
27 Years of Catalytic Carbonylative Coupling Reactions in Hungary (1994–2021)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
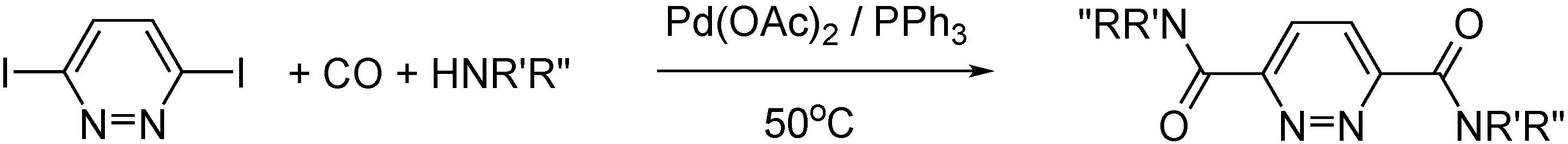{kind=link}
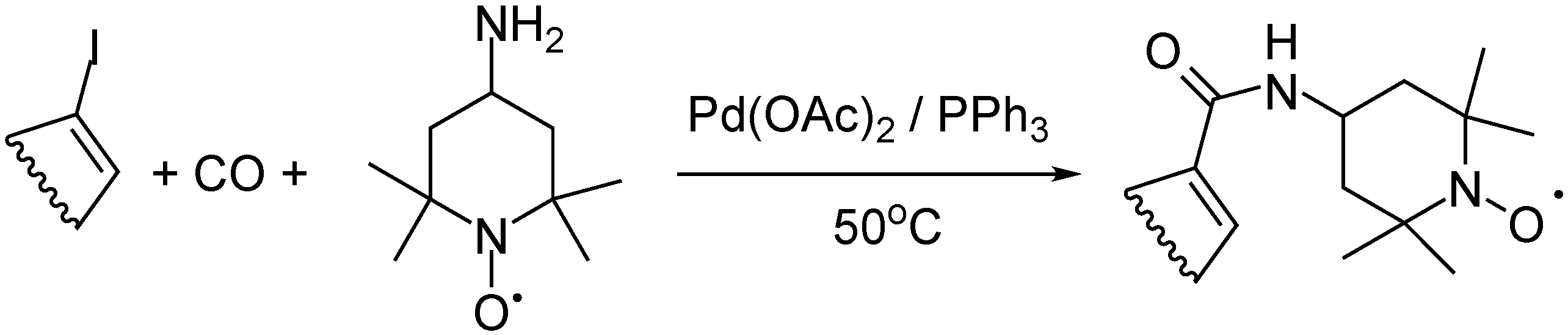{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
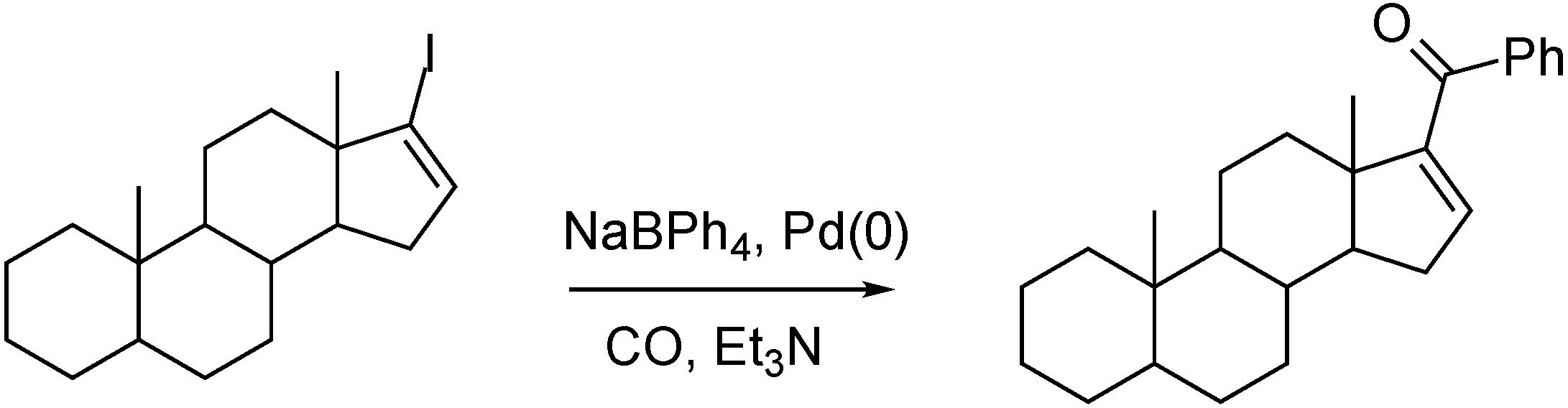{kind=link}
{kind=link}
{kind=link}
{kind=link}
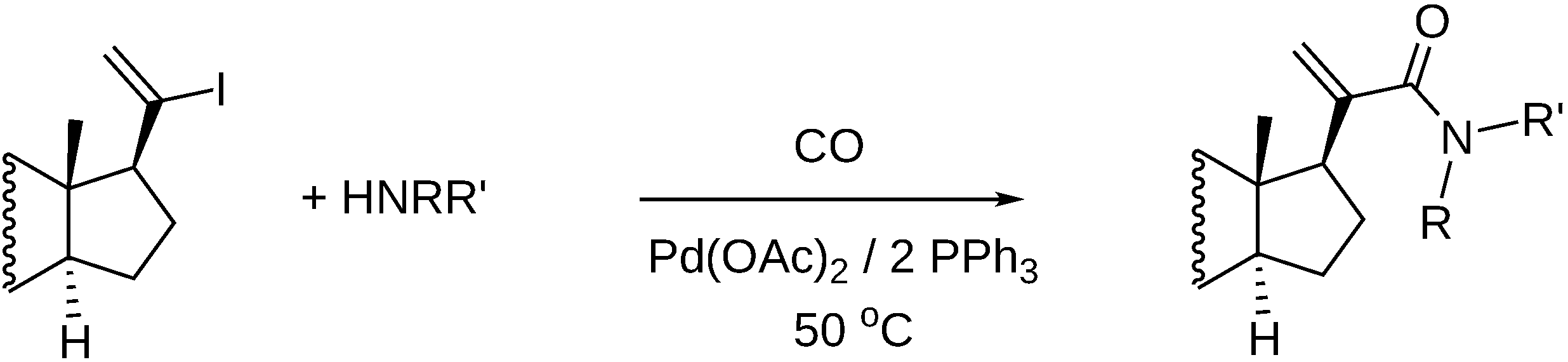{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Formation of the Active Catalyst
3. Reactions in Conventional Solvents
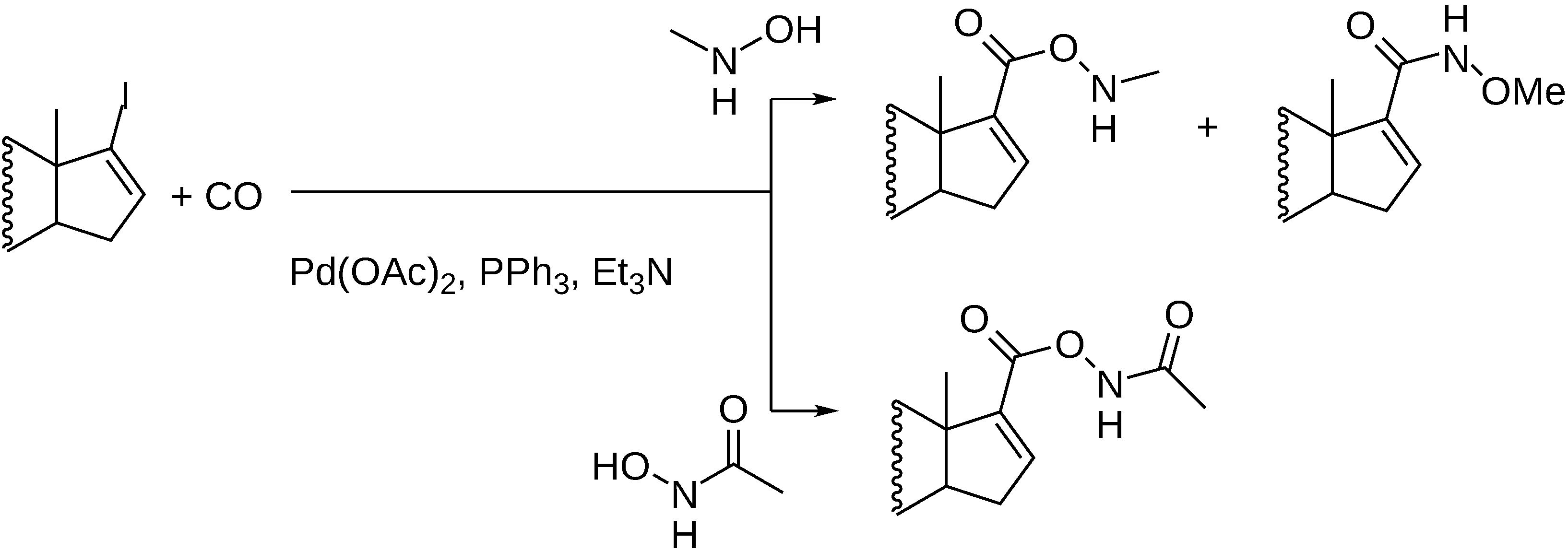
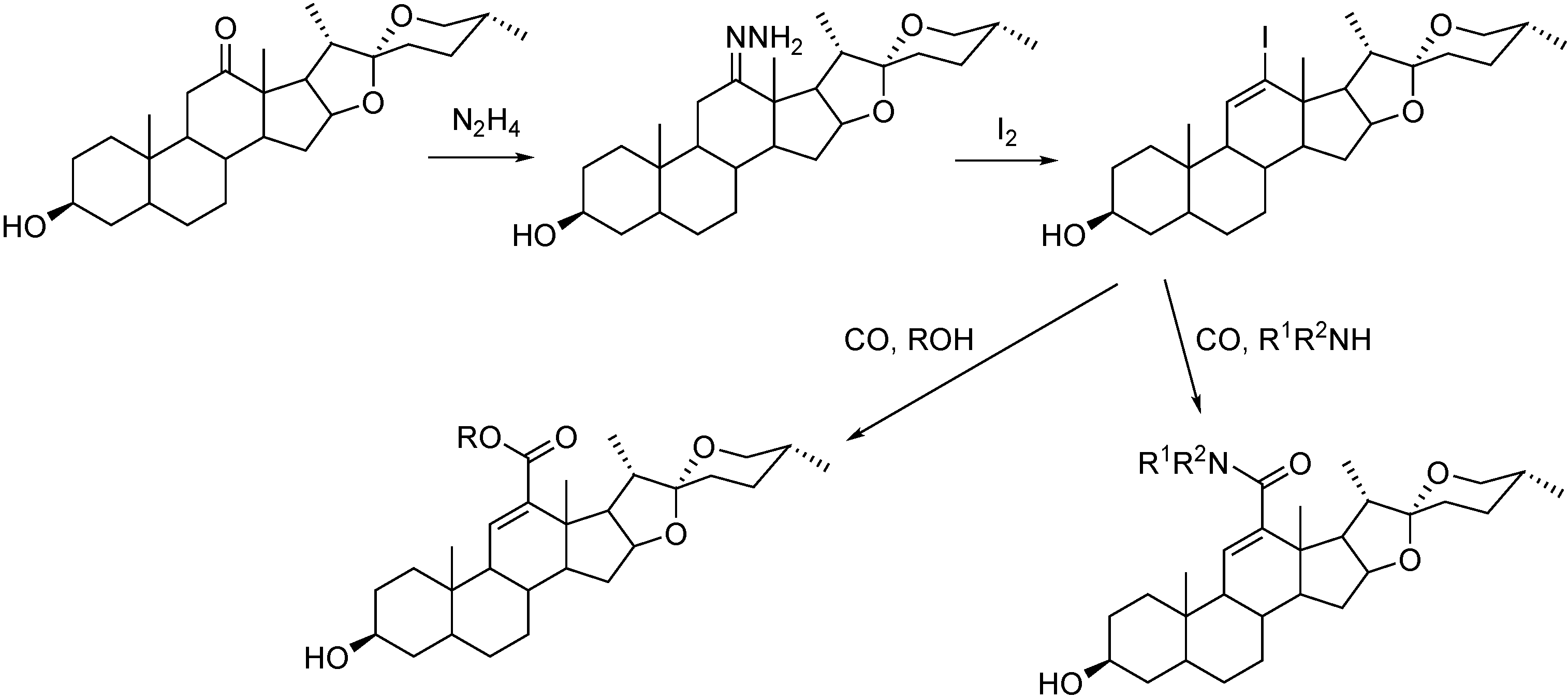
3.1. Preparation of Carbonyl Compounds with Steroid Scaffolds
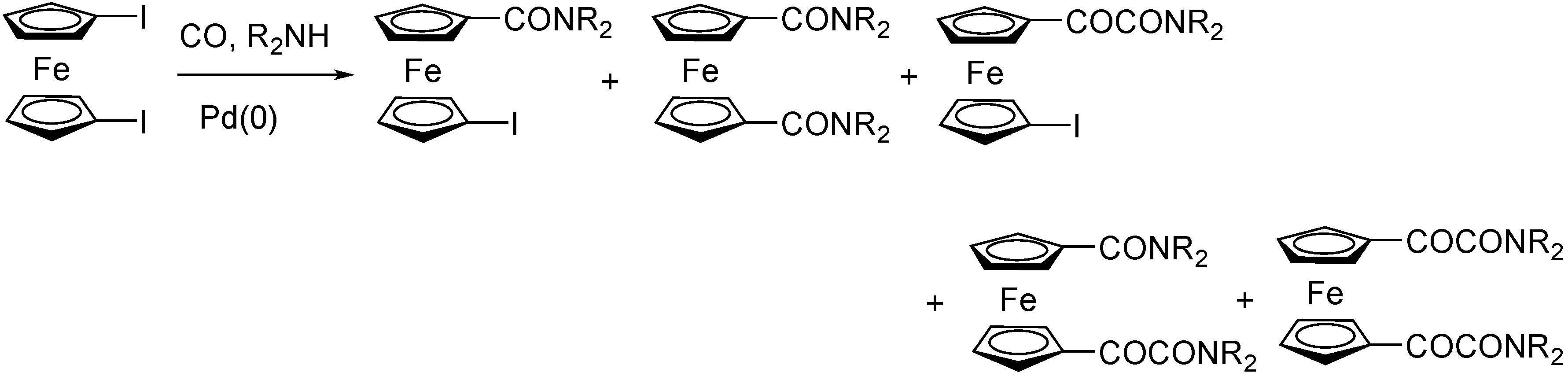
3.2. Ferrocene-Based Substrates
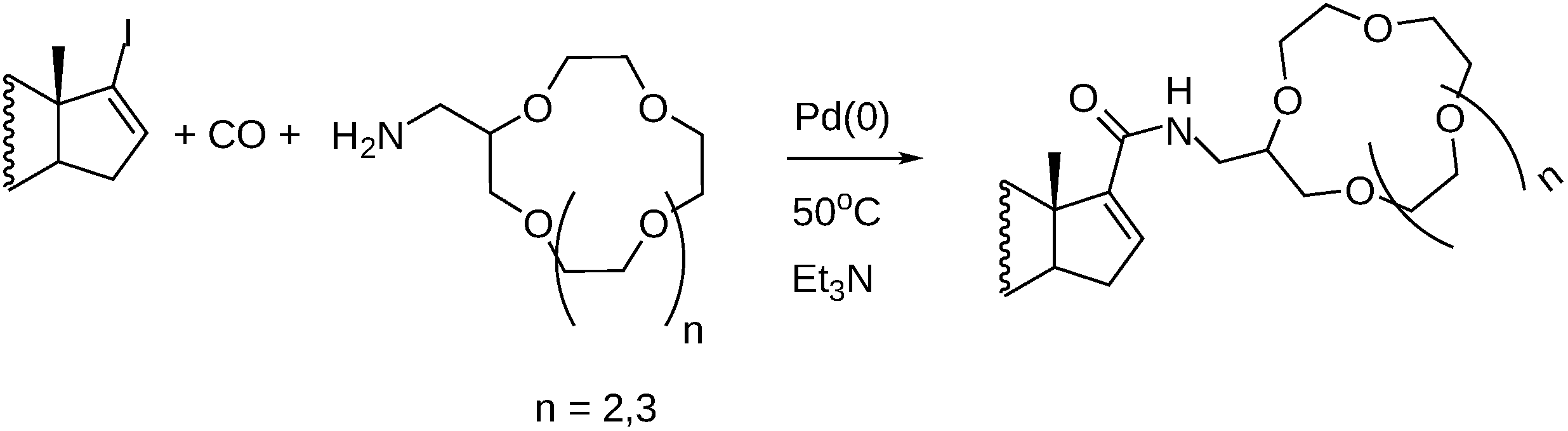
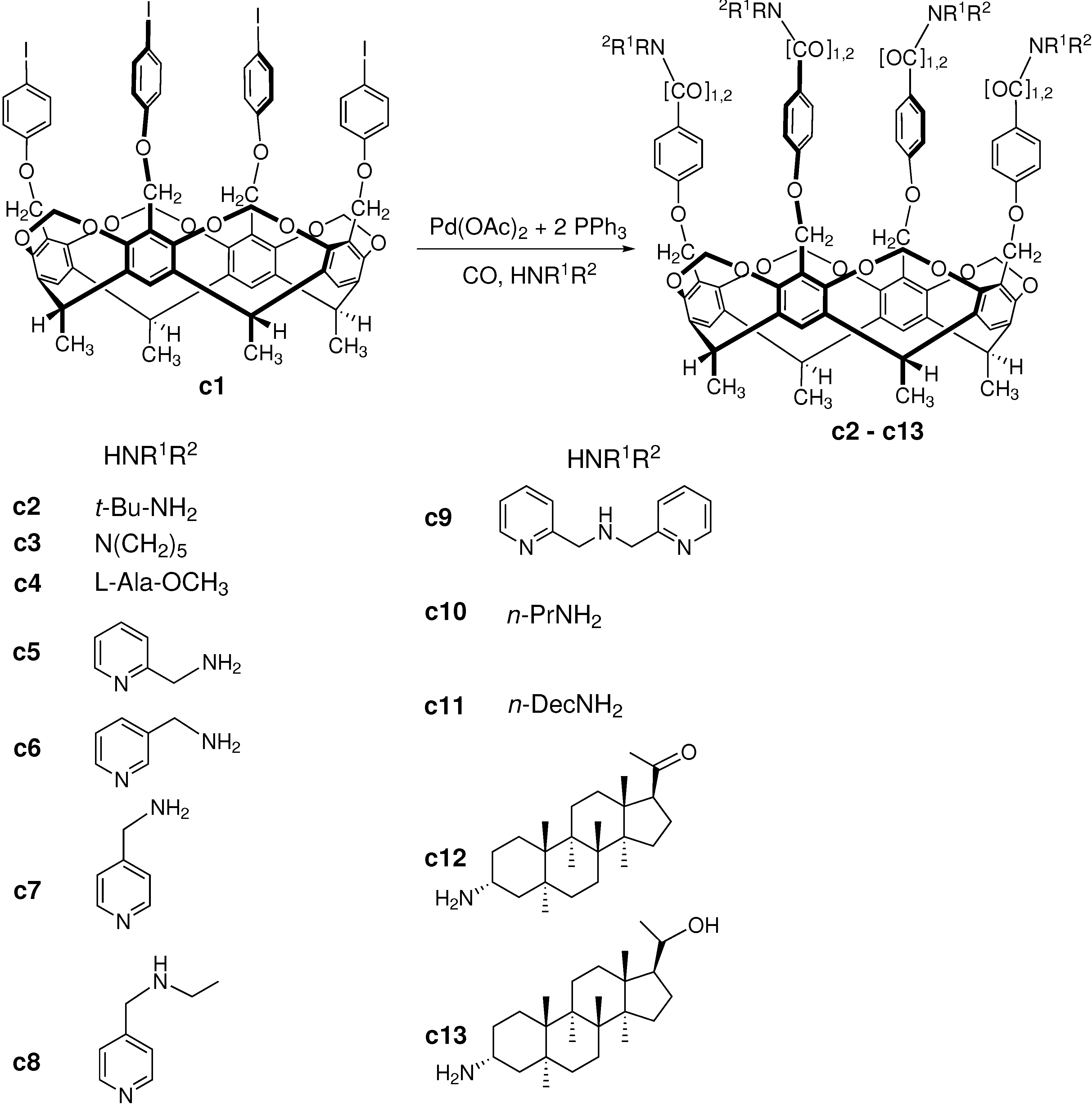
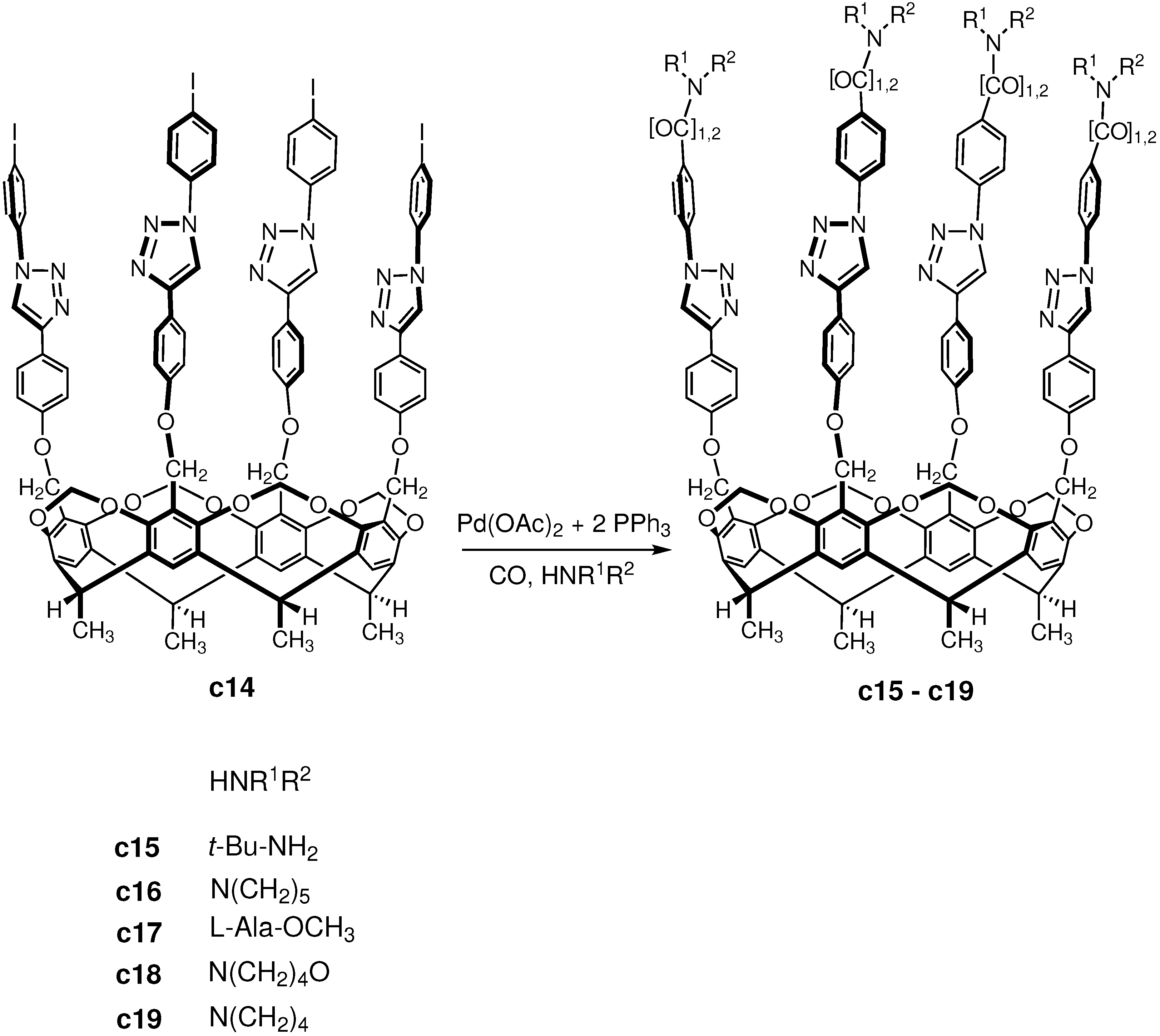
3.3. Carbonylation on Macromolecular Cavitand Scaffolds
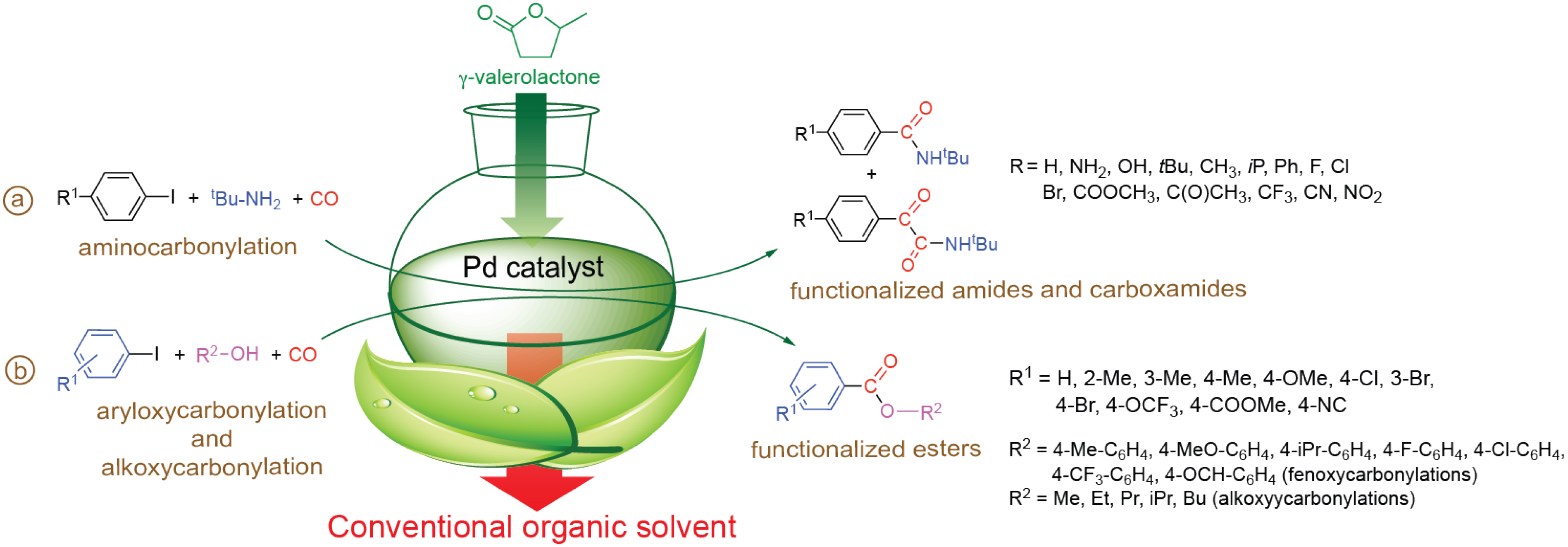
4. Reactions in Biomass-Based Solvents
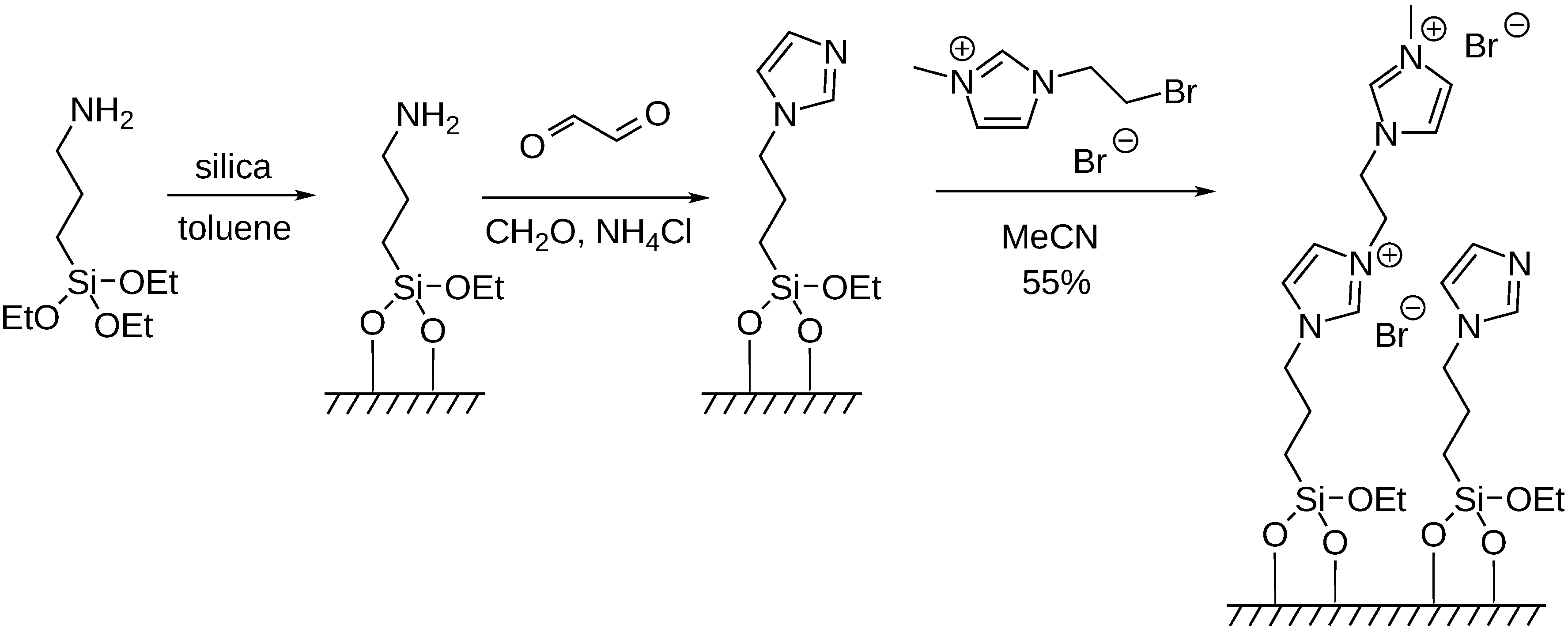
5. Carbonylation Reactions in Ionic Liquid and on Supported Ionic Liquid Phase (SILP)
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef]
- Fortman, G.C.; Nolan, S.P. N-Heterocyclic carbene (NHC) ligands and palladium in homogeneous cross-coupling catalysis: A perfect union. Chem. Soc. Rev. 2011, 40, 5151–5169. [Google Scholar] [CrossRef]
- Gabriele, B. (Ed.) Carbon Monoxide in Organic Synthesis–Carbonylation Chemistry; Wiley-VCH: Weinheim, Germany, 2021. [Google Scholar]
- Skoda-Foldes, R.; Kollár, L. Synthetic applications of palladium catalysed carbonylation of organic halides. Curr. Org. Chem. 2002, 6, 1097–1119. [Google Scholar] [CrossRef]
- Kollár, L. (Ed.) Modern Carbonylation Methods; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Ishiyama, T.; Kizaki, H.; Hayashi, T.; Suzuki, A.; Miyaura, N. Palladium-catalyzed carbonylative cross-coupling reaction of arylboronic acids with aryl electrophiles: Synthesis of biaryl ketones. J. Org. Chem. 1998, 63, 4726–4731. [Google Scholar] [CrossRef]
- Wu, X.F.; Neumann, H.; Spannenberg, A.; Schulz, T.; Jiao, H.; Beller, M. Development of a general palladium-catalyzed carbonylative Heck reaction of aryl halides. J. Am. Chem. Soc 2010, 132, 14596–14602. [Google Scholar] [CrossRef]
- Mohamed Ahmed, M.S.; Mori, A. Carbonylative sonogashira coupling of terminal alkynes with aqueous ammonia. Org. Lett. 2003, 5, 3057–3060. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.F.; Schranck, J.; Neumann, H.; Beller, M. Palladium-Catalyzed Carbonylative Negishi-type Coupling of Aryl Iodides with Benzyl Chlorides. Chem. Asian J. 2012, 7, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, F.; Sugimoto, T.; Yuasa, Y.; Santra, M.; Yamamoto, T.; Yamamoto, A. Palladium-promoted double-carbonylation reactions. Reactions of organopalladium compounds with carbon monoxide and amines to give α-keto amides. Organometallics 1984, 3, 683–692. [Google Scholar] [CrossRef]
- Ozawa, F.; Sugimoto, T.; Yamamoto, T.; Yamamoto, A. Preparation of trans-Pd(COCOR)Cl(PMePh2)2 complexes (R = Ph and Me) and their reactivities relative to double carbonylation promoted by palladium. Organometallics 1984, 3, 692–697. [Google Scholar] [CrossRef]
