
Molybdenum Cofactor Deficiency in Humans



Abstract
1. Introduction
2. Clinical Presentation of Molybdenum Cofactor Deficient Patients
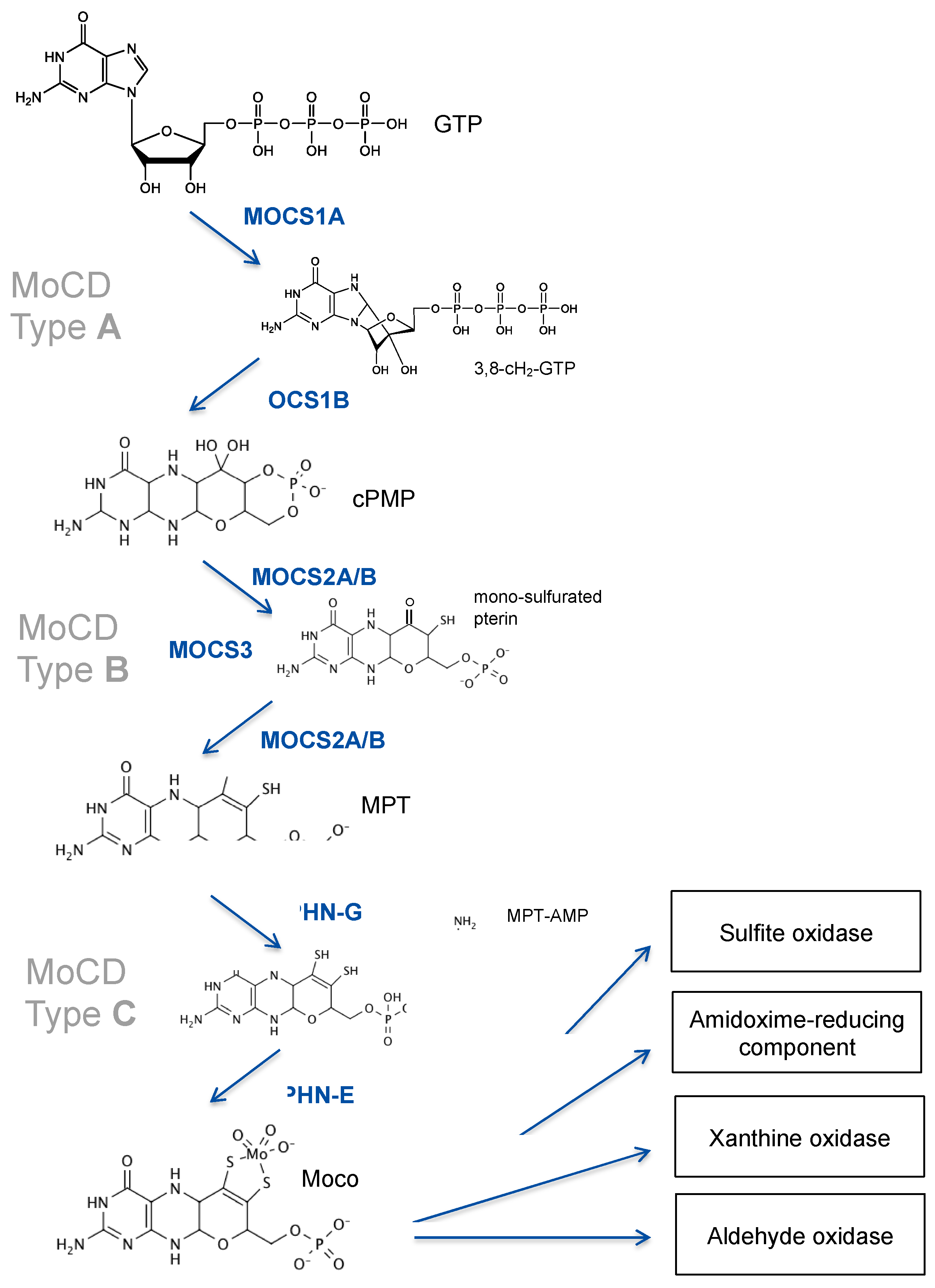
3. Genetics of Moco Deficiency
3.1. MOCS1 Mutations Lead to cPMP Deficiency
3.2. Loss of MPT Synthesis Is Caused by Mutations in MOCS2
3.3. Patients with GPHN Mutations Show a Broad Spectrum of Neurological Disorders
4. Therapies to Treat MoCD
4.1. Disease Mechanisms
4.2. Treatment of MoCD Type A (MOCS1) Patients with cPMP
4.3. Molybdate Treatment
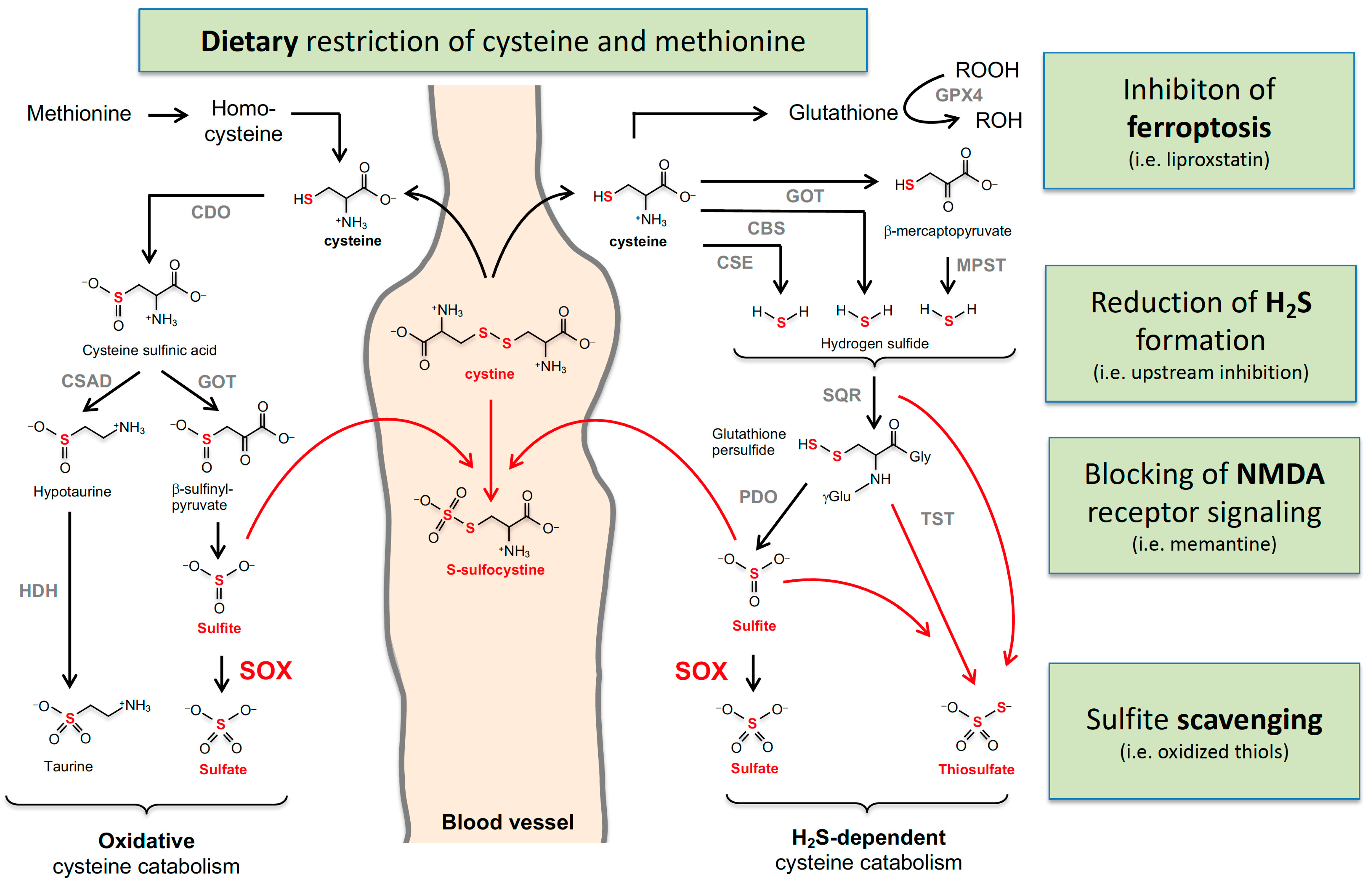
4.4. Dietary Restriction
4.5. Targeting NMDA Receptors
4.6. Sulfite Scavenging
4.7. Ferroptosis Inhibition
4.8. Is H2S Involved in the Pathophysiology of MoCD?
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schwarz, G.; Mendel, R.R.; Ribbe, M.W. Molybdenum Cofactors, Enzymes and Pathways. Nature 2009, 460, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Havemeyer, A.; Bittner, F.; Wollers, S.; Mendel, R.; Kunze, T.; Clement, B. Identification of the Missing Component in the Mitochondrial Benzamidoxime Prodrug-Converting System as a Novel Molybdenum Enzyme. J. Biol. Chem. 2006, 281, 34796–34802. [Google Scholar] [CrossRef]
- Schwarz, G. Molybdenum Cofactor and Human Disease. Curr. Opin. Chem. Biol. 2016, 31, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Mayr, S.J.; Mendel, R.R.; Schwarz, G. Molybdenum Cofactor Biology, Evolution and Deficiency. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118883. [Google Scholar] [CrossRef]
- Wuebbens, M.M.; Rajagopalan, K.V. Investigation of the Early Steps of Molybdopterin Biosynthesis in Escherichia Coli through the Use of in Vivo Labeling Studies. J. Biol. Chem. 1995, 270, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Santamaria-Araujo, J.A.; Fischer, B.; Otte, T.; Nimtz, M.; Mendel, R.R.; Wray, V.; Schwarz, G. The Tetrahydropyranopterin Structure of the Sulfur-Free and Metal-Free Molybdenum Cofactor Precursor. J. Biol. Chem. 2004, 279, 15994–15999. [Google Scholar] [CrossRef]
- Johnson, J.L.; Hainline, B.E.; Rajagopalan, K.V.; Arison, B.H. The Pterin Component of the Molybdenum Cofactor. Structural Characterization of Two Fluorescent Derivatives. J. Biol. Chem. 1984, 259, 5414–5422. [Google Scholar] [CrossRef]
- Reiss, J.; Cohen, N.; Dorche, C.; Mandel, H.; Mendel, R.R.; Stallmeyer, B.; Zabot, M.T.; Dierks, T. Mutations in a Polycistronic Nuclear Gene Associated with Molybdenum Cofactor Deficiency. Nat. Genet. 1998, 20, 51–53. [Google Scholar] [CrossRef]
- Gray, T.A.; Nicholls, R.D. Diverse Splicing Mechanisms Fuse the Evolutionarily Conserved Bicistronic MOCS1A and MOCS1B Open Reading Frames. RNA 2000, 6, 928–936. [Google Scholar] [CrossRef]
- Reiss, J.; Christensen, E.; Kurlemann, G.; Zabot, M.T.; Dorche, C. Genomic Structure and Mutational Spectrum of the Bicistronic MOCS1 Gene Defective in Molybdenum Cofactor Deficiency Type A. Hum. Genet. 1998, 103, 639–644. [Google Scholar] [CrossRef]
- Mayr, S.J.; Röper, J.; Schwarz, G. Alternative Splicing of the Bicistronic Gene Molybdenum Cofactor Synthesis 1 (MOCS1) Uncovers a Novel Mitochondrial Protein Maturation Mechanism. J. Biol. Chem. 2020, 295, 3029–3039. [Google Scholar] [CrossRef] [PubMed]
- Hover, B.M.; Tonthat, N.K.; Schumacher, M.A.; Yokoyama, K. Mechanism of Pyranopterin Ring Formation in Molybdenum Cofactor Biosynthesis. Proc. Natl. Acad. Sci. USA 2015, 112, 6347–6352. [Google Scholar] [CrossRef] [PubMed]
- Stallmeyer, B.; Schwarz, G.; Schulze, J.; Nerlich, A.; Reiss, J.; Kirsch, J.; Mendel, R.R. The Neurotransmitter Receptor-Anchoring Protein Gephyrin Reconstitutes Molybdenum Cofactor Biosynthesis in Bacteria, Plants, and Mammalian Cells. Proc. Natl. Acad. Sci. USA 1999, 96, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Gutzke, G.; Fischer, B.; Mendel, R.R.; Schwarz, G. Thiocarboxylation of Molybdopterin Synthase Provides Evidence for the Mechanism of Dithiolene Formation in Metal-Binding Pterins. J. Biol. Chem. 2001, 276, 36268–36274. [Google Scholar] [CrossRef] [PubMed]
- Wuebbens, M.M.; Rajagopalan, K.V. Mechanistic and Mutational Studies of Escherichia Coli Molybdopterin Synthase Clarify the Final Step of Molybdopterin Biosynthesis. J. Biol. Chem. 2003, 278, 14523–14532. [Google Scholar] [CrossRef] [PubMed]
- Matthies, A.; Rajagopalan, K.V.; Mendel, R.R.; Leimkühler, S. Evidence for the Physiological Role of a Rhodanese-like Protein for the Biosynthesis of the Molybdenum Cofactor in Humans. Proc. Natl. Acad. Sci. USA 2004, 101, 5946–5951. [Google Scholar] [CrossRef]
