
Itraconazole-Induced Increases in Gilteritinib Exposure Are Mediated by CYP3A and OATP1B

 , , , and
, , , and
Abstract
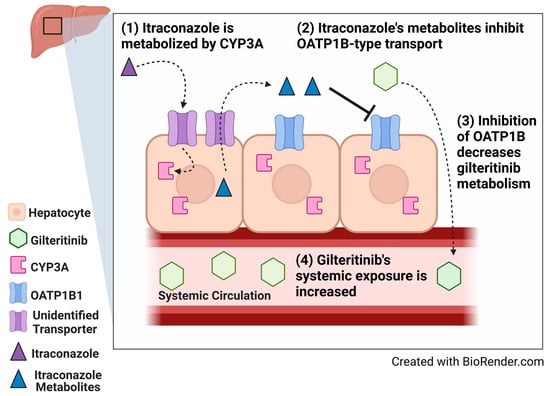
1. Introduction
2. Results
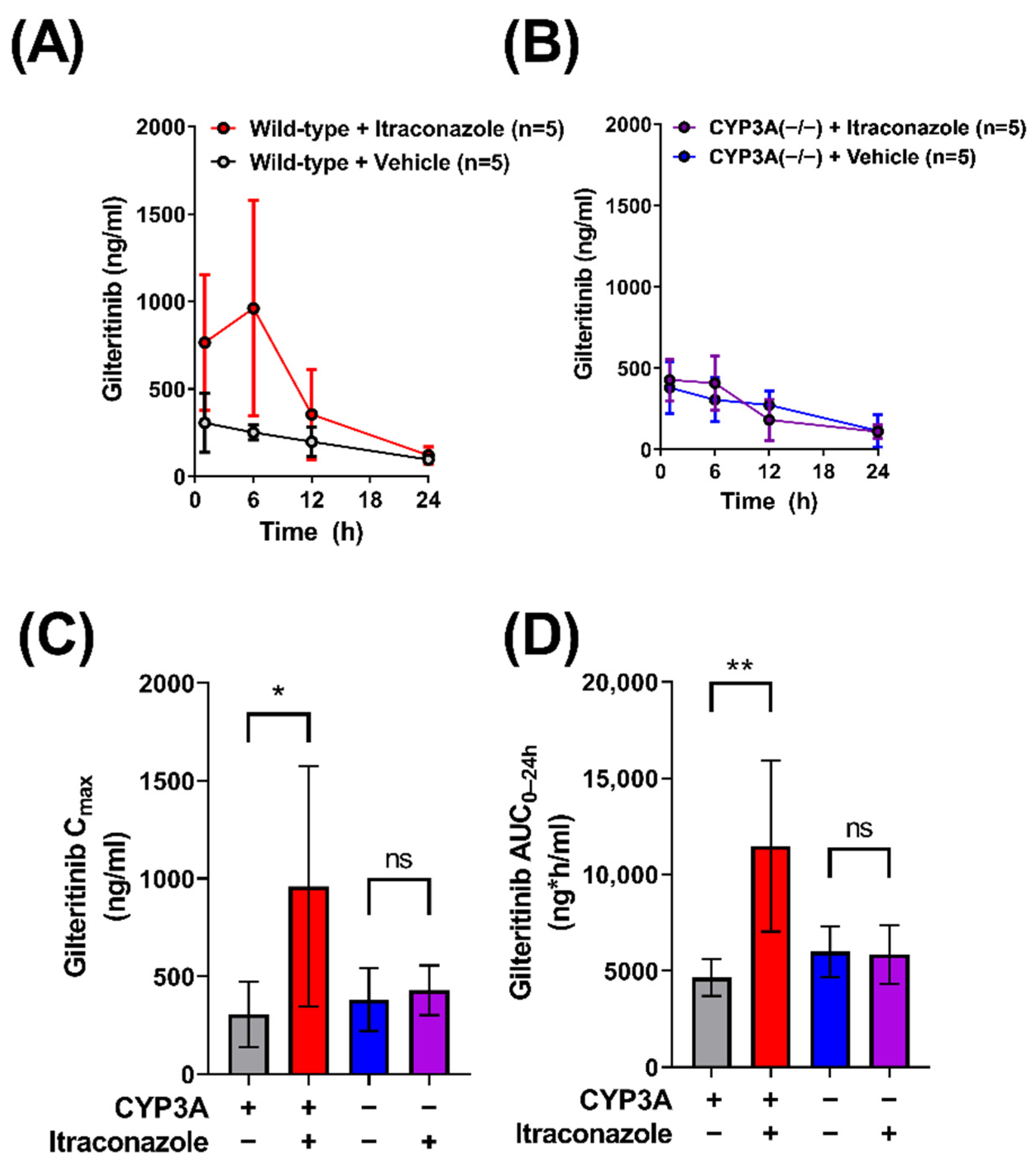
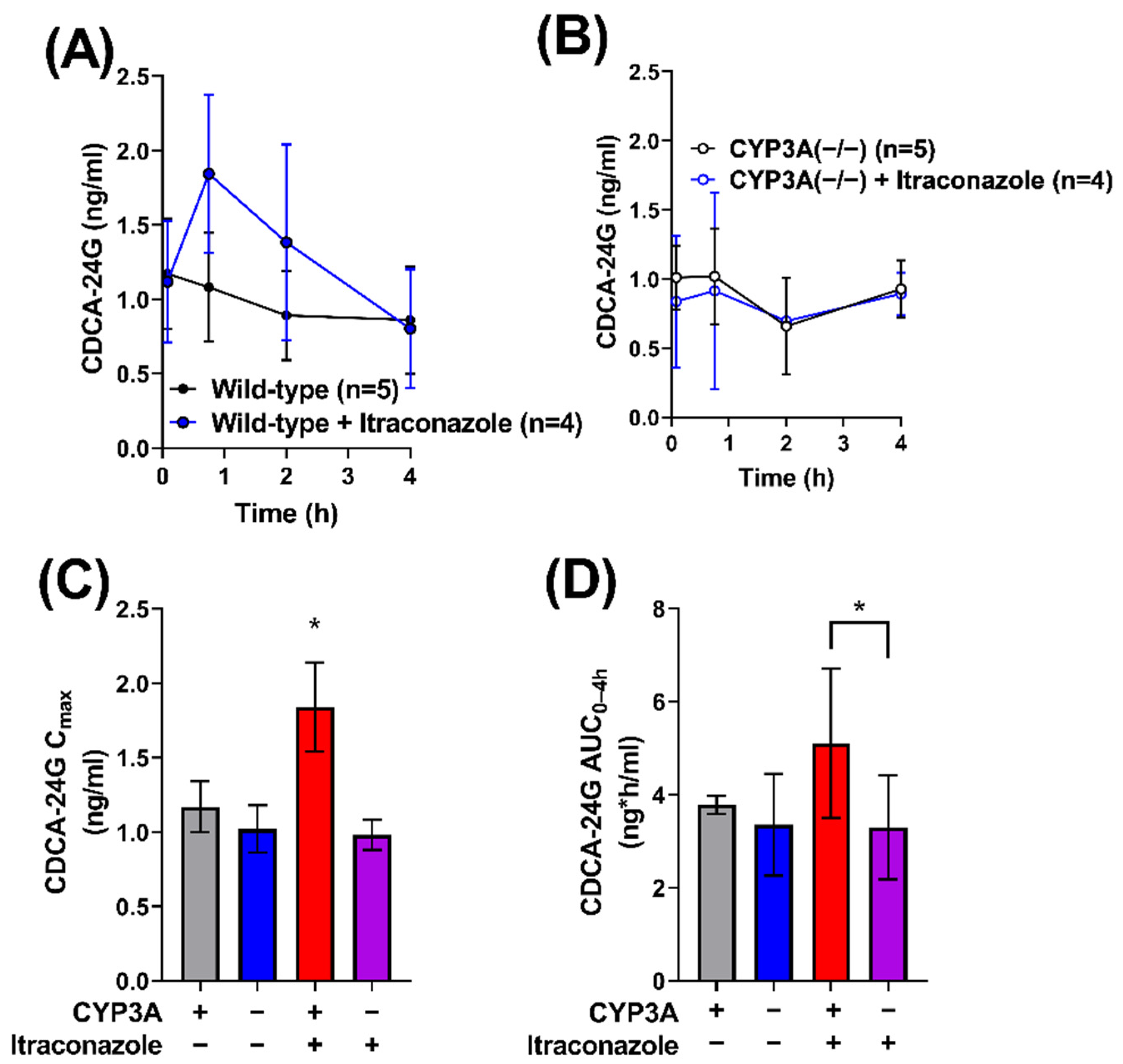
2.1. Contribution of CYP3A to the Interaction between Gilteritinib and Itraconazole
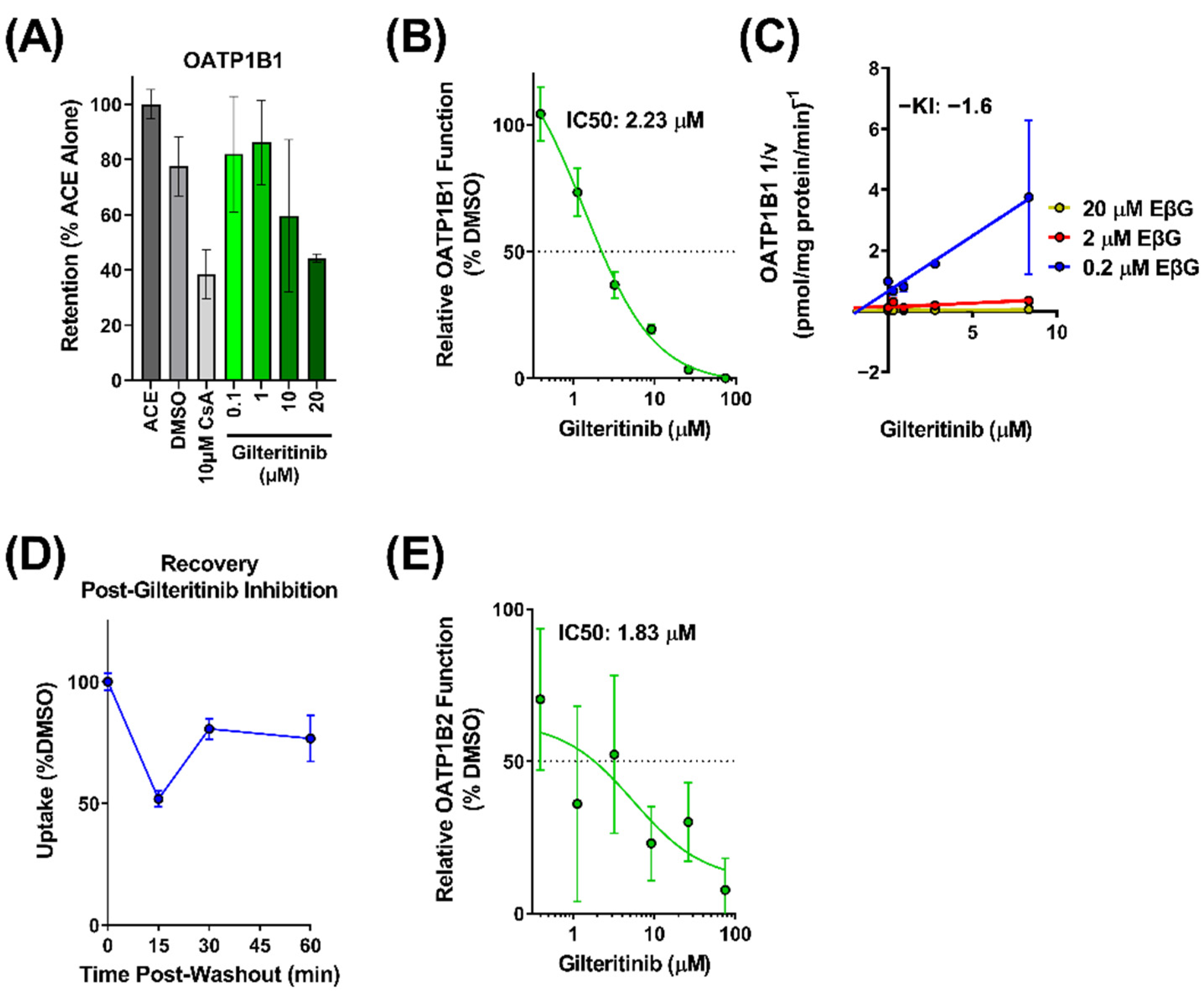
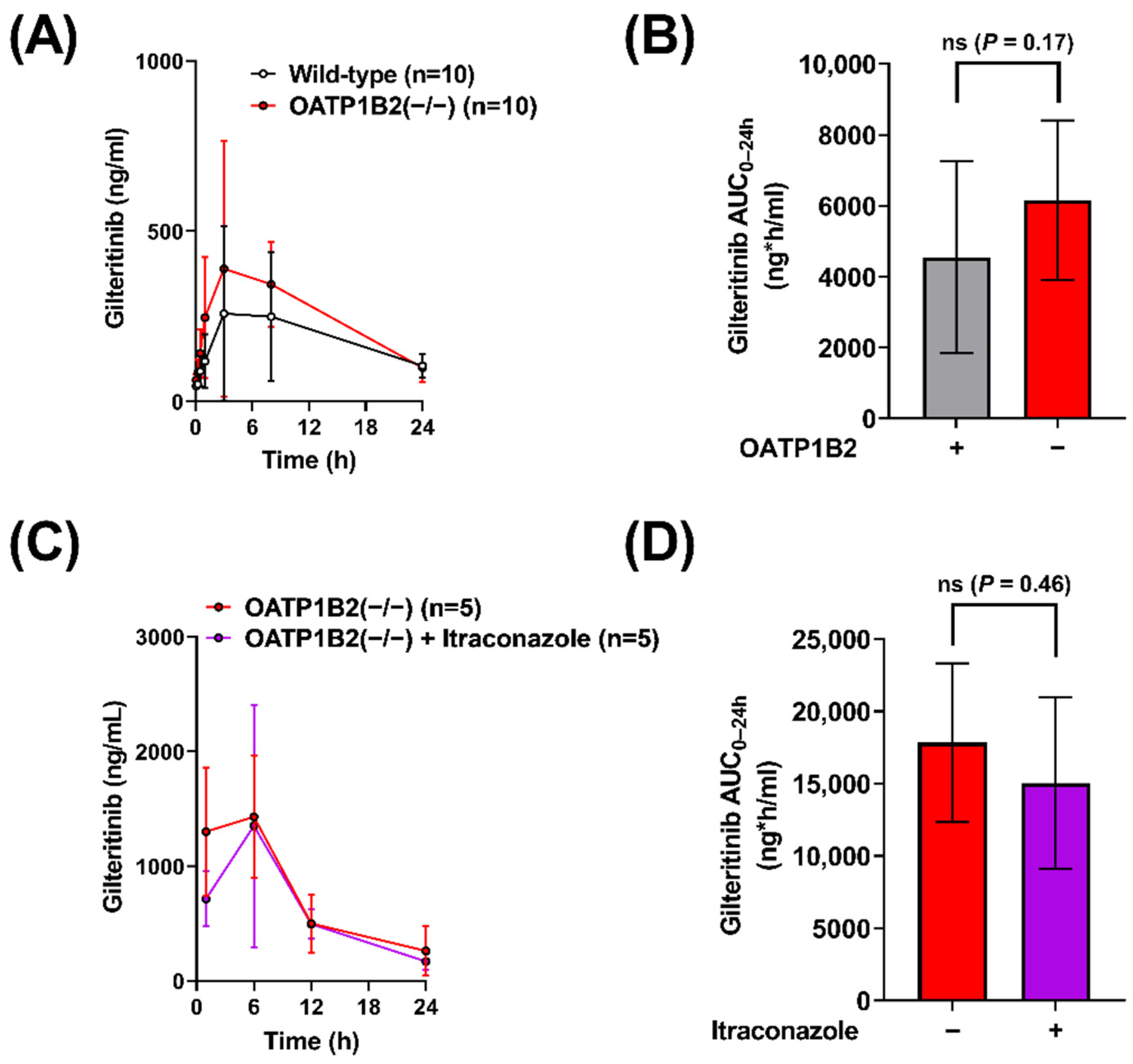
