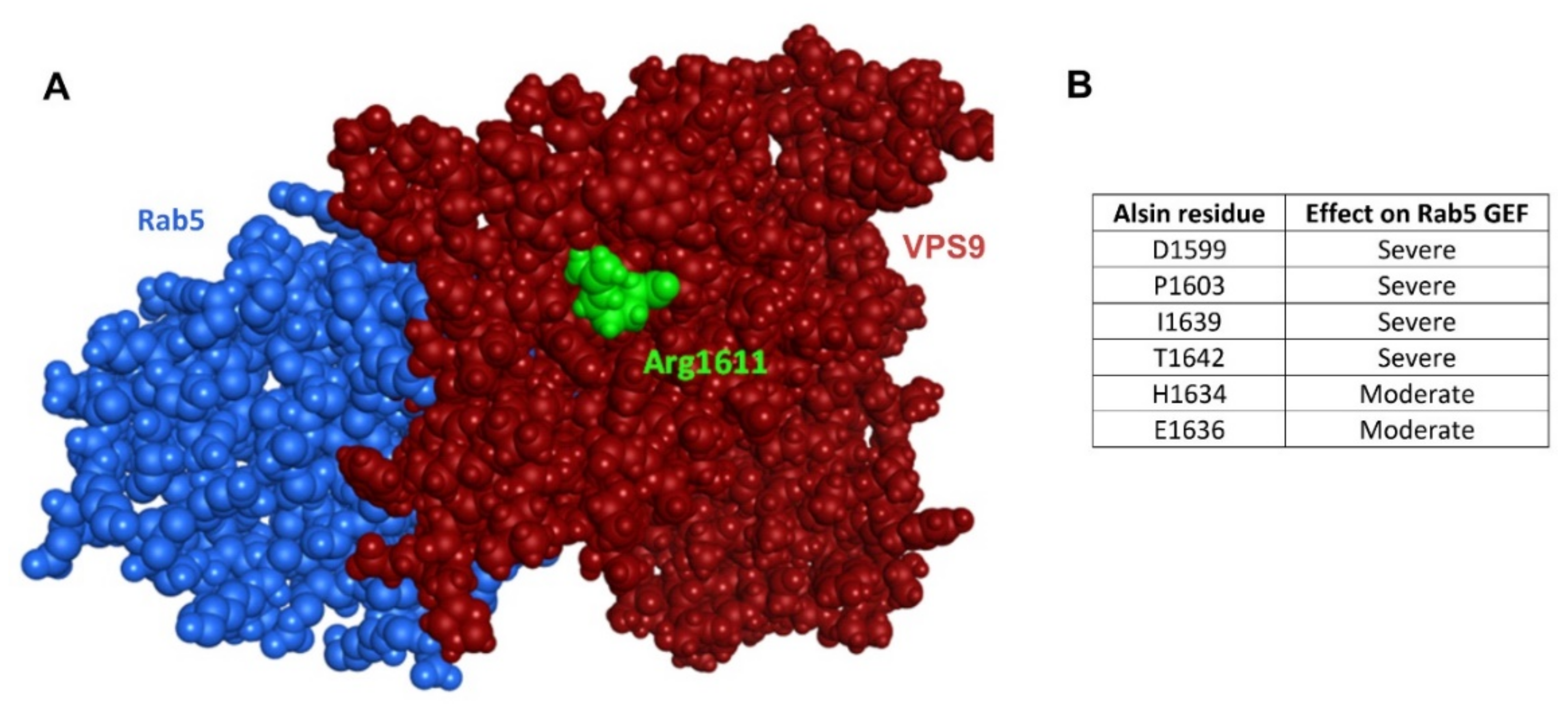
2.5. VPS9 and Rab5
2.6. Analysis of Additional Mutants
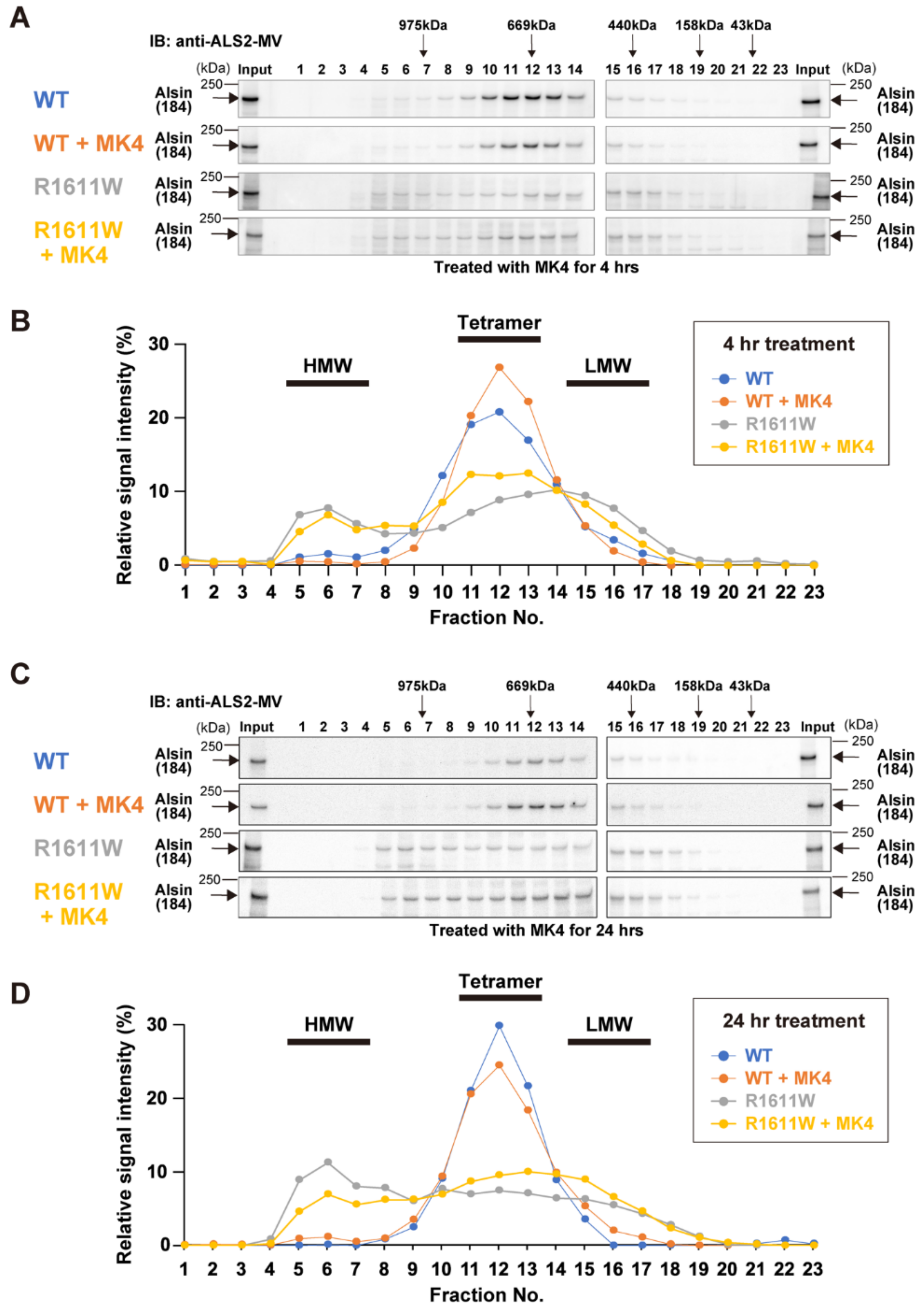
2.7. Virtual Screening to Identify Small Molecules Restoring the Functionality of Alsin
2.8. Experimental Validation
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eymard-Pierre, E.; Lesca, G.; Dollet, S.; Santorelli, F.M.; Di Capua, M.; Bertini, E.; Boespflug-Tanguy, O. Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am. J. Hum. Genet. 2002, 71, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Montiel, M.J.; Chaineau, M.; Durcan, T.M. The Neglected Genes of ALS: Cytoskeletal Dynamics Impact Synaptic Degeneration in ALS. Front. Cell. Neurosci. 2020, 14, 594975. [Google Scholar] [CrossRef] [PubMed]
- Chandran, J.; Ding, J.; Cai, H. Alsin and the molecular pathways of amyotrophic lateral sclerosis. Mol. Neurobiol. 2007, 36, 224–231. [Google Scholar] [PubMed]
- Lesca, G.; Eymard–Pierre, E.; Santorelli, F.M.; Cusmai, R.; Di Capua, M.; Valente, E.M.; Attia–Sobol, J.; Plauchu, H.; Leuzzi, V.; Ponzone, A.; et al. Infantile ascending hereditary spastic paralysis (IAHSP): Clinical features in 11 families. Neurology 2003, 60, 674–682. [Google Scholar] [CrossRef]
- Gautam, M.; Jara, J.H.; Sekerkova, G.; Yasvoina, M.V.; Martina, M.; Özdinler, P.H. Absence of alsin function leads to corticospinal motor neuron vulnerability via novel disease mechanisms. Hum. Mol. Genet. 2015, 25, 1074–1087. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar]
- Beckman, J.S.; Estévez, A.G.; Crow, J.P.; Barbeito, L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001, 24, 15–20. [Google Scholar] [CrossRef]
- Soares, D.C.; Barlow, P.N.; Porteous, D.J.; Devon, R.S. An interrupted beta-propeller and protein disorder: Structural bioinformatics insights into the N-terminus of alsin. J. Mol. Model. 2009, 15, 113–122. [Google Scholar] [CrossRef]
- Shimakura, K.; Sato, K.; Mitsui, S.; Ono, S.; Otomo, A.; Hadano, S. The N-terminal intrinsically disordered region mediates intracellular localization and self-oligomerization of ALS2. Biochem. Biophys. Res. Commun. 2021, 569, 106–111. [Google Scholar] [CrossRef]
- Sato, K.; Otomo, A.; Ueda, M.T.; Hiratsuka, Y.; Suzuki-Utsunomiya, K.; Sugiyama, J.; Murakoshi, S.; Mitsui, S.; Ono, S.; Nakagawa, S.; et al. Altered oligomeric states in pathogenic ALS2 variants associated with juvenile motor neuron diseases cause loss of ALS2-mediated endosomal function. J. Biol. Chem. 2018, 293, 17135–17153. [Google Scholar] [CrossRef]