- Ozawa, F.; Soyama, H.; Yanagihara, H.; Aoyama, I.; Takino, H.; Izawa, K.; Yamamoto, T.; Yamamoto, A. Palladium-catalyzed double carbonylation of aryl halides to give α-keto amides. Mechanistic studies. J. Am. Chem. Soc. 1985, 107, 3235–3245. [Google Scholar] [CrossRef]
- Marosvölgyi-Haskó, D.; Kégl, T.; Kollár, L. Substituent effects in aminocarbonylation of para-substituted iodobenzenes. Tetrahedron 2016, 72, 7509–7516. [Google Scholar] [CrossRef]
- Prasad, A.S.; Satyanarayana, B. Fe3O4 supported Pd(0) nanoparticles catalyzed alkoxycarbonylation of aryl halides. J. Mol. Catal. A Chem. 2013, 370, 205–209. [Google Scholar] [CrossRef]
- Skoda-Földes, R.; Kollár, L.; Marinelli, F.; Arcadi, A. Direct and carbonylative vinylation of steroidal triflates in the presence of homogeneous palladium catalysts. Steroids 1994, 59, 691–695. [Google Scholar] [CrossRef]
- Kollár, L.; Floris, B.; Pino, P. Hydroformylation of Vinylferrocene with Rhodium and Platinum Catalysts. Chimia 1986, 40, 201–202. [Google Scholar]
- Kollár, L.; Consiglio, G.; Pino, P. Asymmetric Hydroformylation of Unsaturated Esters with PtCl(SnCl3)[(R,R)-DIOP] Catalyst. J. Organomet. Chem. 1987, 330, 305–314. [Google Scholar] [CrossRef]
- Kollár, L.; Bakos, J.; Tóth, I.; Heil, B. Temperature dependence of the asymmetric induction in the PtCl(SnCl3)[(-)-(2S,4S)-2,4-bis(diphenylphosphino)pentane]-catalyzed enantioselective hydroformylation reaction. J. Organomet. Chem. 1988, 350, 277–284. [Google Scholar] [CrossRef]
- Kollár, L.; Bakos, J.; Tóth, I.; Heil, B. Asymmetric hydroformylation with Pt-phosphine-SnCl2 and Pt-bisphosphine-CuCl2 (or CuCl) catalytic systems. J. Organomet. Chem. 1989, 370, 257–261. [Google Scholar] [CrossRef]
- Kollár, L.; Bakos, J.; Heil, B.; Sándor, P.; Szalontai, G. Hydroformylation of chiral terpenes with PtCl(SnCl3)-(bis-phosphine) as catalyst. J. Organomet. Chem. 1990, 385, 147–152. [Google Scholar] [CrossRef]
- Kollár, L.; Sándor, P.; Szalontai, G.; Heil, B. The role of additives in platinum-catalyzed hydroformylation. J. Organomet. Chem. 1990, 393, 153–158. [Google Scholar] [CrossRef]
- Skoda-Földes, R.; Kollár, L.; Heil, B.; Gálik, G.; Tuba, Z.; Arcade, A. A possible way for the introduction of α-and β-formyl-ethyl-substituents into the steroid-skeleton via coupling and carbonylation reactions. Tetrahedron Asymmetry 1991, 2, 633–634. [Google Scholar] [CrossRef]
- Kollár, L.; Sándor, P.; Szalontai, G. Temperature dependence of the enantioselective hydroformylation with PtCl2[(S)-BINAP] + SnCl2 catalyst and the dynamic NMR study of the catalytic precursor. J. Mol. Catal. 1991, 67, 191–198. [Google Scholar] [CrossRef]
- Kollár, L.; Floris, B. Highly selective hydroformylation and dimerization reactions of 2-ferrocenylpropene. J. Organomet. Chem. 1992, 441, 117–123. [Google Scholar] [CrossRef]
- Kollár, L.; Wada, T.; Lautens, M. Asymmetric hydroformylation of deltacyclene. Tetrahedron Asymmetry 1992, 3, 1011–1014. [Google Scholar] [CrossRef]
- Kollár, L.; Sándor, P. Highly stereoselective hydroformylation of a (2R)-2-tert-butyl-Δ4-1, 3-oxazoline derivative. J. Organomet. Chem. 1993, 445, 257–259. [Google Scholar] [CrossRef]
- Kollár, L.; Kégl, T.; Bakos, J. Platinum-catalysed enantioselective hydroformylation of styrene. Platinum-diphosphine-tin (II) fluoride catalytic system: A novel asymmetric hydroformylation catalyst. J. Organomet. Chem. 1993, 453, 155–158. [Google Scholar] [CrossRef]