2.2. Interaction of Gilteritinib with OATP1B-Type Transporters
2.3. Contribution of OATP1B to the Interaction between Gilteritinib and Itraconazole
2.4. Impact of Itraconazole on Endogenous Biomarker of OATP1B Function
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Transport Inhibition Assays
4.3. Competitive Counterflow Assays
4.4. Washout Assay
4.5. In Vivo Pharmacokinetic Studies
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chew, S.; Mackey, M.C.; Jabbour, E. Gilteritinib in the Treatment of Relapsed and Refractory Acute Myeloid Leukemia with a FLT3 Mutation. Ther. Adv. Hematol. 2020, 11, 2040620720930614. [Google Scholar] [CrossRef] [PubMed]
- Thol, F. What to Use to Treat AML: The Role of Emerging Therapies. Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.C.; Agarwal, S.; Ahmad, H.; Amin, K.; Bewersdorf, J.P.; Zeidan, A.M. A Review of FLT3 Inhibitors in Acute Myeloid Leukemia. Blood Rev. 2022, 52, 100905. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Song, Y.; Liu, D. Gilteritinib: A Novel FLT3 Inhibitor for Acute Myeloid Leukemia. Biomark. Res. 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trials Using Gilteritinib—National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/gilteritinib (accessed on 16 March 2022).
