
Sequence and Structure-Based Analyses of Human Ankyrin Repeats


Abstract
:1. Introduction
2. Results
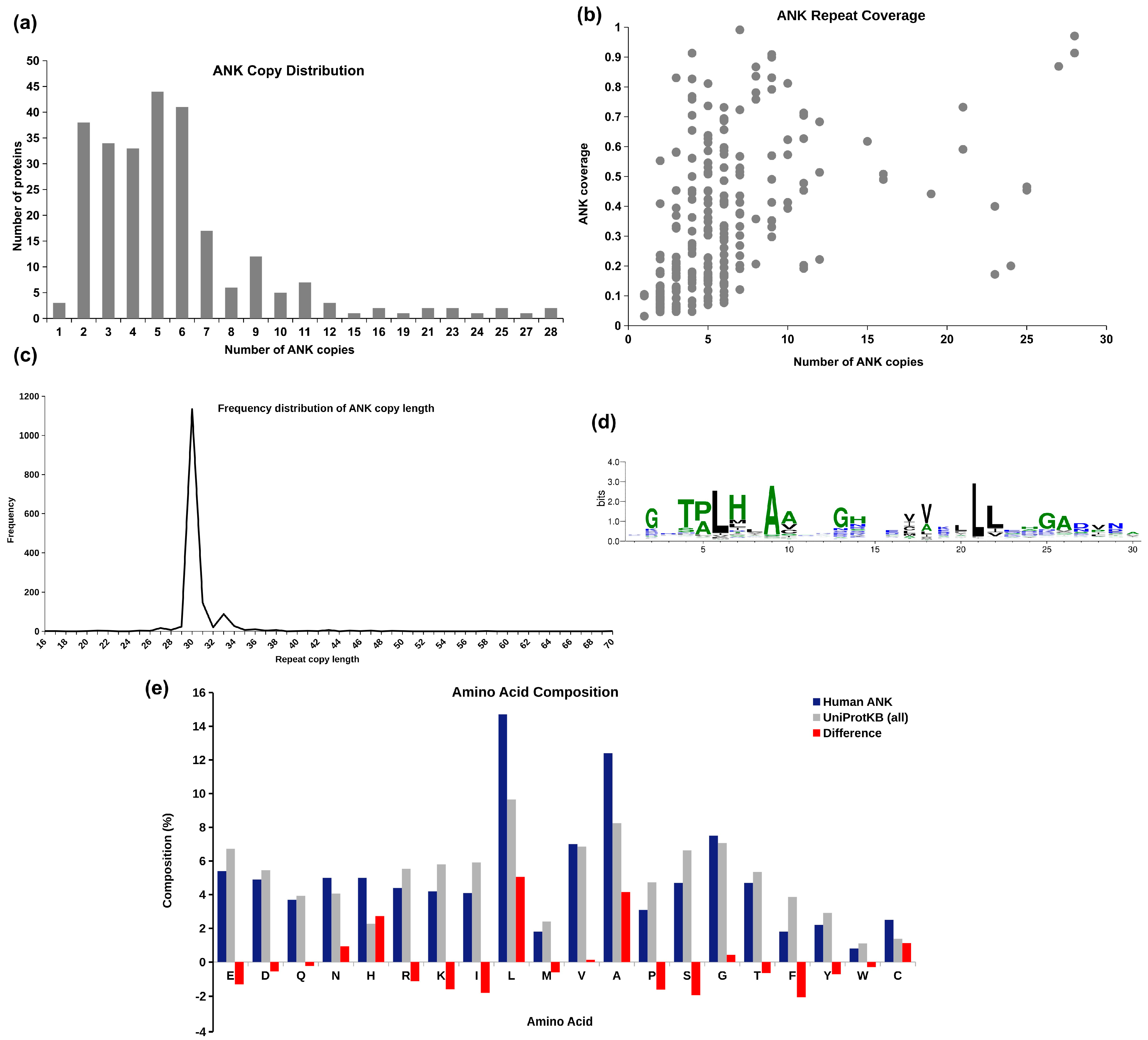
2.1. Human Ankyrins in UniProtKB
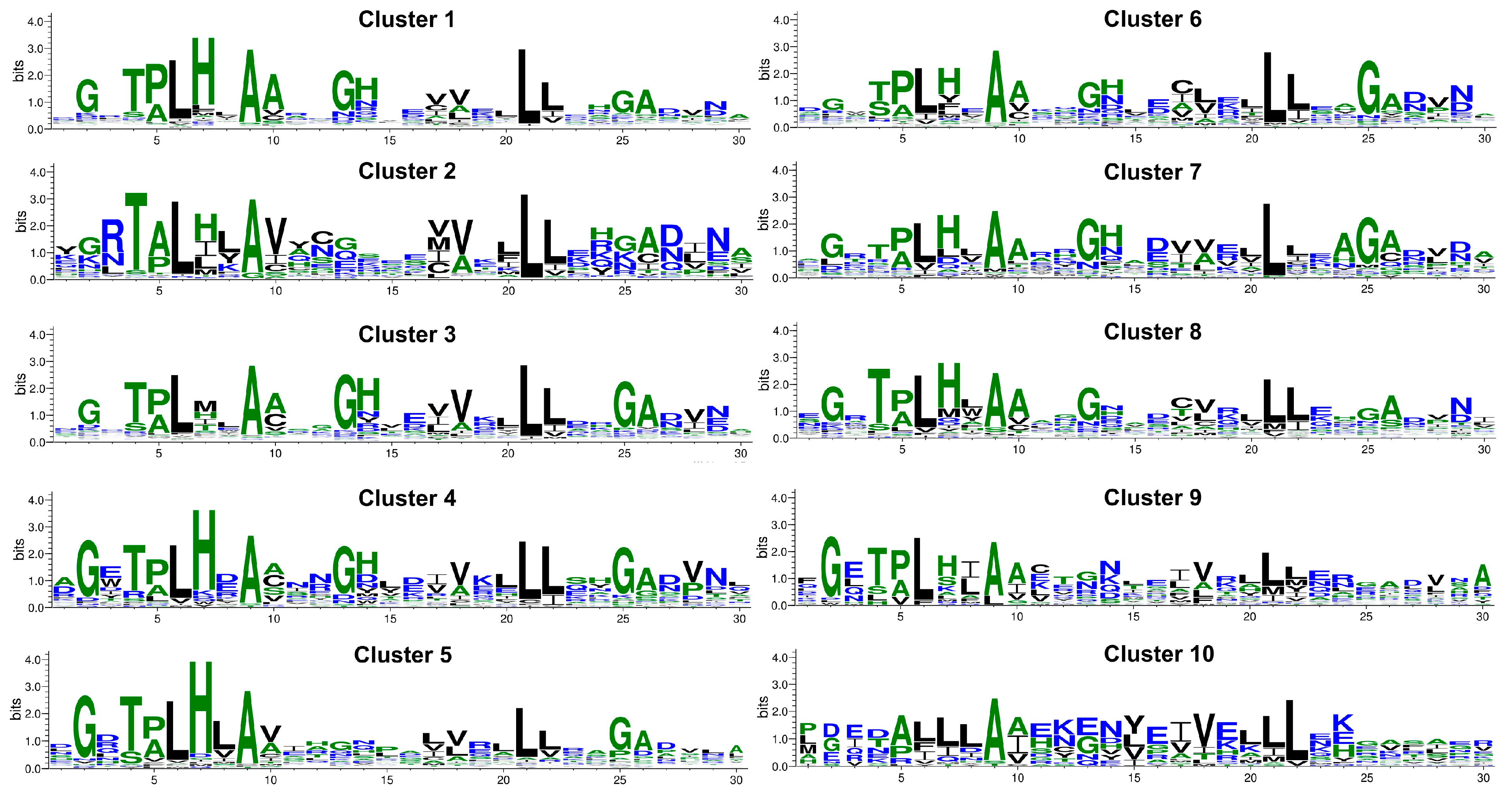
2.2. Sequence Clusters
2.3. Structure Analysis
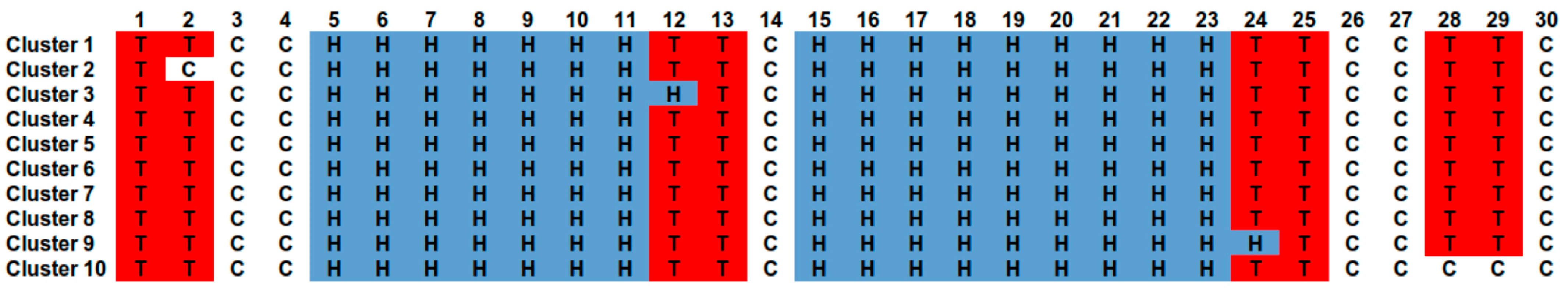
2.3.1. Secondary Structure Architecture
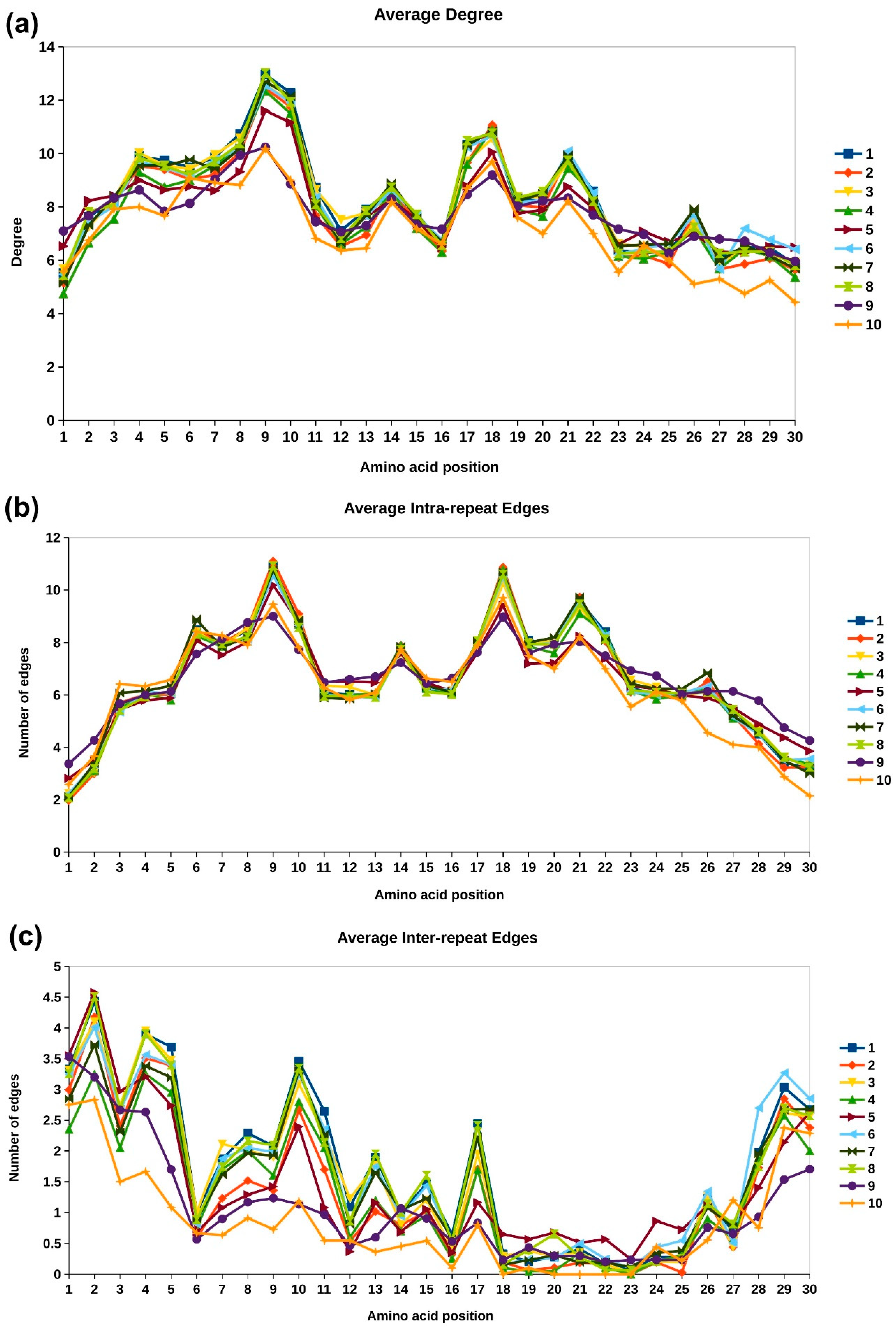
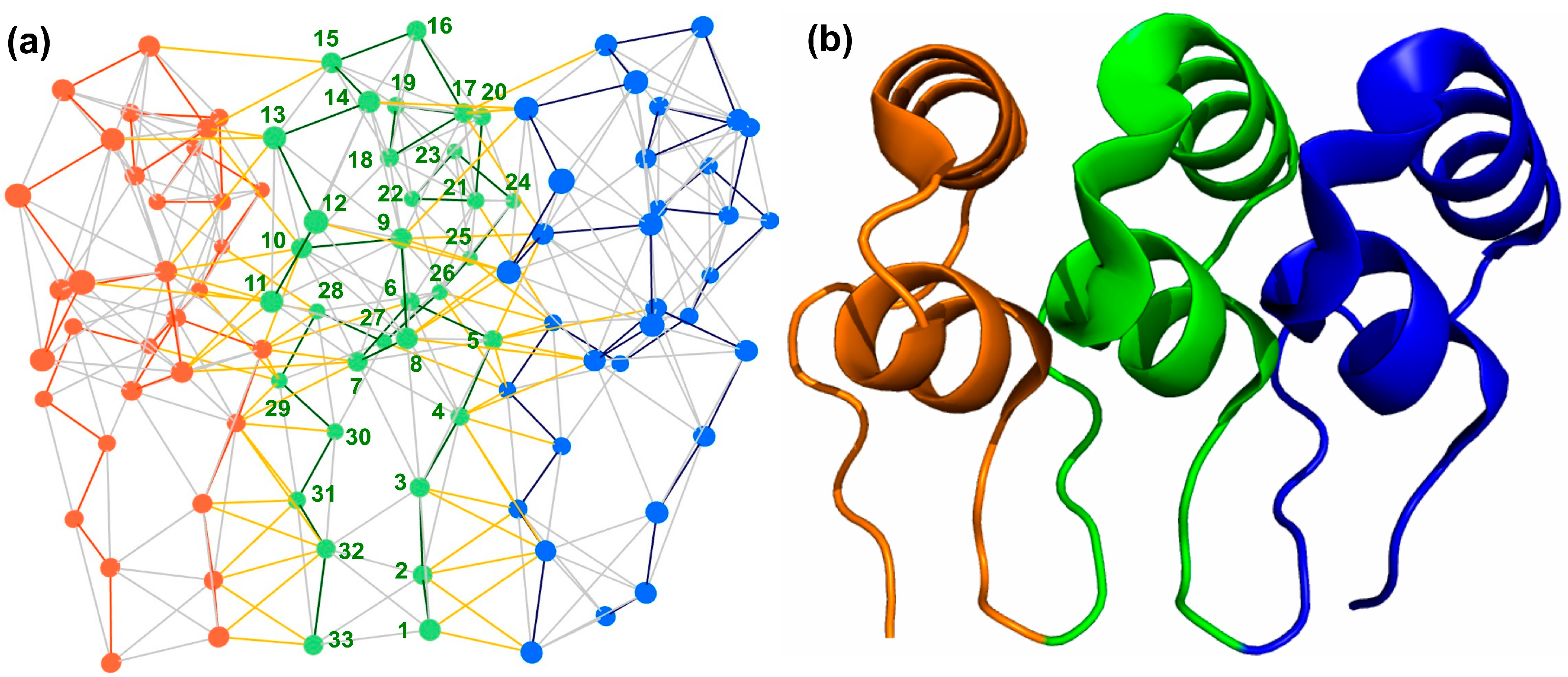
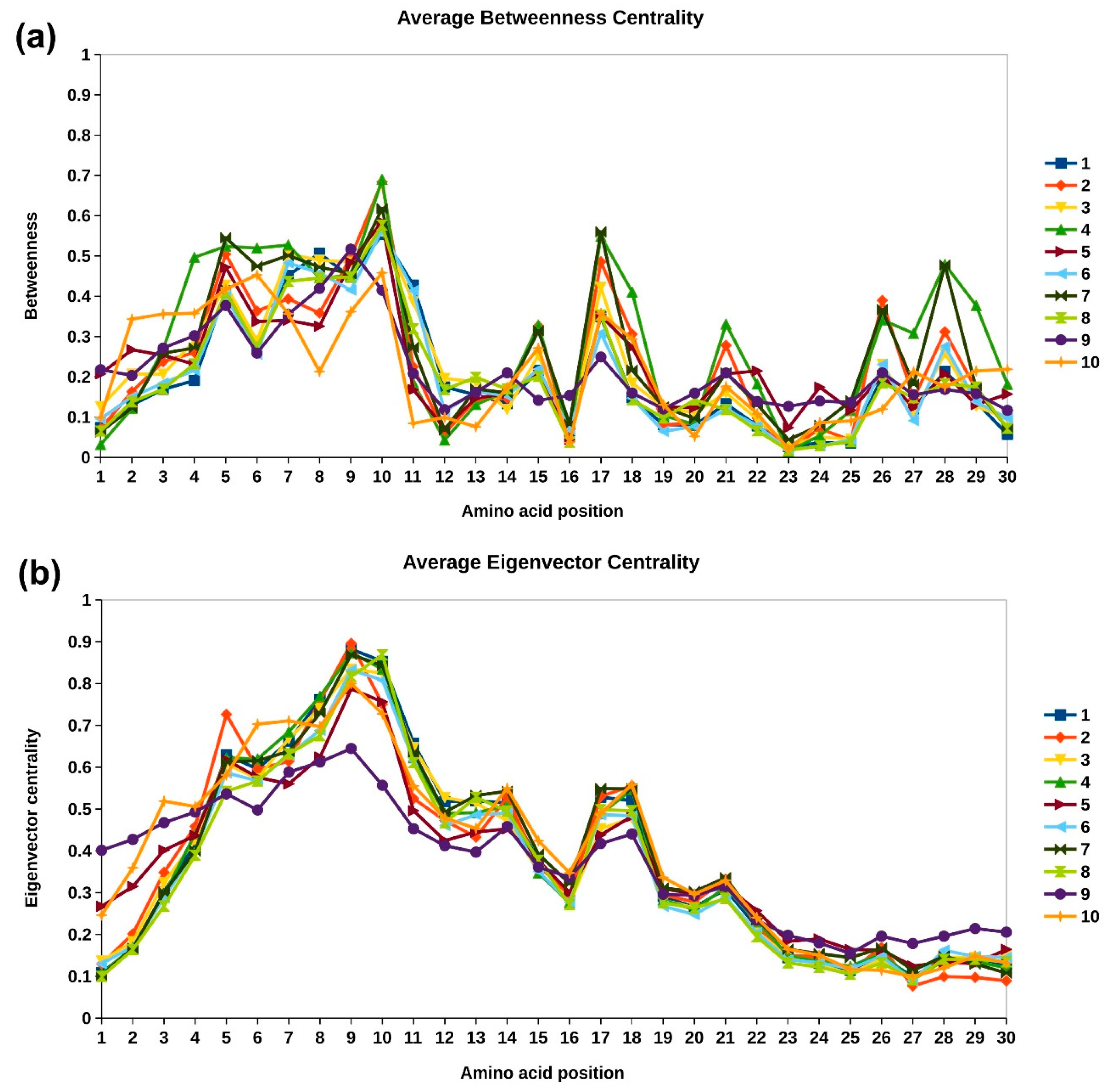
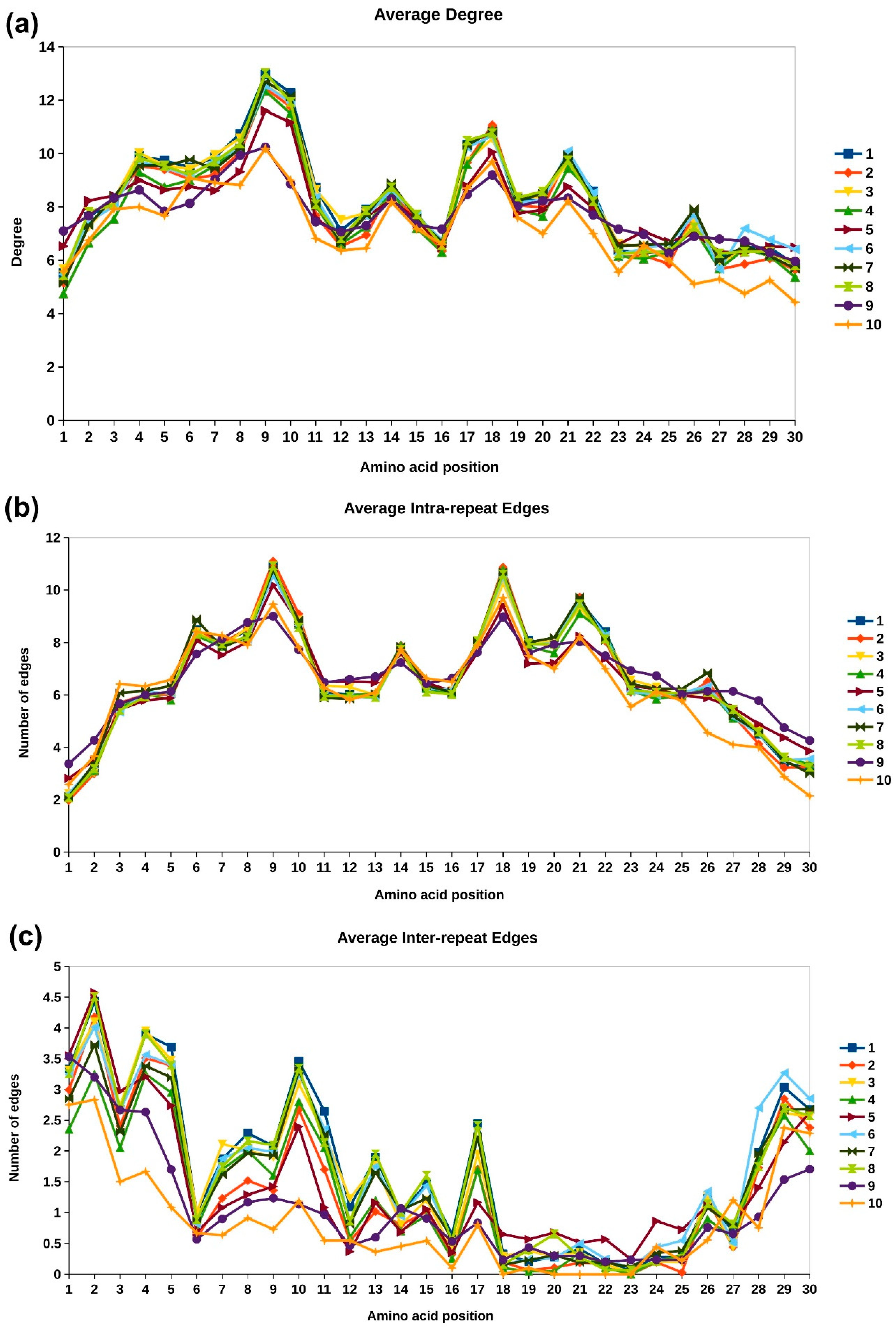
2.3.2. Network Analysis of Protein Structures
3. Discussion
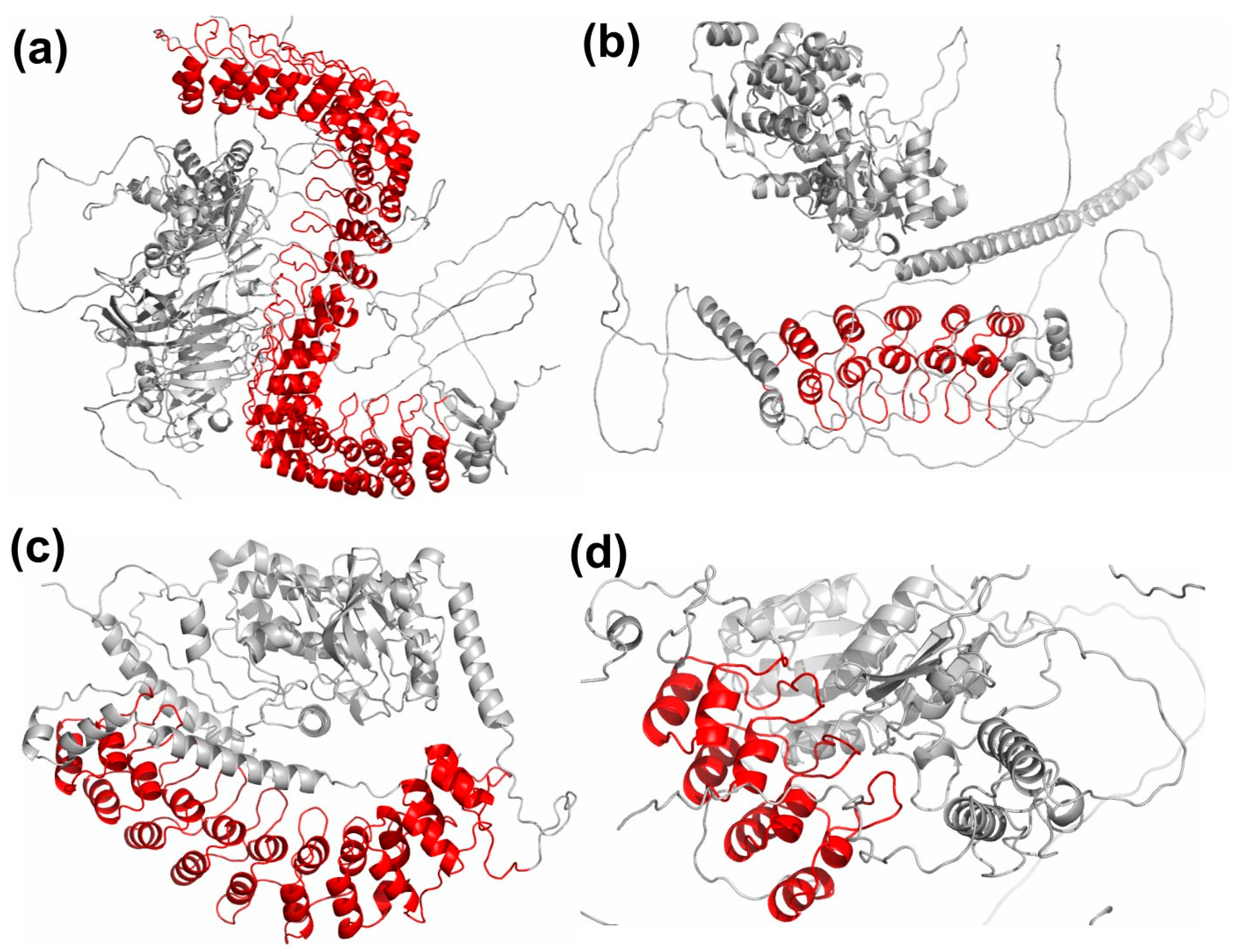
3.1. Variations in ANK Structure
3.2. Disease Associated Variations
3.2.1. Variations at Conserved TPLH Motif
3.2.2. Variations at Conserved Glycine Positions
3.2.3. Variations at Conserved Alanine Positions
3.2.4. Variations at Conserved Leucine Positions
4. Materials and Methods
4.1. Dataset Preparation
4.2. Sequence Analysis
4.2.1. Clustering
4.2.2. Sequence Alignment
4.3. Protein Contact Network
4.3.1. Network Construction
4.3.2. Betweenness Centrality
4.3.3. Eigenvector Centrality
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Bork, P. Hundreds of Ankyrin-like Repeats in Functionally Diverse Proteins: Mobile Modules That Cross Phyla Horizontally? Proteins 1993, 17, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mahajan, A.; Tsai, M.