- Kollár, L.; Skoda-Földes, R.; Mahó, S.; Tuba, Z. Functionalization of the estrone skeleton via homogeneous coupling and hydroformylation reactions. J. Organomet. Chem. 1993, 453, 159–162. [Google Scholar] [CrossRef]
- Tóth, I.; Kégl, T.; Elsevier, C.J.; Kollár, L. CO Insertion in Four-Coordinate cis-Methyl(carbonyl)platinum-Diphosphine Compounds. An Ionic Mechanism for Platinum-Diphosphine-Catalyzed Hydroformylation. Inorg. Chem. 1994, 33, 5708–5712. [Google Scholar] [CrossRef]
- Gladiali, S.; Fabbri, D.; Kollár, L. Asymmetric hydroformylation of styrene catalysed by platinum-tin complexes with chiral bis-binaphthophosphole ligands. J. Organomet. Chem. 1995, 491, 91–96. [Google Scholar] [CrossRef]
- Kollár, L.; Bódi, G. Asymmetric hydroformylation of mono-and sesquiterpenes. Chirality 1995, 7, 121–127. [Google Scholar] [CrossRef]
- Skoda-Földes, R.; Jeges, G.; Kollár, L.; Horváth, J.; Tuba, Z. Synthesis of Pentacyclic Steroids via Tandem Stille Coupling and Diels- Alder Reactions. J. Org. Chem. 1997, 62, 1326–1332. [Google Scholar] [CrossRef]
- Csákai, Z.; Skoda-Földes, R.; Kollár, L. NMR investigation of Pd(II)-Pd(0) reduction in the presence of mono- and ditertiary phosphines. Inorg. Chim. Acta 1999, 286, 93–97. [Google Scholar] [CrossRef]
- Pálinkás, N.; Kollár, L.; Kégl, T. Viable pathways for the oxidative addition of iodobenzene to palladium (0)-triphenylphosphine-carbonyl complexes: A theoretical study. Dalton Trans. 2017, 46, 15789–15802. [Google Scholar] [CrossRef]
- Petz, A.; Pintér, Z.; Kollár, L. Mass spectrometric studies on the coupling model reaction towards alkenyl–aryl ketones. J. Biochem. Biophys. Methods 2004, 61, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Petz, A.; Péczely, G.; Pintér, Z.; Kollár, L. Carbonylative and direct Suzuki–Miyaura cross-coupling reactions with 1-iodo-cyclohexene. J. Mol. Catal. A Chem. 2006, 255, 97–102. [Google Scholar] [CrossRef]
- Takács, E.; Varga, C.; Skoda-Földes, R.; Kollar, L. Facile synthesis of primary amides and ketoamides via a palladium-catalysed carbonylation–deprotection reaction sequence. Tetrahedron Lett. 2007, 48, 2453–2456. [Google Scholar] [CrossRef]
- Takács, A.; Abreu, A.R.; Peixoto, A.F.; Pereira, M.M.; Kollár, L. Synthesis of Ortho-alkoxy-aryl Carboxamides via Palladium-Catalyzed Aminocarbonylation. Synth. Commun. 2009, 39, 1534–1548. [Google Scholar] [CrossRef]
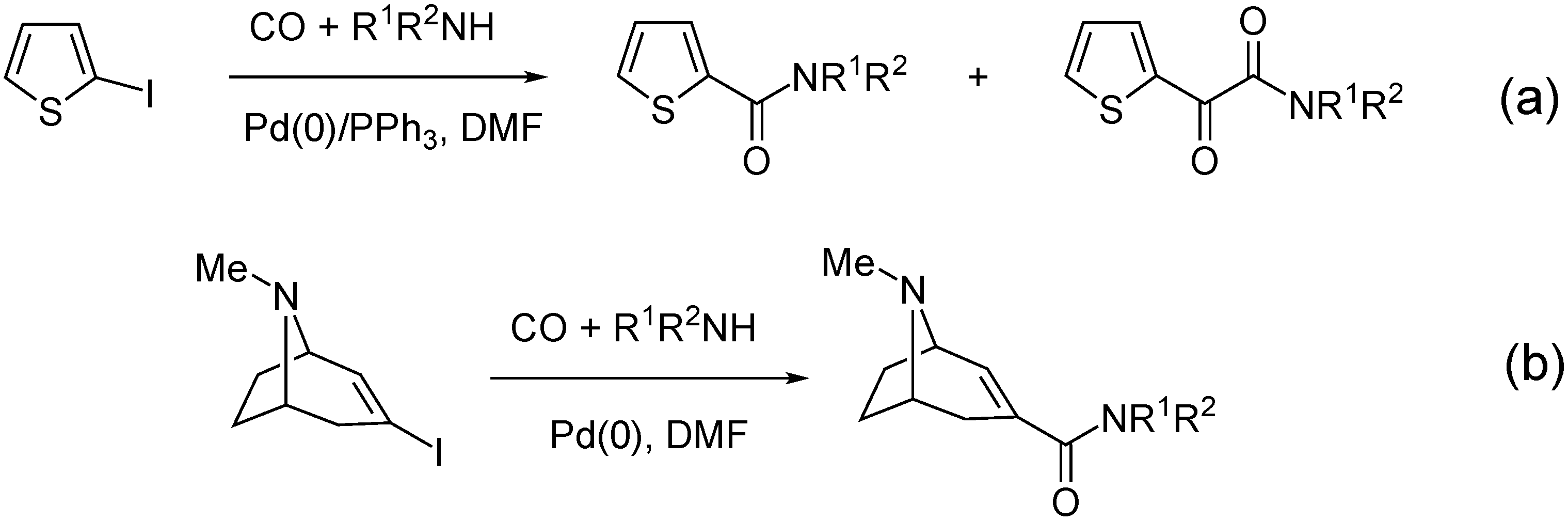
- Takacs, A.; Petz, A.; Jakab, B.; Kollar, L. Aminocarbonylation of 2-iodothiophene: High-yielding synthesis of thiophen-2-yl-glyoxylamides. Lett. Org. Chem. 2007, 4, 590–594. [Google Scholar] [CrossRef]
- Horvath, L.; Berente, Z.; Kollar, L. High-yielding aminocarbonylation of 3-iodo-2-tropene by using amino acid esters as N-nucleophiles. Lett. Org. Chem. 2007, 4, 236–238. [Google Scholar] [CrossRef]
- Takács, E.; Skoda-Földes, R.; Ács, P.; Müller, E.; Kokotos, G.; Kollár, L. Prolinates as secondary amines in aminocarbonylation: Synthesis of N-acylated prolinates. Lett. Org. Chem. 2006, 3, 62–67. [Google Scholar] [CrossRef]
- Kollár, L.; Erdélyi, Á.; Rasheed, H.; Takács, A. Selective Synthesis of N-Acylnortropane Derivatives in Palladium-Catalysed Aminocarbonylation. Molecules 2021, 26, 1813. [Google Scholar] [CrossRef] [PubMed]