- Pratz, K.W.; Cherry, M.; Altman, J.K.; Cooper, B.; Cruz, J.C.; Jurcic, J.G.; Levis, M.J.; Lin, T.L.; Perl, A.E.; Podoltsev, N.A.; et al. Updated Results from a Phase 1 Study of Gilteritinib in Combination with Induction and Consolidation Chemotherapy in Subjects with Newly Diagnosed Acute Myeloid Leukemia (AML). Blood 2018, 132, 564. [Google Scholar] [CrossRef]
- Dhillon, S. Gilteritinib: First Global Approval. Drugs 2019, 79, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Pulte, E.D.; Norsworthy, K.J.; Wang, Y.; Xu, Q.; Qosa, H.; Gudi, R.; Przepiorka, D.; Fu, W.; Okusanya, O.O.; Goldberg, K.B.; et al. FDA Approval Summary: Gilteritinib for Relapsed or Refractory Acute Myeloid Leukemia with a FLT3 Mutation. Clin. Cancer Res. 2021, 27, 3515–3521. [Google Scholar] [CrossRef] [PubMed]
- James, A.J.; Smith, C.C.; Litzow, M.; Perl, A.E.; Altman, J.K.; Shepard, D.; Kadokura, T.; Souda, K.; Patton, M.; Lu, Z.; et al. Pharmacokinetic Profile of Gilteritinib: A Novel FLT-3 Tyrosine Kinase Inhibitor. Clin. Pharmacokinet. 2020, 59, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, L.M.M.; Isringhausen, C.D.; Ogilvie, B.W.; Buckley, D.B. Evaluation of Ketoconazole and Its Alternative Clinical CYP3A4/5 Inhibitors as Inhibitors of Drug Transporters: The In Vitro Effects of Ketoconazole, Ritonavir, Clarithromycin, and Itraconazole on 13 Clinically-Relevant Drug Transporters. Drug Metab. Dispos. 2016, 44, 453–459. [Google Scholar] [CrossRef]
- Megías-Vericat, J.E.; Solana-Altabella, A.; Ballesta-López, O.; Martínez-Cuadrón, D.; Montesinos, P. Drug-Drug Interactions of Newly Approved Small Molecule Inhibitors for Acute Myeloid Leukemia. Ann. Hematol. 2020, 99, 1989–2007. [Google Scholar] [CrossRef] [PubMed]
- Eisenmann, E.D.; Garrison, D.A.; Talebi, Z.; Jin, Y.; Silvaroli, J.A.; Kim, J.-G.; Sparreboom, A.; Savona, M.R.; Mims, A.S.; Baker, S.D. Interaction of Antifungal Drugs with CYP3A- and OATP1B-Mediated Venetoclax Elimination. Pharmaceutics 2022, 14, 694. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.M.; Bock, T.; Meyer zu Schwabedissen, H.E. Establishment and Validation of Competitive Counterflow as a Method To Detect Substrates of the Organic Anion Transporting Polypeptide 2B1. Mol. Pharm. 2018, 15, 5501–5513. [Google Scholar] [CrossRef] [PubMed]
- Ungvári, O.; Király, L.; Bakos, É.; Özvegy-Laczka, C. 8-Acetoxy-Trisulfopyrene as the First Activatable Fluorogenic Probe for Add-and-Read Assessment of Organic Anion-Transporting Polypeptides, OATP1B1, OATP1B3, and OATP2B1. FASEB J. 2021, 35, e21863. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Chu, X.; Gibson, C.; Cui, D.; Yee, K.L.; Ballard, J.; Cabalu, T.; Hochman, J. Drug–Drug Interaction Studies: Regulatory Guidance and An Industry Perspective. AAPS J. 2013, 15, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, M.; Murakami, K. A Graphical Method for Determining Inhibition Constants. J. Enzym. Inhib. Med. Chem. 2009, 24, 1288–1290. [Google Scholar] [CrossRef]
- Roth, M.; Araya, J.J.; Timmermann, B.N.; Hagenbuch, B. Isolation of Modulators of the Liver-Specific Organic Anion-Transporting Polypeptides (OATPs) 1B1 and 1B3 from Rollinia Emarginata Schlecht (Annonaceae). J. Pharmacol. Exp. Ther. 2011, 339, 624–632. [Google Scholar] [CrossRef]
- Lai, Y. Organic Anion-Transporting Polypeptides (OATPs/SLCOs). In Transporters in Drug Discovery and Development; Elsevier: Amsterdam, The Netherlands, 2013; pp. 353–454. ISBN 978-1-907568-21-3. [Google Scholar]
- Uddin, M.E.; Garrison, D.A.; Kim, K.; Jin, Y.; Eisenmann, E.D.; Huang, K.M.; Gibson, A.A.; Hu, Z.; Sparreboom, A.; Hu, S. Influence of YES1 Kinase and Tyrosine Phosphorylation on the Activity of OCT1. Front. Pharmacol. 2021, 12, 644342. [Google Scholar] [CrossRef]