- Kuper, J.; Llamas, A.; Hecht, H.J.; Mendel, R.R.; Schwarz, G. Structure of the Molybdopterin-Bound Cnx1G Domain Links Molybdenum and Copper Metabolism. Nature 2004, 430, 803–806. [Google Scholar] [CrossRef]
- Llamas, A.; Otte, T.; Multhaup, G.; Mendel, R.R.; Schwarz, G. The Mechanism of Nucleotide-Assisted Molybdenum Insertion into Molybdopterin. A Novel Route toward Metal Cofactor Assembly. J. Biol. Chem. 2006, 281, 18343–18350. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Schwarz, G. Metal Insertion into the Molybdenum Cofactor: Product-Substrate Channelling Demonstrates the Functional Origin of Domain Fusion in Gephyrin. Biochem. J. 2013, 450, 149–157. [Google Scholar] [CrossRef]
- Fritschy, J.M.; Harvey, R.J.; Schwarz, G. Gephyrin: Where Do We Stand, Where Do We Go? Trends Neurosci. 2008, 31, 257–264. [Google Scholar] [CrossRef]
- Claerhout, H.; Witters, P.; Régal, L.; Jansen, K.; van Hoestenberghe, M.R.; Breckpot, J.; Vermeersch, P. Isolated Sulfite Oxidase Deficiency. J. Inherit. Metab. Dis. 2018, 41, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Schwahn, B.C.; Squires, L.; Confer, N. Molybdenum Cofactor Deficiency: A Natural History. J. Inherit. Metab. Dis. 2022, 45, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Pristup, J.; Schaeffeler, E.; Arjune, S.; Hofmann, U.; Angel Santamaria-Araujo, J.; Leuthold, P.; Friedrich, N.; Nauck, M.; Mayr, S.; Haag, M.; et al. Molybdenum Cofactor Catabolism Unravels the Physiological Role of the Drug Metabolizing Enzyme Thiopurine S-Methyltransferase. Clin. Pharmacol. Ther. 2022, 112, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Duran, M.; Beemer, F.A.; Heiden, C.V.D.; Korteland, J.; de Bree, P.K.; Brink, M.; Wadman, S.K.; Lombeck, I. Combined Deficiency of Xanthine Oxidase and Sulphite Oxidase: A Defect of Molybdenum Metabolism or Transport? J. Inherit. Metab. Dis. 1978, 1, 175–178. [Google Scholar] [CrossRef]
- Mechler, K.; Mountford, W.K.; Hoffmann, G.F.; Ries, M. Ultra-Orphan Diseases: A Quantitative Analysis of the Natural History of Molybdenum Cofactor Deficiency. Genet. Med. 2015, 17, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, K.; Gunny, R.; Grunewald, S.; Carr, L.; Chong, K.W.; Devile, C.; Robinson, R.; McSweeney, N.; Prabhakar, P. Clinical Neuroimaging Features and Outcome in Molybdenum Cofactor Deficiency. Pediatr. Neurol. 2011, 45, 246–252. [Google Scholar] [CrossRef]
- Arslanoglu, S.; Yalaz, M.; Gökşen, D.; Çoker, M.; Tütüncüoǧlu, S.; Aksu, M.; Darcan, Ş.; Kultursay, N.; Çiriş, M.; Demirtaş, E. Molybdenum Cofactor Deficiency Associated with Dandy-Walker Complex. Brain Dev. 2001, 23, 815–818. [Google Scholar] [CrossRef]
- Bayram, E.; Topcu, Y.; Karakaya, P.; Yis, U.; Cakmakci, H.; Ichida, K.; Kurul, S.H. Molybdenum Cofactor Deficiency: Review of 12 Cases (MoCD and Review). Eur. J. Paediatr. Neurol. 2013, 17, 1–6. [Google Scholar] [CrossRef]
- Scelsa, B.; Gasperini, S.; Righini, A.; Iascone, M.; Brazzoduro, V.G.; Veggiotti, P. Mild Phenotype in Molybdenum Cofactor Deficiency: A New Patient and Review of the Literature. Mol. Genet. Genom. Med. 2019, 7, e657. [Google Scholar] [CrossRef]
- Mayr, S.J.; Sass, J.O.; Vry, J.; Kirschner, J.; Mader, I.; Hövener, J.B.; Reiss, J.; Santamaria-Araujo, J.A.; Schwarz, G.; Grünert, S.C. A Mild Case of Molybdenum Cofactor Deficiency Defines an Alternative Route of MOCS1 Protein Maturation. J. Inherit. Metab. Dis. 2018, 41, 187–196. [Google Scholar] [CrossRef]
- del Rizzo, M.; Burlina, A.P.; Sass, J.O.; Beermann, F.; Zanco, C.; Cazzorla, C.; Bordugo, A.; Giordano, L.; Manara, R.; Burlina, A.B. Metabolic Stroke in a Late-Onset Form of Isolated Sulfite Oxidase Deficiency. Mol. Genet. Metab. 2013, 108, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, B.J.; McCarthy, E.L.; Booker, S.J. Radical S-Adenosylmethionine Enzymes in Human Health and Disease. Annu. Rev. Biochem. 2016, 85, 485–514. [Google Scholar] [CrossRef] [PubMed]
- Walsby, C.J.; Hong, W.; Broderick, W.E.; Cheek, J.; Ortillo, D.; Broderick, J.B.; Hoffman, B.M. Electron-Nuclear Double Resonance Spectroscopic Evidence That S-Adenosylmethionine Binds in Contact with the Catalytically Active [4Fe−4S]+ Cluster of Pyruvate Formate-Lyase Activating Enzyme. J. Am. Chem. Soc. 2002, 124, 3143–3151. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, T.; Yasuno, R.; Wada, H. Do Mammalian Cells Synthesize Lipoic Acid? Identification of a Mouse CDNA Encoding a Lipoic Acid Synthase Located in Mitochondria. FEBS Lett. 2001, 498, 16–21. [Google Scholar] [CrossRef]