-D. Ankyrin Repeat: A Unique Motif Mediating Protein-Protein Interactions. Biochemistry 2006, 45, 15168–15178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, M.A.; Perez-Iratxeta, C.; Ponting, C.P. Protein Repeats: Structures, Functions, and Evolution. J. Struct. Biol. 2001, 134, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosavi, L.K.; Cammett, T.J.; Desrosiers, D.C.; Peng, Z.-Y. The Ankyrin Repeat as Molecular Architecture for Protein Recognition. Protein Sci. 2004, 13, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Utgés, J.S.; Tsenkov, M.I.; Dietrich, N.J.M.; MacGowan, S.A.; Barton, G.J. Ankyrin Repeats in Context with Human Population Variation. PLoS Comput. Biol. 2021, 17, e1009335. [Google Scholar] [CrossRef]
- Sharma, N.; Bham, K.; Senapati, S. Human Ankyrins and Their Contribution to Disease Biology: An Update. J. Biosci. 2020, 45, 146. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Choi, B.Y.; Lee, M.-H.; Bode, A.M.; Dong, Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (P16INK4a) in Cancer. EBioMedicine 2016, 8, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Baron, M.; Trump, D. An Overview of Notch3 Function in Vascular Smooth Muscle Cells. Prog. Biophys. Mol. Biol. 2008, 96, 499–509. [Google Scholar] [CrossRef]