- Topp, J.D.; Gray, N.W.; Gerard, R.D.; Horazdovsky, B.F. Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J. Biol. Chem. 2004, 279, 24612–24623. [Google Scholar] [CrossRef]
- Rossi Sebastiano, M.; Ermondi, G.; Hadano, S.; Caron, G. AI-based protein structure databases have the potential to accelerate rare diseases research: AlphaFoldDB and the case of IAHSP/Alsin. Drug Discov. Today 2021, 27, 1652–1660. [Google Scholar] [CrossRef]
- Gautam, M.; Carratore, R.D.; Helmold, B.; Tessa, A.; Gozutok, O.; Chandel, N.; Idrisoglu, H.; Bongioanni, P.; Battini, R.; Ozdinler, P.H. 2-Year-Old and 3-Year-Old Italian ALS Patients with Novel ALS2 Mutations: Identification of Key Metabolites in Their Serum and Plasma. Metabolites 2022, 12, 174. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Otomo, A.; Hadano, S.; Okada, T.; Mizumura, H.; Kunita, R.; Nishijima, H.; Showguchi-Miyata, J.; Yanagisawa, Y.; Kohiki, E.; Suga, E.; et al. ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum. Mol. Genet. 2003, 12, 1671–1687. [Google Scholar] [CrossRef]
- Delprato, A.; Merithew, E.; Lambright, D.G. Structure, exchange determinants, and family-wide Rab specificity of the tandem helical bundle and Vps9 domains of Rabex-5. Cell 2004, 118, 607–617. [Google Scholar] [CrossRef]
- Kuriata, A.; Gierut, A.M.; Oleniecki, T.; Ciemny, M.P.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. CABS-flex 2.0: A web server for fast simulations of flexibility of protein structures. Nucleic Acids Res. 2018, 46, W338–W343. [Google Scholar] [CrossRef]
- Tiwari, S.P.; Fuglebakk, E.; Hollup, S.M.; Skjærven, L.; Cragnolini, T.; Grindhaug, S.H.; Tekle, K.M.; Reuter, N. WEBnmat v2.0: Web server and services for comparing protein flexibility. BMC Bioinform. 2014, 15, 427. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Caldararu, O.; Blundell, T.L.; Kepp, K.P. Three Simple Properties Explain Protein Stability Change upon Mutation. J. Chem. Inf. Model. 2021, 61, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- Hadano, S.; Benn, S.C.; Kakuta, S.; Otomo, A.; Sudo, K.; Kunita, R.; Suzuki-Utsunomiya, K.; Mizumura, H.; Shefner, J.M.; Cox, G.A.; et al. Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum. Mol. Genet. 2006, 15, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Otomo, A.; Kunita, R.; Suzuki-Utsunomiya, K.; Ikeda, J.-E.; Hadano, S. Defective relocalization of ALS2/alsin missense mutants to Rac1-induced macropinosomes accounts for loss of their cellular function and leads to disturbed amphisome formation. FEBS Lett. 2011, 585, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Kochnev, Y.; Hellemann, E.; Cassidy, K.C.; Durrant, J.D. Webina: An open-source library and web app that runs AutoDock Vina entirely in the web browser. Bioinformatics 2020, 36, 4513–4515. [Google Scholar] [CrossRef]