- Szőke, G.; Takács, A.; Kollár, L. Synthesis of Pyridazine Dicarboxamides via Highly Selective Palladium-catalyzed Aminocarbonylation. J. Heterocycl. Chem. 2015, 53, 2020–2024. [Google Scholar] [CrossRef]
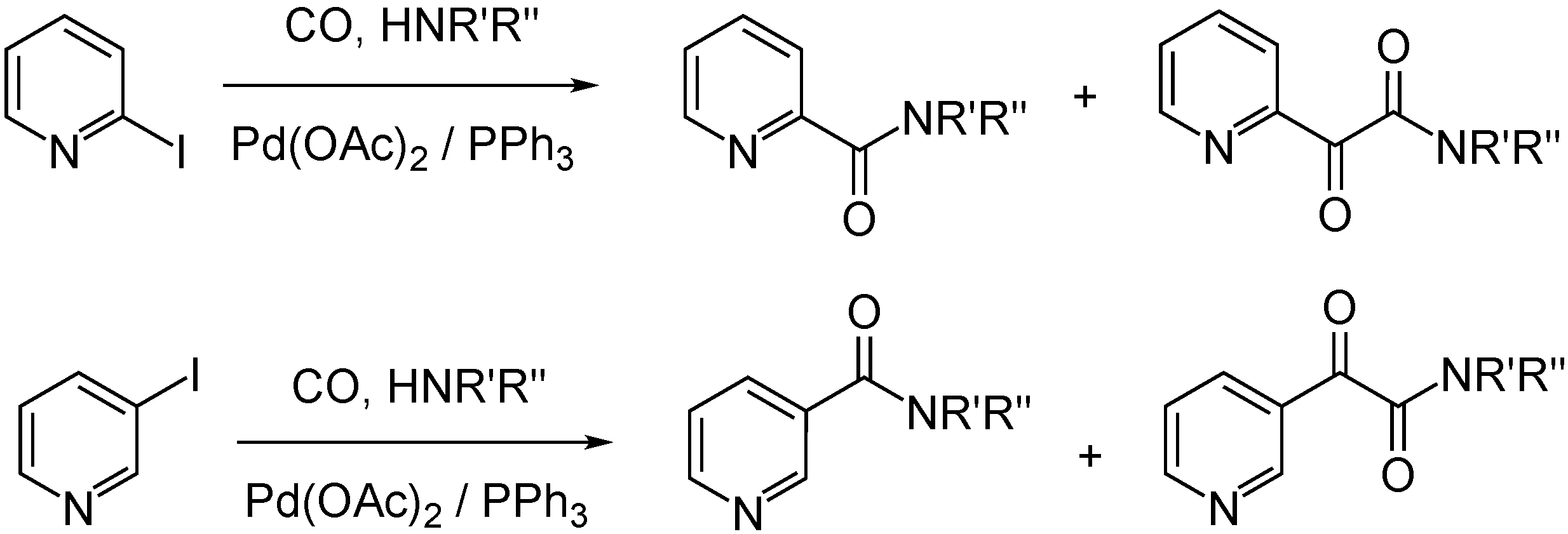
- Gergely, M.; Takács, A.; Kollár, L. 4-Amino-TEMPO asN-Nucleophile in Palladium-Catalyzed Aminocarbonylation. J. Heterocycl. Chem. 2016, 54, 634–640. [Google Scholar] [CrossRef]
- Takács, A.; Jakab, B.; Petz, A.; Kollár, L. Homogeneous catalytic aminocarbonylation of nitrogen-containing iodo-heteroaromatics. Synthesis of N-substituted nicotinamide related compounds. Tetrahedron 2007, 63, 10372–10378. [Google Scholar] [CrossRef]
- Ács, P.; Müller, E.; Rangits, G.; Lóránd, T.; Kollár, L. Palladium-catalysed carbonylation of 4-substituted 2-iodoaniline derivatives: carbonylative cyclisation and aminocarbonylation. Tetrahedron 2006, 62, 12051–12056. [Google Scholar] [CrossRef]
- Takács, A.; Farkas, R.; Kollár, L. High-yielding synthesis of 2-arylacrylamides via homogeneous catalytic aminocarbonylation of α-iodostyrene and α, α′-diiodo-1,4-divinylbenzene. Tetrahedron 2008, 64, 61–66. [Google Scholar] [CrossRef]
- Takács, A.; Ács, P.; Kollár, L. Facile synthesis of 1,8-naphthalimides in palladium-catalysed aminocarbonylation of 1,8-diiodo-naphthalene. Tetrahedron 2008, 64, 983–987. [Google Scholar] [CrossRef]
- Takács, A.; Ács, P.; Farkas, R.; Kokotos, G.; Kollár, L. Homogeneous catalytic aminocarbonylation of 1-iodo-1-dodecene. The facile synthesis of odd-number carboxamides via palladium-catalysed aminocarbonylation. Tetrahedron 2008, 64, 9874–9878. [Google Scholar] [CrossRef]
- Takács, A.; Farkas, R.; Petz, A.; Kollár, L. Synthesis of 2-naphthylacrylamides and 2-naphthylacrylates via homogeneous catalytic carbonylation of 1-iodo-1-naphthylethene derivatives. Tetrahedron 2009, 65, 4795–4800. [Google Scholar] [CrossRef]
- Takács, A.; Petz, A.; Kollár, L. High-yielding synthesis of Weinreb amides via homogeneous catalytic carbonylation of iodoalkenes and iodoarenes. Tetrahedron 2010, 66, 4479–4483. [Google Scholar] [CrossRef]
- Marosvölgyi-Haskó, D.; Takács, A.; Riedl, Z.; Kollár, L. High-yielding synthesis of 1-isoindolinone derivatives via palladium-catalysed cycloaminocarbonylation. Tetrahedron 2011, 67, 1036–1040. [Google Scholar] [CrossRef]
- Takács, A.; Szilágyi, A.; Ács, P.; László, M.; Peixoto, A.F.; Pereira, M.M.; Kollár, L. Palladium-catalysed reactions of 8-hydroxy- and 8-benzyloxy-5,7-diiodoquinoline under aminocarbonylation conditions. Tetrahedron 2011, 67, 2402–2406. [Google Scholar] [CrossRef]
- Marosvölgyi-Haskó, D.; Petz, A.; Takács, A.; Kollár, L. Synthesis of tetrahydrophthalazine and phthalamide (phthalimide) derivatives via palladium-catalysed carbonylation of iodoarenes. Tetrahedron 2011, 67, 9122–9128. [Google Scholar] [CrossRef]
- Takács, A.; Czompa, A.; Krajsovszky, G.; Mátyus, P.; Kollár, L. Functionalization of the pyridazin-3(2H)-one ring via palladium-catalysed aminocarbonylation. Tetrahedron 2012, 68, 7855–7860. [Google Scholar] [CrossRef][Green Version]
- Farkas, R.; Molnár, E.A.; Ács, P.; Takács, A.; Kollár, L. High-yielding synthesis of 1-carboxamido-3,4-dihydronaphthalenes via palladium-catalyzed aminocarbonylation. Tetrahedron 2013, 69, 500–504. [Google Scholar] [CrossRef]