- Sedgwick, S.G.; Smerdon, S.J. The Ankyrin Repeat: A Diversity of Interactions on a Common Structural Framework. Trends Biochem. Sci. 1999, 24, 311–316. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, A.; Binz, H.K.; Forrer, P.; Stumpp, M.T.; Plückthun, A.; Grütter, M.G. Designed to Be Stable: Crystal Structure of a Consensus Ankyrin Repeat Protein. Proc. Natl. Acad. Sci. USA 2003, 100, 1700–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosavi, L.K.; Minor, D.L.; Peng, Z.-Y. Consensus-Derived Structural Determinants of the Ankyrin Repeat Motif. Proc. Natl. Acad. Sci. USA 2002, 99, 16029–16034. [Google Scholar] [CrossRef] [Green Version]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, 439–444. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Chakrabarty, B.; Parekh, N. NAPS: Network Analysis of Protein Structures. Nucleic Acids Res. 2016, 44, 375–382. [Google Scholar] [CrossRef] [Green Version]
- Greene, L.H. Protein Structure Networks. Brief. Funct. Genom. 2012, 11, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, L.; De Ruvo, M.; Paci, P.; Santoni, D.; Giuliani, A. Protein Contact Networks: An Emerging Paradigm in Chemistry. Chem. Rev. 2013, 113, 1598–1613. [Google Scholar] [CrossRef]
- Amitai, G.; Shemesh, A.; Sitbon, E.; Shklar, M.; Netanely, D.; Venger, I.; Pietrokovski, S. Network Analysis of Protein Structures Identifies Functional Residues. J. Mol. Biol. 2004, 344, 1135–1146. [Google Scholar] [CrossRef]