- Reiter, V.; Matschkal, D.M.S.; Wagner, M.; Globisch, D.; Kneuttinger, A.C.; Müller, M.; Carell, T. The CDK5 Repressor CDK5RAP1 Is a Methylthiotransferase Acting on Nuclear and Mitochondrial RNA. Nucleic Acids Res. 2012, 40, 6235–6240. [Google Scholar] [CrossRef] [PubMed]
- Ohara-Imaizumi, M.; Yoshida, M.; Aoyagi, K.; Saito, T.; Okamura, T.; Takenaka, H.; Akimoto, Y.; Nakamichi, Y.; Takanashi-Yanobu, R.; Nishiwaki, C.; et al. Deletion of CDKAL1 Affects Mitochondrial ATP Generation and First-Phase Insulin Exocytosis. PLoS ONE 2010, 5, e15553. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, P.; Hernández, H.L.; Menzel, C.; García-Serres, R.; Huynh, B.H.; Johnson, M.K.; Mendel, R.R.; Schindelin, H. Characterization of MOCS1A, an Oxygen-Sensitive Iron-Sulfur Protein Involved in Human Molybdenum Cofactor Biosynthesis. J. Biol. Chem. 2004, 279, 34721–34732. [Google Scholar] [CrossRef]
- Pang, H.; Lilla, E.A.; Zhang, P.; Zhang, D.; Shields, T.P.; Scott, L.G.; Yang, W.; Yokoyama, K. Mechanism of Rate Acceleration of Radical C-C Bond Formation Reaction by a Radical SAM GTP 3′,8-Cyclase. J. Am. Chem. Soc. 2020, 142, 9314–9326. [Google Scholar] [CrossRef]
- Pang, H.; Yokoyama, K. Lessons From the Studies of a CC Bond Forming Radical SAM Enzyme in Molybdenum Cofactor Biosynthesis. Methods Enzymol. 2018, 606, 485–522. [Google Scholar] [CrossRef]
- Hover, B.M.; Lilla, E.A.; Yokoyama, K. Mechanistic Investigation of CPMP Synthase in Molybdenum Cofactor Biosynthesis Using an Uncleavable Substrate Analogue. Biochemistry 2015, 54, 7229–7236. [Google Scholar] [CrossRef]
- Gross-Hardt, S.; Reiss, J. The bicistronic MOCS1 gene has alternative start codons on two mutually exclusive exons. Mol Genet Metab. 2002, 76, 340–343. [Google Scholar] [CrossRef]
- Reiss, J.; Hahnewald, R. Molybdenum Cofactor Deficiency: Mutations in GPHN, MOCS1, and MOCS2. Hum. Mutat. 2011, 32, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.; Mahtani, K.; Abbott, J.; Schwarz, G.; Atwal, P. Molybdenum Cofactor Deficiency. GeneReviews 2021, 117, 1–4. [Google Scholar]
- Macaya, A.; Brunso, L.; Fernández-Castillo, N.; Arranz, J.A.; Ginjaar, H.B.; Cuenca-León, E.; Corominas, R.; Roig, M.; Cormand, B. Molybdenum Cofactor Deficiency Presenting as Neonatal Hyperekplexia: A Clinical, Biochemical and Genetic Study. Neuropediatrics 2005, 36, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Kingsmore, S.F.; Ramchandar, N.; James, K.; Niemi, A.K.; Feigenbaum, A.; Ding, Y.; Benson, W.; Hobbs, C.; Nahas, S.; Chowdhury, S.; et al. Mortality in a Neonate with Molybdenum Cofactor Deficiency Illustrates the Need for a Comprehensive Rapid Precision Medicine System. Cold Spring Harb. Mol. Case Stud. 2020, 6, a004705. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Kibe, T.; Hasegawa, H.; Ichida, K.; Koshimizu, E.; Miyatake, S.; Matsumoto, N.; Yokochi, K. The Persistent Generalized Muscle Contraction in Siblings with Molybdenum Cofactor Deficiency Type A. Neuropediatrics 2019, 50, 126–129. [Google Scholar] [CrossRef]
- Abe, Y.; Aihara, Y.; Endo, W.; Hasegawa, H.; Ichida, K.; Uematsu, M.; Kure, S. The Effect of Dietary Protein Restriction in a Case of Molybdenum Cofactor Deficiency with MOCS1 Mutation. Mol. Genet. Metab. Rep. 2021, 26, 100716. [Google Scholar] [CrossRef]
- Rudolph, M.J.; Wuebbens, M.M.; Turque, O.; Rajagopalan, K.V.; Schindelin, H. Structural Studies of Molybdopterin Synthase Provide Insights into Its Catalytic Mechanism. J. Biol. Chem. 2003, 278, 14514–14522. [Google Scholar] [CrossRef]
- Stallmeyer, B.; Drugeon, G.; Reiss, J.; Haenni, A.L.; Mendel, R.R. Human Molybdopterin Synthase Gene: Identification of a Bicistronic Transcript with Overlapping Reading Frames. Am. J. Hum. Genet. 1999, 64, 698–705. [Google Scholar] [CrossRef]
- Hahnewald, R.; Leimkühler, S.; Vilaseca, A.; Acquaviva-Bourdain, C.; Lenz, U.; Reiss, J. A Novel MOCS2 Mutation Reveals Coordinated Expression of the Small and Large Subunit of Molybdopterin Synthase. Mol. Genet. Metab. 2006, 89, 210–213. [Google Scholar] [CrossRef]