- Hayden, E.R.; Chen, M.; Pasquariello, K.Z.; Gibson, A.A.; Petti, J.J.; Shen, S.; Qu, J.; Ong, S.S.; Chen, T.; Jin, Y.; et al. Regulation of OATP1B1 Function by Tyrosine Kinase–Mediated Phosphorylation. Clin. Cancer Res. 2021, 27, 4301–4310. [Google Scholar] [CrossRef]
- Hove, V.N.; Anderson, K.; Hayden, E.R.; Pasquariello, K.Z.; Gibson, A.A.; Shen, S.; Qu, J.; Jin, Y.; Miecznikowski, J.C.; Hu, S.; et al. Influence of Tyrosine Kinase Inhibition on Organic Anion Transporting Polypeptide 1B3-Mediated Uptake. Mol. Pharmacol. 2022, 101, 381–389. [Google Scholar] [CrossRef]
- Bednarczyk, D. Fluorescence-Based Assays for the Assessment of Drug Interaction with the Human Transporters OATP1B1 and OATP1B3. Anal. Biochem. 2010, 405, 50–58. [Google Scholar] [CrossRef] [PubMed]
- van Hoppe, S.; Rood, J.J.M.; Buil, L.; Wagenaar, E.; Sparidans, R.W.; Beijnen, J.H.; Schinkel, A.H. P-Glycoprotein (MDR1/ABCB1) Restricts Brain Penetration of the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib, While Cytochrome P450-3A (CYP3A) Limits Its Oral Bioavailability. Mol. Pharm. 2018, 15, 5124–5134. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Hu, S.; Li, Y.; Gibson, A.A.; Fu, Q.; Baker, S.D.; Sparreboom, A. Role of Oatp2b1 in Drug Absorption and Drug-Drug Interactions. Drug Metab. Dispos. 2020, 48, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Chen, M.; Hu, S.; McElroy, C.A.; Mathijssen, R.H.; Sparreboom, A.; Baker, S.D. Development and Validation of an Analytical Method for Regorafenib and Its Metabolites in Mouse Plasma. J. Chromatogr. B 2018, 1090, 43–51. [Google Scholar] [CrossRef]
- Takehara, I.; Terashima, H.; Nakayama, T.; Yoshikado, T.; Yoshida, M.; Furihata, K.; Watanabe, N.; Maeda, K.; Ando, O.; Sugiyama, Y.; et al. Investigation of Glycochenodeoxycholate Sulfate and Chenodeoxycholate Glucuronide as Surrogate Endogenous Probes for Drug Interaction Studies of OATP1B1 and OATP1B3 in Healthy Japanese Volunteers. Pharm. Res. 2017, 34, 1601–1614. [Google Scholar] [CrossRef]
- Jin, Y.; Li, Y.; Eisenmann, E.D.; Figg, W.D.; Baker, S.D.; Sparreboom, A.; Hu, S. Determination of the Endogenous OATP1B Biomarkers Glycochenodeoxycholate-3-Sulfate and Chenodeoxycholate-24-Glucuronide in Human and Mouse Plasma by a Validated UHPLC-MS/MS Method. J. Chromatogr. B 2022, 1210, 123437. [Google Scholar] [CrossRef]
- Usuki, K.; Sakura, T.; Kobayashi, Y.; Miyamoto, T.; Iida, H.; Morita, S.; Bahceci, E.; Kaneko, M.; Kusano, M.; Yamada, S.; et al. Clinical Profile of Gilteritinib in Japanese Patients with Relapsed/Refractory Acute Myeloid Leukemia: An Open-Label Phase 1 Study. Cancer Sci. 2018, 109, 3235–3244. [Google Scholar] [CrossRef]
- Daver, N.; Perl, A.E.; Maly, J.; Levis, M.; Ritchie, E.; Litzow, M.; McCloskey, J.; Smith, C.C.; Schiller, G.; Bradley, T.; et al. Venetoclax Plus Gilteritinib for FLT3-Mutated Relapsed/Refractory Acute Myeloid Leukemia. JCO 2022. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Z.; Chen, D.; Ye, X.-Y. An LC-MS/MS Bioanalytical Assay for the Determination of Gilteritinib in Rat Plasma and Application to a Drug-Drug Interaction Study. Drug Des. Devel. Ther. 2020, 14, 2061–2067. [Google Scholar] [CrossRef]
- Garrison, D.A.; Jin, Y.; Uddin, M.E.; Sparreboom, A.; Baker, S.D. Development, Validation, and Application of an LC-MS/MS Method for the Determination of the AXL/FLT3 Inhibitor Gilteritinib in Mouse Plasma. J. Chromatogr. B 2021, 1179, 122882. [Google Scholar] [CrossRef]
- Alluri, R.; Li, R.; Varma, M. Transporter–Enzyme Interplay and the Hepatic Drug Clearance: What Have We Learned so Far? Available online: https://www.tandfonline.com/doi/epub/10.1080/17425255.2020.1749595?needAccess=true (accessed on 17 March 2022).