- Brinda, K.V.; Vishveshwara, S. A Network Representation of Protein Structures: Implications for Protein Stability. Biophys. J. 2005, 89, 4159–4170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böde, C.; Kovács, I.A.; Szalay, M.S.; Palotai, R.; Korcsmáros, T.; Csermely, P. Network Analysis of Protein Dynamics. FEBS Lett. 2007, 581, 2776–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blacklock, K.; Verkhivker, G.M. Computational Modeling of Allosteric Regulation in the Hsp90 Chaperones: A Statistical Ensemble Analysis of Protein Structure Networks and Allosteric Communications. PLoS Comput. Biol. 2014, 10, e1003679. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, B.; Das, D.; Bung, N.; Roy, A.; Bulusu, G. Network Analysis of Hydroxymethylbilane Synthase Dynamics. J. Mol. Graph. Model. 2020, 99, 107641. [Google Scholar] [CrossRef]
- Chakrabarty, B.; Parekh, N. PRIGSA: Protein Repeat Identification by Graph Spectral Analysis. J. Bioinform. Comput. Biol. 2014, 12, 1442009. [Google Scholar] [CrossRef]
- Freeman, L.C. A Set of Measures of Centrality Based on Betweenness. Sociometry 1977, 40, 35–41. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Dokholyan, N.V.; Paci, E.; Karplus, M. Small-World View of the Amino Acids That Play a Key Role in Protein Folding. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2002, 65, 061910. [Google Scholar] [CrossRef] [Green Version]