- Gergely, M.; Farkas, R.; Takács, A.; Petz, A.; Kollár, L. Synthesis of N-picolylcarboxamides via palladium-catalysed aminocarbonylation of iodobenzene and iodoalkenes. Tetrahedron 2014, 70, 218–224. [Google Scholar] [CrossRef]
- Szőke, G.; Takács, A.; Berente, Z.; Petz, A.; Kollár, L. Synthesis of amino-substituted pyridylglyoxylamides via palladium-catalysed aminocarbonylation. Tetrahedron 2016, 72, 3063–3067. [Google Scholar] [CrossRef]
- Mikle, G.; Bede, F.; Kollár, L. Synthesis of N-picolylcarboxamides in aminocarbonylation. Tetrahedron 2021, 88, 132128. [Google Scholar] [CrossRef]
- Takács, A.; Varga, G.M.; Kardos, J.; Kollár, L. Palladium-catalysed aminocarbonylation of diiodopyridines. Tetrahedron 2017, 73, 2131–2138. [Google Scholar] [CrossRef]
- Kollár, L.; Varga, M.G.; Dörnyei, Á.; Takács, A. Functionalisation of the uracil ring via palladium-catalysed aminocarbonylation. Tetrahedron 2019, 75, 4632–4639. [Google Scholar] [CrossRef]
- Kollár, L.; Takács, A. Novel synthesis of 3-carboxamidolactam derivatives via palladium-catalysed aminocarbonylation. Tetrahedron 2018, 74, 6116–6128. [Google Scholar] [CrossRef]
- Skoda-Földes, R.; Szarka, Z.; Kollár, L.; Dinya, Z.; Horváth, J.; Tuba, Z. Synthesis of N-substituted steroidal hydrazides in homogeneous catalytic hydrazinocarbonylation reaction. J. Org. Chem. 1999, 64, 2134–2136. [Google Scholar] [CrossRef]
- Kollár, L.; Szarka, Z.; Horváth, J.; Tuba, Z. Facile, high-yielding synthesis of steroidal hydrazides via homogeneous hydrazinocarbonylation reaction. Tetrahedron Lett. 1997, 38, 4467–4468. [Google Scholar] [CrossRef]
- Skoda-Földes, R.; Horváth, J.; Tuba, Z.; Kollár, L. Homogeneous coupling and carbonylation reactions of steroids possessing iodoalkene moieties. Catalytic and mechanistic aspects. J. Organomet. Chem. 1999, 586, 94–100. [Google Scholar] [CrossRef]
- Skoda-Földes, R.; Székvölgyi, Z.; Kollár, L.; Berente, Z.; Horváth, J.; Tuba, Z. Facile synthesis of steroidal phenyl ketones via homogeneous catalytic carbonylation. Tetrahedron 2000, 56, 3415–3418. [Google Scholar] [CrossRef]
- Szarka, Z.; Skoda-Földes, R.; Kollar, L.; Horváth, J.; Tuba, Z. Novel method for the high-yielding synthesis of steroidal hydroxamic acid derivatives. Synth. Commun. 2000, 30, 1945–1953. [Google Scholar] [CrossRef]
- Szarka, Z.; Skoda-Földes, R.; Horváth, J.; Tuba, Z.; Kollár, L. Synthesis of steroidal diacyl hydrazines and their 1,3,4-oxadiazole derivatives. Steroids 2002, 67, 581–586. [Google Scholar] [CrossRef]
- Szarka, Z.; Skoda-Földes, R.; Kollár, L.; Berente, Z.; Horváth, J.; Tuba, Z. Highly efficient synthesis of steroidal hydroxamic acid derivatives via homogeneous catalytic carbonylation reaction. Tetrahedron 2000, 56, 5253–5257. [Google Scholar] [CrossRef]
- Ács, P.; Takács, A.; Szilágyi, A.; Wölfling, J.; Schneider, G.; Kollár, L. The synthesis of 17-alkoxycarbonyl-and 17-carboxamido-13α-estra-1,3,5(10),16-tetraene derivatives via palladium-catalyzed carbonylation reactions. Steroids 2008, 73, 669–675. [Google Scholar] [CrossRef]