- Arican, P.; Gencpinar, P.; Kirbiyik, O.; Bozkaya Yilmaz, S.; Ersen, A.; Oztekin, O.; Olgac Dundar, N. The Clinical and Molecular Characteristics of Molybdenum Cofactor Deficiency Due to MOCS2 Mutations. Pediatr. Neurol. 2019, 99, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Megahed, H.; Nicouleau, M.; Barcia, G.; Medina-Cano, D.; Siquier-Pernet, K.; Bole-Feysot, C.; Parisot, M.; Masson, C.; Nitschké, P.; Rio, M.; et al. Utility of Whole Exome Sequencing for the Early Diagnosis of Pediatric-Onset Cerebellar Atrophy Associated with Developmental Delay in an Inbred Population. Orphanet J. Rare Dis. 2016, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Reiss, J.; Dorche, C.; Stallmeyer, B.; Mendel, R.R.; Cohen, N.; Zabot, M.T. Human Molydopterin Synthase Gene: Genomic Structure and Mutations in Molybdenum Cofactor Deficiency Type B. Am. J. Hum. Genet. 1999, 64, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Coyne, K.E.; Rajagopalan, K.V.; van Hove, J.L.K.; Mackay, M.; Pitt, J.; Boneh, A. Molybdopterin Synthase Mutations in a Mild Case of Molybdenum Cofactor Defciency. Am. J. Med. Genet. 2001, 104, 169–173. [Google Scholar] [CrossRef]
- Tian, X.J.; Li, X.; Fang, F.; Liu, Z.M.; Wu, W.J.; Liu, K.; Sun, S.Z. Molybdenum Cofactor Deficiency Type B Manifested as Leigh-like Syndrome: A Case Report and Literature Review. Zhonghua Er Ke Za Zhi 2021, 59, 119–124. [Google Scholar] [CrossRef]
- Leimkühler, S.; Charcosset, M.; Latour, P.; Dorche, C.; Kleppe, S.; Scaglia, F.; Szymczak, I.; Schupp, P.; Hahnewald, R.; Reiss, J. Ten Novel Mutations in the Molybdenum Cofactor Genes MOCS1 and MOCS2 and in Vitro Characterization of a MOCS2 Mutation That Abolishes the Binding Ability of Molybdopterin Synthase. Hum. Genet. 2005, 117, 565–570. [Google Scholar] [CrossRef]
- Reiss, J.; Johnson, J.L. Mutations in the Molybdenum Cofactor Biosynthetic Genes MOCS1, MOCS2, and GEPH. Hum. Mutat. 2003, 21, 569–576. [Google Scholar] [CrossRef]
- Per, H.; Gümüş, H.; Ichida, K.; Çaǧlayan, O.; Kumandaş, S. Molybdenum Cofactor Deficiency: Clinical Features in a Turkish Patient. Brain Dev. 2007, 29, 365–368. [Google Scholar] [CrossRef]
- Yoganathan, S.; Sudhahakar, V.; Sv, S.; Thomas, M.; Kumar Dutta, K.; Danda, A.; Chandran, S. Novel Imaging Finding and Novel Mutation in an Infant with Molybdenum Cofactor Deficiency, a Mimicker of Hypoxic-Ischaemic Encephalopathy. Iran J. Child Neurol. Spring 2018, 12, 107–112. [Google Scholar]
- Nagappa, M.; Bindu, P.S.; Taly, A.B.; Sinha, S.; Bharath, R.D. Child Neurology: Molybdenum Cofactor Deficiency. Neurology 2015, 85, e175–e178. [Google Scholar] [CrossRef]
- Kikuchi, K.; Hamano, S.I.; Mochizuki, H.; Ichida, K.; Ida, H. Molybdenum Cofactor Deficiency Mimics Cerebral Palsy: Differentiating Factors for Diagnosis. Pediatr. Neurol. 2012, 47, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.; Roeper, J.; Allgood, C.; Chin, R.; Santamaria, J.; Schwarz, G.; Whitehall, J. Investigation of Molybdenum Cofactor Deficiency Due to MOCS2 Deficiency in a Newborn Baby. Metab. Enceph. Meta Gene 2015, 3, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Dandamudi, R.; Granadillo, J.L.; Grange, D.K.; Kakajiwala, A. Rare Cause of Xanthinuria: A Pediatric Case of Molybdenum Cofactor Deficiency B. CEN Case Rep. 2021, 10, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Alkufri, F.; Harrower, T.; Rahman, Y.; Hughes, E.; Mundy, H.; Knibb, J.A.; Moriarty, J.; Connor, S.; Samuel, M. Molybdenum Cofactor Deficiency Presenting with a Parkinsonism-Dystonia Syndrome. Mov. Disord. 2013, 28, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Poulter, J.A.; Mao, X.; Wang, H.; Tian, Q.; Cao, Y.; Shu, L.; Chen, Y.; Peng, Y.; Wang, Y.; et al. Case Report: Compound Heterozygous Variants in MOCS3 Identified in a Chinese Infant With Molybdenum Cofactor Deficiency. Front. Genet. 2021, 1, 651878. [Google Scholar] [CrossRef]
- Huijmans, J.G.M.; Schot, R.; de Klerk, J.B.C.; Williams, M.; de Coo, R.F.M.; Duran, M.; Verheijen, F.W.; van Slegtenhorst, M.; Mancini, G.M.S. Molybdenum Cofactor Deficiency: Identification of a Patient with Homozygote Mutation in the MOCS3 Gene. Am. J. Med. Genet. A 2017, 173, 1601–1606. [Google Scholar] [CrossRef]
- Leimkühler, S.; Freuer, A.; Araujo, J.A.S.; Rajagopalan, K.V.; Mendel, R.R. Mechanistic Studies of Human Molybdopterin Synthase Reaction and Characterization of Mutants Identified in Group B Patients of Molybdenum Cofactor Deficiency. J. Biol. Chem. 2003, 278, 26127–26134. [Google Scholar] [CrossRef]
- Jezela-Stanek, A.; Blaz, W.; Gora, A.; Bochenska, M.; Kusmierska, K.; Sykut-Cegielska, J. Proteins Structure Models in the Evaluation of Novel Variant (C.472_477del) in the MOSC2 Gene. Diagnostics 2020, 10, 821. [Google Scholar] [CrossRef]
- Nichols, J.; Rajagopalan, K.V. Escherichia Coli MoeA and MogA. Function in Metal Incorporation Step of Molybdenum Cofactor Biosynthesis. J. Biol. Chem. 2002, 277, 24995–25000. [Google Scholar] [CrossRef]
- Kim, E.Y.; Schrader, N.; Smolinsky, B.; Bedet, C.; Vannier, C.; Schwarz, G.; Schindelin, H. Deciphering the Structural Framework of Glycine Receptor Anchoring by Gephyrin. EMBO J. 2006, 25, 1385–1395. [Google Scholar] [CrossRef]