- Czyrski, A.; Resztak, M.; Świderski, P.; Brylak, J.; Główka, F.K. The Overview on the Pharmacokinetic and Pharmacodynamic Interactions of Triazoles. Pharmaceutics 2021, 13, 1961. [Google Scholar] [CrossRef]
- Isoherranen, N.; Kunze, K.L.; Allen, K.E.; Nelson, W.L.; Thummel, K.E. Role of Itraconazole Metabolites in CYP3A4 Inhibition. Drug Metab. Dispos. 2004, 32, 1121–1131. [Google Scholar] [CrossRef]
- Niwa, T.; Shiraga, T.; Takagi, A. Effect of Antifungal Drugs on Cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 Activities in Human Liver Microsomes. Biol. Pharm. Bull. 2005, 28, 1805–1808. [Google Scholar] [CrossRef]
- Templeton, I.E.; Thummel, K.E.; Kharasch, E.D.; Kunze, K.L.; Hoffer, C.; Nelson, W.L.; Isoherranen, N. Contribution of Itraconazole Metabolites to Inhibition of CYP3A4 in Vivo. Clin. Pharmacol. Ther. 2008, 83, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Zhao, P.; Zhang, L. Physiologically Based Pharmacokinetic (PBPK) Modeling of Pitavastatin and Atorvastatin to Predict Drug-Drug Interactions (DDIs). Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 689–705. [Google Scholar] [CrossRef] [PubMed]
- Sane, R.S.; Cheung, K.W.K.; Cho, E.; Liederer, B.M.; Hanover, J.; Malhi, V.; Plise, E.; Wong, S.; Musib, L. Evaluation of Ipatasertib Interactions with Itraconazole and Coproporphyrin I and III in a Single Drug Interaction Study in Healthy Subjects. J. Pharmacol. Exp. Ther. 2021, 378, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Christopher, L.; Lai, Y.; Gong, J.; Kandoussi, H.; Garonzik, S.; Perera, V.; Garimella, T.; Humphreys, W.G. Further Studies to Support the Use of Coproporphyrin I and III as Novel Clinical Biomarkers for Evaluating the Potential for Organic Anion Transporting Polypeptide 1B1 and OATP1B3 Inhibition. Drug Metab. Dispos. 2018, 46, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Ikeda, Y.; Fujita, T.; Yoshida, K.; Azuma, Y.; Haruyama, Y.; Yamane, N.; Kumagai, Y.; Sugiyama, Y. Identification of the Rate-Determining Process in the Hepatic Clearance of Atorvastatin in a Clinical Cassette Microdosing Study. Clin. Pharmacol. Ther. 2011, 90, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Gilibili, R.R.; Chatterjee, S.; Bagul, P.; Mosure, K.W.; Murali, B.V.; Mariappan, T.T.; Mandlekar, S.; Lai, Y. Coproporphyrin-I: A Fluorescent, Endogenous Optimal Probe Substrate for ABCC2 (MRP2) Suitable for Vesicle-Based MRP2 Inhibition Assay. Drug Metab. Dispos. 2017, 45, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Bezençon, J.; Saran, C.; Hussner, J.; Beaudoin, J.J.; Zhang, Y.; Shen, H.; Fallon, J.K.; Smith, P.C.; Meyer Zu Schwabedissen, H.E.; Brouwer, K.L.R. Endogenous Coproporphyrin I and III Are Altered in Multidrug Resistance-Associated Protein 2-Deficient (TR-) Rats. J. Pharm. Sci. 2021, 110, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.C.J.; Hawksworth, G.M.; Weaver, R.J. ATP-Dependent Transport of Statins by Human and Rat MRP2/Mrp2. Toxicol. Appl. Pharmacol. 2013, 269, 187–194. [Google Scholar] [CrossRef]