- Inoue, T.; Sugiyama, T.; Matsubara, T.; Kawai, S.; Furukawa, S. Inverse correlation between the changes of lumbar bone mineral density and serum undercarboxylated osteocalcin after vitamin K2 (menatetrenone) treatment in children treated with glucocorticoid and alfacalcidol. Endocr. J. 2001, 48, 11–18. [Google Scholar] [CrossRef][Green Version]
- Simes, D.C.; Viegas, C.S.B.; Araújo, N.; Marreiros, C. Vitamin K as a diet supplement with impact in human health: Current evidence in age-related diseases. Nutrients 2020, 12, 138. [Google Scholar] [CrossRef]
- Popa, D. The Role of Vitamin K in Humans: Implication in Aging and Age-Associated Diseases. Antioxidants 2021, 10, 566. [Google Scholar] [CrossRef]
- Westhofen, P.; Watzka, M.; Marinova, M.; Hass, M.; Kirfel, G.; Müller, J.; Bevans, C.G.; Müller, C.R.; Oldenburg, J. Human Vitamin K 2, 3-Epoxide Reductase Complex Subunit 1-like 1 (VKORC1L1) Mediates Vitamin K-dependent Intracellular Antioxidant Function. J. Biol. Chem. 2011, 286, 15085–15094. [Google Scholar] [CrossRef]
- Kiryu-seo, S.; Tamada, H.; Kato, Y.; Yasud, K.; Ishihara, N. Mitochondrial fission is an acute and adaptive response in injured motor neurons. Sci. Rep. 2016, 6, 28331. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Review Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Hsu, F.; Spannl, S.; Ferguson, C.; Hyman, A.A.; Parton, R.G.; Zerial, M. Rab5 and Alsin regulate stress-activated cytoprotective signaling on mitochondria. eLife 2018, 7, e32282. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Desta, I.T.; Porter, K.A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and Its Limits in Rigid Body Protein-Protein Docking. Structure 2020, 28, 1071–1081.e3. [Google Scholar] [CrossRef]
- Mitternacht, S. FreeSASA: An open source C library for solvent accessible surface area calculations. F1000Research 2016, 5, 189. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 7855, 33–38. [Google Scholar] [CrossRef]
- Ribeiro, J.V.; Bernardi, R.C.; Rudack, T.; Stone, J.E.; Phillips, J.C.; Freddolino, P.L.; Schulten, K. QwikMD—Integrative Molecular Dynamics Toolkit for Novices and Experts. Sci. Rep. 2016, 6, 26536. [Google Scholar] [CrossRef]