- Schwarz, G.; Schrader, N.; Mendel, R.R.; Hecht, H.J.; Schindelin, H. Crystal Structures of Human Gephyrin and Plant Cnx1 G Domains: Comparative Analysis and Functional Implications. J. Mol. Biol. 2001, 312, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Sander, B.; Tria, G.; Shkumatov, A.V.; Kim, E.Y.; Grossmann, J.G.; Tessmer, I.; Svergun, D.I.; Schindelin, H. Structural Characterization of Gephyrin by AFM and SAXS Reveals a Mixture of Compact and Extended States. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 2050–2060. [Google Scholar] [CrossRef] [PubMed]
- Prior, P.; Schmitt, B.; Grenningloh, G.; Pribilla, I.; Multhaup, G.; Beyreuther, K.; Maulet, Y.; Werner, P.; Langosch, D.; Kirsch, J.; et al. Primary Structure and Alternative Splice Variants of Gephyrin, a Putative Glycine Receptor-Tubulin Linker Protein. Neuron 1992, 8, 1161–1170. [Google Scholar] [CrossRef]
- dos Reis, R.; Kornobis, E.; Pereira, A.; Tores, F.; Carrasco, J.; Gautier, C.; Jahannault-Talignani, C.; Nitschké, P.; Muchardt, C.; Schlosser, A.; et al. Complex Regulation of Gephyrin Splicing Is a Determinant of Inhibitory Postsynaptic Diversity. Nat. Commun. 2022, 13, 3507. [Google Scholar] [CrossRef]
- Rees, M.I.; Harvey, K.; Ward, H.; White, J.H.; Evans, L.; Duguid, I.C.; Hsu, C.C.H.; Coleman, S.L.; Miller, J.; Baer, K.; et al. Isoform Heterogeneity of the Human Gephyrin Gene (GPHN), Binding Domains to the Glycine Receptor, and Mutation Analysis in Hyperekplexia. J. Biol. Chem. 2003, 278, 24688–24696. [Google Scholar] [CrossRef]
- Reiss, J.; Gross-Hardt, S.; Christensen, E.; Schmidt, P.; Mendel, R.R.; Schwarz, G. A Mutation in the Gene for the Neurotransmitter Receptor-Clustering Protein Gephyrin Causes a Novel Form of Molybdenum Cofactor Deficiency. Am. J. Hum. Genet. 2001, 68, 208–213. [Google Scholar] [CrossRef]
- Feng, G.; Tintrup, H.; Kirsch, J.; Nichol, M.C.; Kuhse, J.; Betz, H.; Sanes, J.R. Dual Requirement for Gephyrin in Glycine Receptor Clustering and Molybdoenzyme Activity. Science 1998, 282, 1321–1324. [Google Scholar] [CrossRef]
- Lee, H.J.; Adham, I.M.; Schwarz, G.; Kneussel, M.; Sass, J.O.; Engel, W.; Reiss, J. Molybdenum Cofactor-Deficient Mice Resemble the Phenotype of Human Patients. Hum. Mol. Genet. 2002, 11, 3309–3317. [Google Scholar] [CrossRef]
- Jakubiczka-Smorag, J.; Santamaria-Araujo, J.A.; Metz, I.; Kumar, A.; Hakroush, S.; Brueck, W.; Schwarz, G.; Burfeind, P.; Reiss, J.; Smorag, L. Mouse Model for Molybdenum Cofactor Deficiency Type B Recapitulates the Phenotype Observed in Molybdenum Cofactor Deficient Patients. Hum. Genet. 2016, 135, 813–826. [Google Scholar] [CrossRef]
- MacHa, A.; Liebsch, F.; Fricke, S.; Hetsch, F.; Neuser, F.; Johannes, L.; Kress, V.; Djemie, T.; Santamaria-Araujo, J.A.; Vilain, C.; et al. Biallelic Gephyrin Variants Lead to Impaired GABAergic Inhibition in a Patient with Developmental and Epileptic Encephalopathy. Hum. Mol. Genet. 2022, 31, 901–913. [Google Scholar] [CrossRef]
- Dejanovic, B.; Lal, D.; Catarino, C.B.; Arjune, S.; Belaidi, A.A.; Trucks, H.; Vollmar, C.; Surges, R.; Kunz, W.S.; Motameny, S.; et al. Exonic Microdeletions of the Gephyrin Gene Impair GABAergic Synaptic Inhibition in Patients with Idiopathic Generalized Epilepsy. Neurobiol. Dis. 2014, 67, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Lionel, A.C.; Vaags, A.K.; Sato, D.; Gazzellone, M.J.; Mitchell, E.B.; Chen, H.Y.; Costain, G.; Walker, S.; Egger, G.; Thiruvahindrapuram, B.; et al. Rare Exonic Deletions Implicate the Synaptic Organizer Gephyrin (GPHN) in Risk for Autism, Schizophrenia and Seizures. Hum. Mol. Genet. 2013, 22, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dejanovic, B.; Hetsch, F.; Semtner, M.; Fusca, D.; Arjune, S.; Santamaria-Araujo, J.A.; Winkelmann, A.; Ayton, S.; Bush, A.I.; et al. S-Sulfocysteine/NMDA Receptor-Dependent Signaling Underlies Neurodegeneration in Molybdenum Cofactor Deficiency. J. Clin. Investig. 2017, 127, 4365–4378. [Google Scholar] [CrossRef] [PubMed]
- Footitt, E.J.; Heales, S.J.; Mills, P.B.; Allen, G.F.G.; Oppenheim, M.; Clayton, P.T. Pyridoxal 5′-Phosphate in Cerebrospinal Fluid; Factors Affecting Concentration. J. Inherit. Metab. Dis. 2011, 34, 529–538. [Google Scholar] [CrossRef]
- Mellis, A.T.; Misko, A.L.; Arjune, S.; Liang, Y.; Erdélyi, K.; Ditrói, T.; Kaczmarek, A.T.; Nagy, P.; Schwarz, G. The Role of Glutamate Oxaloacetate Transaminases in Sulfite Biosynthesis and H2S Metabolism. Redox Biol. 2021, 38, 101800. [Google Scholar] [CrossRef]