- Lau, Y.Y.; Okochi, H.; Huang, Y.; Benet, L.Z. Multiple Transporters Affect the Disposition of Atorvastatin and Its Two Active Hydroxy Metabolites: Application of in Vitro and Ex Situ Systems. J. Pharmacol. Exp. Ther. 2006, 316, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.I.; Hu, S.; Roberts, J.L.; Gibson, A.A.; Orwick, S.J.; Li, L.; Sparreboom, A.; Baker, S.D. Contribution of OATP1B1 and OATP1B3 to the Disposition of Sorafenib and Sorafenib-Glucuronide. Clin. Cancer Res. 2013, 19, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- van Herwaarden, A.E.; Wagenaar, E.; van der Kruijssen, C.M.M.; van Waterschoot, R.A.B.; Smit, J.W.; Song, J.-Y.; van der Valk, M.A.; van Tellingen, O.; van der Hoorn, J.W.A.; Rosing, H.; et al. Knockout of Cytochrome P450 3A Yields New Mouse Models for Understanding Xenobiotic Metabolism. J. Clin. Investig. 2007, 117, 3583–3592. [Google Scholar] [CrossRef] [PubMed]
- Ly, J.Q.; Messick, K.; Qin, A.; Takahashi, R.H.; Choo, E.F. Utility of CYP3A4 and PXR-CAR-CYP3A4/3A7 Transgenic Mouse Models To Assess the Magnitude of CYP3A4 Mediated Drug-Drug Interactions. Mol. Pharm. 2017, 14, 1754–1759. [Google Scholar] [CrossRef]
- Leblanc, A.F.; Huang, K.M.; Uddin, M.E.; Anderson, J.T.; Chen, M.; Hu, S. Murine Pharmacokinetic Studies. Bio. Protoc. 2018, 8, e3056. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Genotype | Co-Treatment Dose (mg/kg) | Gilteritinib Cmax (ng/mL) | Gilteritinib AUC0–24h (ng ∗ h/mL) | Gilteritinib AUC Fold Increase |
|---|---|---|---|---|
| Wild-type FVB | None | 310 (±180) | 4650 (±960) | |
| Wild-type FVB | Itraconazole (100) | 960 (±630) * | 11,480 (±4430) ** | 2.5 (vs. Vehicle) |
| CYP3A(−/−) | None | 380 (±160) | 5990 (±1320) | 1.3 (vs. WT) |
| CYP3A(−/−) | Itraconazole (100) | 430 (±130) | 5840 (±1520) | 0.97 (vs. Vehicle) |
| Wild-type DBA/1J | None | 260 (±250) | 4550 (±2720) | |
| OATP1B2(−/−) | None | 390 (±380) | 6160 (±2250) | 1.4 (vs. WT) |
| OATP1B2(−/−) | None | 1430 (±540) | 17,840 (±5500) | |
| OATP1B2(−/−) | Itraconazole (100) | 1350 (±1050) | 15,050 (±5930) | 0.84 (vs. Vehicle) |
| Mouse Genotype | Treatment (Dose mg/kg) | CDCA-24G Cmax (ng/mL) | CDCA-24G AUC0–4h (ng ∗ h/mL) | CDCA-24G AUC Fold Increase |
|---|---|---|---|---|
| Wild-type FVB | None | 1.17 (±0.38) | 3.78 (±0.20) | |
| Wild-type FVB | Itraconazole (100) | 1.84 (±0.60) * | 5.10 (±1.60) * | 1.3 (vs. WT) 1.5 (vs. CYP3A (−/−) + Itraconazole) |
| CYP3A(−/−) | None | 1.02 (±0.36) | 3.35 (±1.10) | 0.9 (vs. WT) |
| CYP3A(−/−) | Itraconazole (100) | 0.98 (±0.20) | 3.30 (±1.00) | 1 (vs. CYP3A(−/−)) 0.9 (vs. WT Alone) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrison, D.A.; Jin, Y.; Talebi, Z.; Hu, S.; Sparreboom, A.; Baker, S.D.; Eisenmann, E.D. Itraconazole-Induced Increases in Gilteritinib Exposure Are Mediated by CYP3A and OATP1B. Molecules 2022, 27, 6815. https://doi.org/10.3390/molecules27206815
Garrison DA, Jin Y, Talebi Z, Hu S, Sparreboom A, Baker SD, Eisenmann ED. Itraconazole-Induced Increases in Gilteritinib Exposure Are Mediated by CYP3A and OATP1B. Molecules. 2022; 27(20):6815. https://doi.org/10.3390/molecules27206815
Chicago/Turabian StyleGarrison, Dominique A., Yan Jin, Zahra Talebi, Shuiying Hu, Alex Sparreboom, Sharyn D. Baker, and Eric D. Eisenmann. 2022. "Itraconazole-Induced Increases in Gilteritinib Exposure Are Mediated by CYP3A and OATP1B" Molecules 27, no. 20: 6815. https://doi.org/10.3390/molecules27206815
APA StyleGarrison, D. A., Jin, Y., Talebi, Z., Hu, S., Sparreboom, A., Baker, S. D., & Eisenmann, E. D. (2022). Itraconazole-Induced Increases in Gilteritinib Exposure Are Mediated by CYP3A and OATP1B. Molecules, 27(20), 6815. https://doi.org/10.3390/molecules27206815