- Kannan, N.; Vishveshwara, S. Identification of Side-Chain Clusters in Protein Structures by a Graph Spectral Method. J. Mol. Biol. 1999, 292, 441–464. [Google Scholar] [CrossRef]
- Vishveshwara, S.; Brinda, K.V.; Kannan, N. Protein Structure: Insights from Graph Theory. J. Theor. Comput. Chem. 2002, 1, 187–211. [Google Scholar] [CrossRef]
- Chakrabarty, B.; Parekh, N. Identifying Tandem Ankyrin Repeats in Protein Structures. BMC Bioinform. 2014, 15, 6599. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarty, B.; Parekh, N. PRIGSA2: Improved Version of Protein Repeat Identification by Graph Spectral Analysis. J. Biosci. 2020, 45, 95. [Google Scholar] [CrossRef]
- Chakrabarty, B.; Parekh, N. DbStRiPs: Database of Structural Repeats in Proteins. Protein Sci. 2021. [Google Scholar] [CrossRef]
- Kajava, A.V. Tandem Repeats in Proteins: From Sequence to Structure. J. Struct Biol. 2012, 179, 279–288. [Google Scholar] [CrossRef]
- Schilling, J.; Jost, C.; Ilie, I.M.; Schnabl, J.; Buechi, O.; Eapen, R.S.; Truffer, R.; Caflisch, A.; Forrer, P. Thermostable Designed Ankyrin Repeat Proteins (DARPins) as Building Blocks for Innovative Drugs. J. Biol. Chem. 2021, 298, 101403. [Google Scholar] [CrossRef]