- Schwarz, G.; Santamaria-Araujo, J.A.; Wolf, S.; Lee, H.J.; Adham, I.M.; Grone, H.J.; Schwegler, H.; Sass, J.O.; Otte, T.; Hanzelmann, P.; et al. Rescue of Lethal Molybdenum Cofactor Deficiency by a Biosynthetic Precursor from Escherichia Coli. Hum. Mol. Genet. 2004, 13, 1249–1255. [Google Scholar] [CrossRef]
- Veldman, A.; Santamaria-Araujo, J.A.; Sollazzo, S.; Pitt, J.; Gianello, R.; Yaplito-Lee, J.; Wong, F.; Ramsden, C.A.; Reiss, J.; Cook, I.; et al. Successful Treatment of Molybdenum Cofactor Deficiency Type A With CPMP. Pediatrics 2010, 125, 1249–1254. [Google Scholar] [CrossRef]
- Schwahn, B.C.; van Spronsen, F.J.; Belaidi, A.A.; Bowhay, S.; Christodoulou, J.; Derks, T.G.; Hennermann, J.B.; Jameson, E.; Konig, K.; McGregor, T.L.; et al. Efficacy and Safety of Cyclic Pyranopterin Monophosphate Substitution in Severe Molybdenum Cofactor Deficiency Type A: A Prospective Cohort Study. Lancet 2015, 386, 1955–1963. [Google Scholar] [CrossRef]
- Clinch, K.; Watt, D.K.; Dixon, R.A.; Baars, S.M.; Gainsford, G.J.; Tiwari, A.; Schwarz, G.; Saotome, Y.; Storek, M.; Belaidi, A.A.; et al. Synthesis of Cyclic Pyranopterin Monophosphate, a Biosynthetic Intermediate in the Molybdenum Cofactor Pathway. J. Med. Chem. 2013, 56, 1730–1738. [Google Scholar] [CrossRef]
- Farrell, S.; Karp, J.; Hager, R.; Wang, Y.; Adeniyi, O.; Wang, J.; Li, L.; Ma, L.; Peretz, J.; Summan, M.; et al. Regulatory News: Nulibry (Fosdenopterin) Approved to Reduce the Risk of Mortality in Patients with Molybdenum Cofactor Deficiency Type A: FDA Approval Summary. J. Inherit. Metab. Dis. 2021, 44, 1085–1087. [Google Scholar] [CrossRef]
- Schwahn, B. Fosdenopterin: A First-in-Class Synthetic Cyclic Pyranopterin Monophosphate for the Treatment of Molybdenum Cofactor Deficiency Type A. Neurology 2021, 17, 85–91. [Google Scholar] [CrossRef]
- Endres, W.; Shin, Y.S.; Günther, R.; Ibel, H.; Duran, M.; Wadman, S.K. Report on a New Patient with Combined Deficiencies of Sulphite Oxidase and Xanthine Dehydrogenase Due to Molybdenum Cofactor Deficiency. Eur. J. Pediatr. 1988, 148, 246–249. [Google Scholar] [CrossRef]
- Reiss, J.; Lenz, U.; Aquaviva-Bourdain, C.; Joriot-Chekaf, S.; Mention-Mulliez, K.; Holder-Espinasse, M. A GPHN Point Mutation Leading to Molybdenum Cofactor Deficiency. Clin. Genet. 2011, 80, 598–599. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.M.; Schwarz, G. Cofactor-Dependent Maturation of Mammalian Sulfite Oxidase Links Two Mitochondrial Import Pathways. J. Cell Sci. 2012, 125, 4876–4885. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.M.; Busch, J.D.; Pottings, C.; Baker, M.J.; Langers, T.; Schwarz, G. The Mitochondrial Amidoxime-Reducing Component (MARC1) Is a Novel Signal-Anchored Protein of the Outer Mitochondrial Membrane. J. Biol. Chem. 2012, 287, 42795–42803. [Google Scholar] [CrossRef]
- Bender, D.; Kaczmarek, A.T.; Santamaria-Araujo, J.A.; Stueve, B.; Waltz, S.; Bartsch, D.; Kurian, L.; Cirak, S.; Schwarz, G. Impaired Mitochondrial Maturation of Sulfite Oxidase in a Patient with Severe Sulfite Oxidase Deficiency. Hum. Mol. Genet. 2019, 28, 2885–2899. [Google Scholar] [CrossRef]
- Kaczmarek, A.T.; Bahlmann, N.; Thaqi, B.; May, P.; Schwarz, G. Machine Learning-Based Identification and Characterization of 15 Novel Pathogenic SUOX Missense Mutations. Mol. Genet. Metab. 2021, 134, 188–194. [Google Scholar] [CrossRef]
- Kaczmarek, A.T.; Bender, D.; Gehling, T.; Kohl, J.B.; Daimagüler, H.S.; Santamaria-Araujo, J.A.; Liebau, M.C.; Koy, A.; Cirak, S.; Schwarz, G. A Defect in Molybdenum Cofactor Binding Causes an Attenuated Form of Sulfite Oxidase Deficiency. J. Inherit. Metab. Dis. 2022, 45, 169–182. [Google Scholar] [CrossRef]
- Boles, R.G.; Ment, L.R.; Meyn, M.S.; Horwich, A.L.; Kratz, L.E.; Rinaldo, P. Short-Term Response to Dietary Therapy in Molybdenum Cofactor Deficiency. Ann. Neurol. 1993, 34, 742–744. [Google Scholar] [CrossRef]
- Touati, G.; Rusthoven, E.; Depondt, E.; Dorche, C.; Duran, M.; Heron, B.; Rabier, D.; Russo, M.; Saudubray, J.M. Dietary Therapy in Two Patients with a Mild Form of Sulphite Oxidase Deficiency. Evidence for Clinical and Biological Improvement. J. Inherit. Metab. Dis. 2000, 23, 45–53. [Google Scholar] [CrossRef]