- Ács, P.; Takács, A.; Szilágyi, A.; Wölfling, J.; Schneider, G.; Kollár, L. The synthesis of 13α-androsta-5,16-diene derivatives with carboxylic acid, ester and carboxamido functionalities at position-17 via palladium-catalyzed carbonylation. Steroids 2009, 74, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Takács, A.; Ács, P.; Berente, Z.; Wölfling, J.; Schneider, G.; Kollár, L. Novel 13β- and 13α-D-homo steroids: 17a-carboxamido-D-homoestra-1,3,5(10),17-tetraene derivatives via palladium-catalyzed aminocarbonylations. Steroids 2010, 75, 1075–1081. [Google Scholar] [CrossRef]
- Kiss, M.; Pálinkás, N.; Takács, A.; Mahó, S.; Kollár, L. A systematic approach to the synthesis of androstane-based 3,17-dicarboxamides (homo- and mixed dicarboxamides) via palladium-catalyzed aminocarbonylation. Steroids 2013, 78, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Petz, A.; Gálik, G.; Horváth, J.; Tuba, Z.; Berente, Z.; Pintér, Z.; Kollár, L. Facile, high-yielding synthesis of steroidal crown ethers via palladium-catalyzed carbonylation reaction. Synth. Commun. 2001, 31, 335–341. [Google Scholar] [CrossRef]
- Ács, P.; Müller, E.; Czira, G.; Mahó, S.; Perreira, M.; Kollár, L. Facile synthesis of 12-carboxamido-11-spirostenes via palladium-catalyzed carbonylation reactions. Steroids 2006, 71, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Ács, P.; Jakab, B.; Takács, A.; Kollár, L. Facile synthesis of 11-carboxamido-androst-4, 9(11)-dienes via palladium-catalyzed aminocarbonylation. Steroids 2007, 72, 627–632. [Google Scholar] [CrossRef]
- Mikle, G.; Zugó, A.; Szatnik, E.; Maxim, A.; Mahó, S.; Kollár, L. Synthesis of novel pregnane-based 20-carboxamides via palladium-catalysed aminocarbonylation. Chem. Pap. 2021, 75, 1861–1867. [Google Scholar] [CrossRef]
- Petz, A.; Horváth, J.; Tuba, Z.; Pintér, Z.; Kollár, L. Facile synthesis of 17-formyl steroids via palladium-catalyzed homogeneous carbonylation reaction. Steroids 2002, 67, 777–781. [Google Scholar] [CrossRef]
- Kuik, Á.; Skoda-Földes, R.; Balogh, J.; Kollár, L. Synthesis of ferrocenoyl amino acid derivatives via homogeneous catalytic aminocarbonylation. J. Organomet. Chem. 2005, 690, 3237–3242. [Google Scholar] [CrossRef]
- Szarka, Z.; Kuik, Á.; Skoda-Földes, R.; Kollár, L. Aminocarbonylation of 1,1′-diiodoferrocene, two-step synthesis of heterodisubstituted ferrocene derivatives via homogeneous catalytic carbonylation/coupling reactions. J. Organomet. Chem. 2004, 689, 2770–2775. [Google Scholar] [CrossRef]
- Kuik, Á.; Skoda-Földes, R.; Bényei, A.C.; Rangits, G.; Kollár, L. Formation of intramolecular hydrogen bonds in heterodisubstituted ferrocene diamides with a secondary and a tertiary amido group: X-ray structure of 1′-(N′-butyl-carbamoyl)-morpholino ferrocenecarboxamide. J. Organomet. Chem. 2006, 691, 3037–3042. [Google Scholar] [CrossRef]
- Szarka, Z.; Skoda-Földes, R.; Kuik, Á.; Berente, Z.; Kollár, L. Synthesis of ferrocene amides and α-ketoamides via palladium-catalyzed homogeneous carbonylation reaction. Synthesis 2003, 2003, 0545–0550. [Google Scholar]
- Szarka, Z.; Skoda-Földes, R.; Kollár, L. Facile synthesis of novel ferrocene α-ketoamides via homogeneous catalytic carbonylation. Tetrahedron Lett. 2001, 42, 739–741. [Google Scholar] [CrossRef]
- Kuik, Á.; Skoda-Földes, R.; Jánosi, L.; Kollar, L. Facile Synthesis of Unsymmetrical 1, n′-Disubstituted Ferrocenoyl Amino Acid Derivatives by Palladium-Catalyzed Aminocarbonylation. Synthesis 2007, 2007, 1456–1458. [Google Scholar]