- Guo, Y.; Yuan, C.; Tian, F.; Huang, K.; Weghorst, C.M.; Tsai, M.-D.; Li, J. Contributions of Conserved TPLH Tetrapeptides to the Conformational Stability of Ankyrin Repeat Proteins. J. Mol. Biol. 2010, 399, 168–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Nakanishi, M.; Kusakabe, Y.; Goto, Y.; Kitade, Y.; Nakamura, K.T. Structural Basis for Recognition of 2’,5’-Linked Oligoadenylates by Human Ribonuclease L. EMBO J. 2004, 23, 3929–3938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro Protein Families and Domains Database: 20 Years On. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to Accessing Data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster No. | No. of Proteins | Average Copy No. | Std. Dev. Copy No. | InterPro Domains |
|---|---|---|---|---|
| 1 | 34 | 10.9 | 8.1 | Ankyrin repeat-containing domain, ZU5 domain, Death domain, EGF-like domain, Notch domain, Sterile alpha motif domain |
| 2 | 32 | 5.2 | 0.8 | Ankyrn repeat-containing domain, CCDC144C-like coiled-coil domain |
| 3 | 13 | 10.7 | 6.8 | Ankyrin repeat-containing domain, K Homology domain, Sterile alpha motif domain, Protein kinase domain |
| 4 | 9 | 3.7 | 1 | Ankyrin repeat-containing domain, BRCT domain, BCL-6 corepressor, PCGF1 binding domain |
| 5 | 8 | 6.4 | 0.7 | Ankyrin repeat-containing domain, NFkappaB IPT domain, Rel homology domain (RHD), Death domain |
| 6 | 7 | 9.1 | 2.4 | Ankyrin repeat-containing domain, SOCS box domain |
| 7 | 6 | 6.0 | 2.4 | Ankyrin repeat-containing domain |
| 8 | 6 | 7.8 | 4.4 | Ankyrin repeat-containing domain |