- Tan, W.H.; Eichler, F.S.; Hoda, S.; Lee, M.S.; Baris, H.; Hanley, C.A.; Grant, P.E.; Krishnamoorthy, K.S.; Shih, V.E. Isolated Sulfite Oxidase Deficiency: A Case Report with a Novel Mutation and Review of the Literature. Pediatrics 2005, 116, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Misra, C.H.; Gubareff, T. de Cysteine-S-Sulfate: Brain Damaging Metabolite in Sulfite Oxidase Deficiency. J. Neuropathol. Exp. Neurol. 1975, 34, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Schuierer, G.; Kurlemann, G.; Bick, U.; Stephani, U. Molybdenum-Cofactor Deficiency: CT and MR Findings. Neuropediatrics 1995, 26, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Kohl, J.B.; Mellis, A.T.; Schwarz, G. Homeostatic Impact of Sulfite and Hydrogen Sulfide on Cysteine Catabolism. Br. J. Pharmacol. 2019, 176, 554–570. [Google Scholar] [CrossRef]
- Jocelyn, P.C. Chemical Reduction of Disulfides. Methods Enzymol. 1987, 143, 246–256. [Google Scholar] [CrossRef]
- Smolin, L.A.; Clark, K.F.; Thoene, J.G.; Gahl, W.A.; Schneider, J.A. A Comparison of the Effectiveness of Cysteamine and Phosphocysteamine in Elevating Plasma Cysteamine Concentration and Decreasing Leukocyte Free Cystine in Nephropathic Cystinosis. Pediatr. Res. 1988, 23, 616–620. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef]
- Yang, W.S.; Sriramaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Fujii, J.; Homma, T.; Kobayashi, S. Ferroptosis Caused by Cysteine Insufficiency and Oxidative Insult. Free. Radic. Res. 2019, 54, 969–980. [Google Scholar] [CrossRef]
- Reed, M.C.; Thomas, R.L.; Pavisic, J.; James, S.J.; Ulrich, C.M.; Nijhout, H.F. A Mathematical Model of Glutathione Metabolism. Theor. Biol. Med. Model. 2008, 5, 8. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Valente, M.; Ferri, L.; Gregolin, C. Purification from Pig Liver of a Protein Which Protects Liposomes and Biomembranes from Peroxidative Degradation and Exhibits Glutathione Peroxidase Activity on Phosphatidylcholine Hydroperoxides. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1982, 710, 197–211. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Gao, X.; Xu, H.; Cui, Y.; Zhang, Y.; Gou, X. The Emerging Roles of Ferroptosis in Huntington’s Disease. Neuromol. Med. 2019, 21, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, Y.; Shen, Y.; Wang, Y.; Zhao, M.; Sun, L. The Potential Role of Ferroptosis in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 80, 907–925. [Google Scholar] [CrossRef]
- Mahoney-Sanchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.C. Ferroptosis and Its Potential Role in the Physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2021, 196, 101890. [Google Scholar] [CrossRef]
- Schwarz, G.; Belaidi, A.A. Molybdenum in Human Health and Disease. In Interrelations between Essential Metal Ions and Human Diseases; Springer: Dordrecht, The Netherlands, 2013; Volume 13, pp. 415–450. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Arjune, S.; Santamaria-Araujo, J.A.; Sass, J.O.; Schwarz, G. Molybdenum Cofactor Deficiency: A New HPLC Method for Fast Quantification of s-Sulfocysteine in Urine and Serum. JIMD Rep. 2012, 5, 35–43. [Google Scholar] [CrossRef]
- Niknahad, H.; O’Brien, P.J. Mechanism of Sulfite Cytotoxicity in Isolated Rat Hepatocytes. Chem. Biol. Interact. 2008, 174, 147–154. [Google Scholar] [CrossRef]
- Pereira, C.M.; Oliveira, C.R. Glutamate Toxicity on a PC12 Cell Line Involves Glutathione (GSH) Depletion and Oxidative Stress. Free. Radic. Biol. Med. 1997, 23, 637–647. [Google Scholar] [CrossRef]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152–1170. [Google Scholar] [CrossRef]
- Viscomi, C.; Burlina, A.B.; Dweikat, I.; Savoiardo, M.; Lamperti, C.; Hildebrandt, T.; Tiranti, V.; Zeviani, M. Combined Treatment with Oral Metronidazole and N-Acetylcysteine Is Effective in Ethylmalonic Encephalopathy. Nat. Med. 2010, 16, 869–871. [Google Scholar] [CrossRef]
- Nicholls, P.; Kim, J.K. Sulphide as an Inhibitor and Electron Donor for the Cytochrome c Oxidase System. Can. J. Biochem. 1982, 60, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Carballal, S.; Vitvitsky, V.; Kumar, R.; Hanna, D.A.; Libiad, M.; Gupta, A.; Jones, J.W.; Banerjee, R. Hydrogen Sulfide Stimulates Lipid Biogenesis from Glutamine That Is Dependent on the Mitochondrial NAD(P)H Pool. J. Biol. Chem. 2021, 297, 100950. [Google Scholar] [CrossRef] [PubMed]
- Grings, M.; Wajner, M.; Leipnitz, G. Mitochondrial Dysfunction and Redox Homeostasis Impairment as Pathomechanisms of Brain Damage in Ethylmalonic Encephalopathy: Insights from Animal and Human Studies. Cell. Mol. Neurobiol. 2022, 42, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Grings, M.; Seminotti, B.; Karunanidhi, A.; Ghaloul-Gonzalez, L.; Mohsen, A.W.; Wipf, P.; Palmfeldt, J.; Vockley, J.; Leipnitz, G. ETHE1 and MOCS1 Deficiencies: Disruption of Mitochondrial Bioenergetics, Dynamics, Redox Homeostasis and Endoplasmic Reticulum-Mitochondria Crosstalk in Patient Fibroblasts. Sci. Rep. 2019, 9, 12651. [Google Scholar] [CrossRef]