- Csók, Z.; Kégl, T.; Li, Y.; Skoda-Földes, R.; Kiss, L.; Kunsági-Máté, S.; Todd, M.H.; Kollár, L. Synthesis of elongated cavitands via click reactions and their use as chemosensors. Tetrahedron 2013, 69, 8186–8190. [Google Scholar] [CrossRef]
- Filotás, D.; Nagy, L.; Kégl, T.R.; Csók, Z.; Kollár, L.; Nagy, G. Synthesis and Electrochemical Properties of the Tetraferrocenyl-Cavitand in Dimethyl Formamide Solvent Using Platinum and Carbon Working Electrodes. Electroanalysis 2015, 27, 799–807. [Google Scholar] [CrossRef]
- Janosi, T.Z.; Makkai, G.; Kegl, T.; Matyus, P.; Kollar, L.; Erostyak, J. Light-enhanced fluorescence of multi-level cavitands possessing pyridazine upper rim. J. Fluoresc. 2016, 26, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Kégl, T.; Csekő, G.; Mikle, G.; Takátsy, A.; Kollár, L.; Kégl, T. The Role of Weak Interactions in Supramolecular Compounds: A Synthetic and Theoretical Study of Novel Elongated Cavitands. ChemistrySelect 2017, 2, 8337–8345. [Google Scholar] [CrossRef]
- Csók, Z.; Takátsy, A.; Kollár, L. Highly selective palladium-catalyzed aminocarbonylation and cross-coupling reactions on a cavitand scaffold. Tetrahedron 2012, 68, 2657–2661. [Google Scholar] [CrossRef]
- Nagymihály, Z.; Kollár, L. High-yielding synthesis of deepened cavitands bearing picolyl moieties on the upper rim. Tetrahedron 2015, 71, 2555–2560. [Google Scholar] [CrossRef]
- Nagymihály, Z.; Wölfling, J.; Schneider, G. Synthesis of 2-Methylresorcinol-Based Deepened Cavitands with Chiral Inlet Bearing Steroidal Moieties on the Upper Rim. ChemistrySelect 2020, 5, 6933–6938. [Google Scholar] [CrossRef]
- Kégl, T.R.; Kégl, T. Palladium-catalyzed carbonylative synthesis and theoretical study of elongated tubular cavitands. J. Organomet. Chem. 2020, 923, 121387. [Google Scholar] [CrossRef]
- Nagymihály, Z.; Csók, Z.; Kollár, L. Influence of base additives on the selectivity of palladium-catalysed aminocarbonylation: Highly selective functionalization of a cavitand scaffold. Mol. Catal. 2018, 444, 70–75. [Google Scholar] [CrossRef]
- Reichardt, C. Solvents and Solvent Effects: An Introduction. Org. Process Res. Dev. 2006, 11, 105–113. [Google Scholar] [CrossRef]
- Horváth, I.T. Solvents from nature. Green Chem. 2008, 10, 1024. [Google Scholar] [CrossRef]
- Horváth, I.T.; Mehdi, H.; Fábos, V.; Boda, L.; Mika, L.T. Γ-Valerolactone—A sustainable liquid for energy and carbon-based chemicals. Green Chem. 2008, 10, 238–242. [Google Scholar] [CrossRef]
- Mika, L.T.; Cséfalvay, E.; Németh, Á. Catalytic Conversion of Carbohydrates to Initial Platform Chemicals: Chemistry and Sustainability. Chem. Rev. 2018, 118, 505–613. [Google Scholar] [CrossRef]
- Tukacs, J.M.; Novák, M.; Dibó, G.; Mika, L.T. An improved catalytic system for the reduction of levulinic acid to γ-valerolactone. Catal. Sci. Technol. 2014, 4, 2908–2912. [Google Scholar] [CrossRef]
- Pongrácz, P.; Bartal, B.; Kollár, L.; Mika, L.T. Rhodium-catalyzed hydroformylation in γ-valerolactone as a biomass-derived solvent. J. Organomet. Chem. 2017, 847, 140–145. [Google Scholar] [CrossRef]
- Pongrácz, P.; Kollár, L.; Mika, L.T. A step towards hydroformylation under sustainable conditions: platinum-catalysed enantioselective hydroformylation of styrene in gamma-valerolactone. Green Chem. 2016, 18, 842–847. [Google Scholar] [CrossRef]