| 9 | 6 | 5.0 | 1.5 | Ankyrin repeat-containing domain, Ion transport domain |
| 10 | 6 | 4.0 | 0 | Ankyrin repeat-containing domain, Transient receptor ion channel domain, Ion transport domain |
| Cluster No. | Consensus (50%) |
|---|---|
| 1 | XGXTPLHXAAXXGXXXXXXXLLXXGAXXXX |
| 2 | XXRTALXXAVXXXXXXXVXXLLXXXXXXXX |
| 3 | XGXTXLXXAXXXGHXXXVXXLLXXGAXXXX |
| 4 | XGXTPLHXAXXXGXXXXXXXLLXXGAXXXX |
| 5 | XGXTXLHLAXXXXXXXXXXXLXXXGXXXXX |
| 6 | XXXXPLXXAXXXGXXXXXXXLLXXGAXXXX |
| 7 | XGXXXLHXAAXXGXXXXXXXLXXAGXXXXX |
| 8 | XGXTXLHXAAXXGXXXXXXXLLXXXAXXXX |
| 9 | XGXTXLXXAXXXXXXXXXXXLXXXXXXXXX |
| 10 | XXXXAXLLAXXXXXXXXVXXLLXXXXXXXX |
| Residue Position | Gene Name | Copy No. | Cluster No. | Variation | Condition(s) |
|---|---|---|---|---|---|
| Gly2 | ANKRD29 | 4 | 1 | G112E | G -> E (in dbSNP:rs17855552) |
| RNASEL | 2 | 3 | G59S | G -> S (in dbSNP:rs151296858) | |
| BARD1 | 1 | 4 | G428* | Hereditary cancer-predisposing syndrome | |
| CDKN2B | 2 | 7 | G47E | Lung adenocarcinoma | |
| Thr4 | ANKK1 | 8 | 1 | T595I | T -> I (in dbSNP:rs55787008) |
| Pro5 | ANKK1 | 8 | 1 | P596L | P -> L (in dbSNP:rs7104979) |
| ASB2 | 4 | 6 | P160S | P -> S (in dbSNP:rs2295213) | |
| CDKN2A | 4 | 7 | P114L | Non-small cell lung carcinoma | |
| CDKN2A | 4 | 7 | P114S | Melanoma; lossof CDK4 binding; dbSNP:rs104894104 | |
| CDKN2A | 2 | 7 | P48L | CMM2 $; also found in head and neck tumor | |