- Mellis, A.T.; Roeper, J.; Misko, A.L.; Kohl, J.; Schwarz, G. Sulfite Alters the Mitochondrial Network in Molybdenum Cofactor Deficiency. Front. Genet. 2021, 11, 594828. [Google Scholar] [CrossRef]
- Warnhoff, K.; Ruvkun, G. Molybdenum Cofactor Transfer from Bacteria to Nematode Mediates Sulfite Detoxification. Nat. Chem. Biol. 2019, 15, 480–488. [Google Scholar] [CrossRef]
- Warnhoff, K.; Hercher, T.W.; Mendel, R.R.; Ruvkun, G. Protein-Bound Molybdenum Cofactor Is Bioavailable and Rescues Molybdenum Cofactor-Deficient C. Elegans. Genes Dev. 2021, 35, 212–217. [Google Scholar] [CrossRef]
- Koehler, F.C.; Fu, C.-Y.; Späth, M.R.; Hoyer-Allo, K.J.R.; Bohl, K.; Göbel, H.; Lackmann, J.-W.; Grundmann, F.; Osterholt, T.; Gloistein, C.; et al. A Systematic Analysis of Diet-Induced Nephroprotection Reveals Overlapping Changes in Cysteine Catabolism. Transl. Res. 2022, 244, 32–46. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Rosales, C.; McIntosh, J.; Rastegarlari, G.; Nathwani, D.; Raj, D.; Nawathe, S.; Waddington, S.N.; Bronson, R.; Jackson, S.; et al. Long-Term Safety and Efficacy Following Systemic Administration of a Self-Complementary AAV Vector Encoding Human FIX Pseudotyped with Serotype 5 and 8 Capsid Proteins. Mol. Ther. 2011, 19, 876–885. [Google Scholar] [CrossRef]
- Stroes, E.S.; Nierman, M.C.; Meulenberg, J.J.; Franssen, R.; Twisk, J.; Henny, C.P.; Maas, M.M.; Zwinderman, A.H.; Ross, C.; Aronica, E.; et al. Intramuscular Administration of AAV1-Lipoprotein Lipase S447X Lowers Triglycerides in Lipoprotein Lipase-Deficient Patients. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2303–2304. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed]
- McCain, J. The Future of Gene Therapy. Biotechnol. Healthc. 2005, 2, 52. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Gene | Nucleotide Change | Amino Acid Change/ Predicted Effect | Onset | Reference |
|---|---|---|---|---|
| MOCS2A | c.-9_14del23 | Initiation failure | Early | [50] |
| c.-9G > C | - | Early | [51] | |
| c.1A > G | Initiation failure | - | [42] | |
| c.3G > A | p.M1I (Initiation failure) | Early | [52,53] | |
| c.16C > T (c.44C > T) | p.Q6X | Early | [54] | |
| c.19G > T (c.47G > T) | p.V7F * | Early | [54,55] | |
| c.33T > G | p.Y11X | - | [56] | |
| c.45T > A | p.S15R * | - | [57] | |
| c.88C > T | p.Q30X | - | [57] | |
| c.106C > T | p.Q36X | - | [57] | |
| c.130C > T | p.R44X | Early | [58] | |
| MOCS2A/B | c.218T > C | p.L73P (MOCS2A) | Early | [59] |
| c.220C > T | p.Q74X (MOCS2A) | Late | [42] | |
| c.226G > A | p.G76R (MOCS2A) | Early | [26] | |
| c.252insC | Premature termination | Early | [60] | |
| c.265T > C | p.X89Q (MOCS2A), silenced p.D26D (MOCS2B) | Late | [61] | |
| c.266A > G | p.X89W (MOCS2A), p.D26G (MOCS2B) | Late | [61] | |
| MOCS2B | c.413G > A | p.G76R | Early | [57] |
| c.419C > T | p.S140F * | Early | [62] | |
| c.493 T > C | p.W165R | Late | [63] | |
| c.501delA | p.K105fs | - | [42] | |
| c.501 + 2delT | Disruption of splice site | Early | [62] | |
| c.533_536delGTCA | p.V116fs(Premature termination) | - | [53] | |
| c.564G > C | p.G126A | - | [57,64] | |
| c.564 + 1G > A | Skipping exon 5 | Early | [42] | |
| c.635_637delGCT | p.A150del * | Early | [57] | |
| c.658_664delTTTAAAAinsG | p.L158_K159del | - | [57] | |
| c.689G > A | p.E168K * | - | [57] | |
| c.714_718delGGAAA | p.G178fs (Premature termination) | - | [57] | |
| c.726_727delAA | p.K180fs (Premature termination) | Late | [57,64] | |
| c.754A > C | p.X189Y | - | [56] | |
| MOCS3 | c.325C > G | p.L109V | Early | [65] |
| c.769G > A | p.A257T | Late | [66] | |
| c.1375C > T | p.Q459X | Early | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johannes, L.; Fu, C.-Y.; Schwarz, G. Molybdenum Cofactor Deficiency in Humans. Molecules 2022, 27, 6896. https://doi.org/10.3390/molecules27206896
Johannes L, Fu C-Y, Schwarz G. Molybdenum Cofactor Deficiency in Humans. Molecules. 2022; 27(20):6896. https://doi.org/10.3390/molecules27206896
Chicago/Turabian StyleJohannes, Lena, Chun-Yu Fu, and Günter Schwarz. 2022. "Molybdenum Cofactor Deficiency in Humans" Molecules 27, no. 20: 6896. https://doi.org/10.3390/molecules27206896
APA StyleJohannes, L., Fu, C.-Y., & Schwarz, G. (2022). Molybdenum Cofactor Deficiency in Humans. Molecules, 27(20), 6896. https://doi.org/10.3390/molecules27206896