- Marosvölgyi-Haskó, D.; Lengyel, B.; Tukacs, J.M.; Kollár, L.; Mika, L.T. Application of γ-Valerolactone as an Alternative Biomass-Based Medium for Aminocarbonylation Reactions. ChemPlusChem 2016, 81, 1224–1229. [Google Scholar] [CrossRef]
- Tukacs, J.M.; Marton, B.; Albert, E.; Tóth, I.; Mika, L.T. Palladium-catalyzed aryloxy- and alkoxycarbonylation of aromatic iodides in γ-valerolactone as bio-based solvent. J. Organomet. Chem. 2020, 923, 121407. [Google Scholar] [CrossRef]
- Skoda-Földes, R.; Takács, E.; Horváth, J.; Tuba, Z.; Kollár, L. Palladium-catalysed aminocarbonylation of steroidal 17-iodo-androst-16-ene derivatives in N,N′-dialkyl-imidazolium-type ionic liquids. Green Chem. 2003, 5, 643–645. [Google Scholar] [CrossRef]
- Müller, E.; Péczely, G.; Skoda-Földes, R.; Takács, E.; Kokotos, G.; Bellis, E.; Kollár, L. Homogeneous catalytic aminocarbonylation of iodoalkenes and iodobenzene with amino acid esters under conventional conditions and in ionic liquids. Tetrahedron 2005, 61, 797–802. [Google Scholar] [CrossRef]
- Takács, E.; Berente, Z.; Háda, V.; Mahó, S.; Kollár, L.; Skoda-Földes, R. Synthesis of new steroidal derivatives by the reaction of steroid–amino acid conjugates with N,N′-dicyclohexyl-carbodiimide. Unusual formation of steroidal imide derivatives. Tetrahedron 2009, 65, 4659–4663. [Google Scholar] [CrossRef]
- Papp, M.; Szabó, P.; Srankó, D.; Sáfrán, G.; Kollár, L.; Skoda-Földes, R. Mono- and double carbonylation of aryl iodides with amine nucleophiles in the presence of recyclable palladium catalysts immobilised on a supported dicationic ionic liquid phase. RSC Adv. 2017, 7, 44587–44597. [Google Scholar] [CrossRef]
- Urbán, B.; Nagy, E.; Nagy, P.; Papp, M.; Skoda-Földes, R. Double carbonylation of iodoarenes in the presence of a pyridinium SILP-Pd catalyst. J. Organomet. Chem. 2020, 918, 121287. [Google Scholar] [CrossRef]
- Urbán, B.; Skoda-Földes, R. Development of palladium catalysts immobilized on supported phosphonium ionic liquid phases. Phosphorus Sulfur Silicon Relat. Elem. 2018, 194, 302–306. [Google Scholar] [CrossRef]
- Adamcsik, B.; Nagy, E.; Urbán, B.; Szabó, P.; Pekker, P.; Skoda-Földes, R. Palladium nanoparticles on a pyridinium supported ionic liquid phase: A recyclable and low-leaching palladium catalyst for aminocarbonylation reactions. RSC Adv. 2020, 10, 23988–23998. [Google Scholar] [CrossRef]






















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kégl, T.R.; Mika, L.T.; Kégl, T. 27 Years of Catalytic Carbonylative Coupling Reactions in Hungary (1994–2021). Molecules 2022, 27, 460. https://doi.org/10.3390/molecules27020460
Kégl TR, Mika LT, Kégl T. 27 Years of Catalytic Carbonylative Coupling Reactions in Hungary (1994–2021). Molecules. 2022; 27(2):460. https://doi.org/10.3390/molecules27020460
Chicago/Turabian StyleKégl, Tímea R., László T. Mika, and Tamás Kégl. 2022. "27 Years of Catalytic Carbonylative Coupling Reactions in Hungary (1994–2021)" Molecules 27, no. 2: 460. https://doi.org/10.3390/molecules27020460
APA StyleKégl, T. R., Mika, L. T., & Kégl, T. (2022). 27 Years of Catalytic Carbonylative Coupling Reactions in Hungary (1994–2021). Molecules, 27(2), 460. https://doi.org/10.3390/molecules27020460