- Koes, D.R.; Camacho, C.J. Small-molecule inhibitor starting points learned from protein-protein interaction inhibitor structure. Bioinformatics 2012, 28, 784–791. [Google Scholar] [CrossRef]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Schöning-Stierand, K.; Diedrich, K.; Fährrolfes, R.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Steinegger, R.; Rarey, M. ProteinsPlus: Interactive analysis of protein–ligand binding interfaces. Nucleic Acids Res. 2020, 48, W48–W53. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15-Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein-Ligand Docking Using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Cross, S.; Cruciani, G. Molecular fields in drug discovery: Getting old or reaching maturity? Drug Discov. Today 2010, 15, 23–32. [Google Scholar] [CrossRef]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef]
- Kunita, R.; Otomo, A.; Mizumura, H.; Suzuki, K.; Showguchi-Miyata, J.; Yanagisawa, Y.; Hadano, S.; Ikeda, J.E. Homo-oligomerization of ALS2 through its unique carboxyl-terminal regions is essential for the ALS2-associated Rab5 guanine nucleotide exchange activity and its regulatory function on endosome trafficking. J. Biol. Chem. 2004, 279, 38626–38635. [Google Scholar] [CrossRef]
- Kunita, R.; Otomo, A.; Mizumura, H.; Suzuki-Utsunomiya, K.; Hadano, S.; Ikeda, J.E. The Rab5 activator ALS2/alsin acts as a novel Rac1 effector through Rac1-activated endocytosis. J. Biol. Chem. 2007, 282, 16599–16611. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi Sebastiano, M.; Ermondi, G.; Sato, K.; Otomo, A.; Hadano, S.; Caron, G. Personalized Treatment for Infantile Ascending Hereditary Spastic Paralysis Based on In Silico Strategies. Molecules 2022, 27, 7063. https://doi.org/10.3390/molecules27207063
Rossi Sebastiano M, Ermondi G, Sato K, Otomo A, Hadano S, Caron G. Personalized Treatment for Infantile Ascending Hereditary Spastic Paralysis Based on In Silico Strategies. Molecules. 2022; 27(20):7063. https://doi.org/10.3390/molecules27207063
Chicago/Turabian StyleRossi Sebastiano, Matteo, Giuseppe Ermondi, Kai Sato, Asako Otomo, Shinji Hadano, and Giulia Caron. 2022. "Personalized Treatment for Infantile Ascending Hereditary Spastic Paralysis Based on In Silico Strategies" Molecules 27, no. 20: 7063. https://doi.org/10.3390/molecules27207063
APA StyleRossi Sebastiano, M., Ermondi, G., Sato, K., Otomo, A., Hadano, S., & Caron, G. (2022). Personalized Treatment for Infantile Ascending Hereditary Spastic Paralysis Based on In Silico Strategies. Molecules, 27(20), 7063. https://doi.org/10.3390/molecules27207063