| CDKN2A | 2 | 7 | P48T | Hereditary melanoma|Hereditary cancer-predisposing syndrome | |
| CDKN2A | 3 | 7 | P81L | Melanoma; impairs the function; | |
| CDKN2A | 3 | 7 | P81T | CMM2; loss of CDK4 binding; | |
| Leu6 | ANK1 | 8 | 1 | L276R | Spherocytosis type 1 |
| ANK1 | 17 | 1 | L573fs | Spherocytosis type 1 | |
| ANKK1 | 1 | 1 | L366F | L -> F (in dbSNP:rs56339158) | |
| NFKBIA | 2 | 5 | 115..120 | LHLAVI->AHAAVA: Greatly reduced nuclearlocalization. Great reduction in its ability to inhibit DNA binding of RELA. | |
| CDKN2A | 1 | 7 | L16fs | Hereditary cancer-predisposing syndrome | |
| CDKN2A | 1 | 7 | L16fs | Squamous cell lung carcinoma; Hereditary melanoma; Hereditary cancer-predisposing syndrome | |
| CDKN2A | 1 | 7 | L16P | Biliary tract tumor; familial melanoma | |
| CDKN2A | 1 | 7 | L16P | Hereditary melanoma | |
| CDKN2A | 1 | 7 | L16R | Hereditary melanoma; Hereditary cancer-predisposing syndrome | |
| INVS | 15 | 8 | L493S | NPHP2; impairs ability to target DVL1 fordegradation | |
| His7 | ANKK1 | 1 | 1 | H367Q | H -> Q (in dbSNP:rs34298987) |
| CDKN2A | 3 | 7 | H83N | Lung tumor | |
| CDKN2A | 3 | 7 | H83Q | H -> Q (in dbSNP:rs34968276) | |
| CDKN2A | 3 | 7 | H83Y | Pancreas tumor; head andneck tumor | |
| Ala9 | CDKN2A | 1 | 7 | A19T | CMM2; loss of CDK4 binding |
| CDKN2A | 4 | 7 | A118T | CMM2 | |
| CDKN2A | 3 | 7 | A85T | A -> T (in dbSNP:rs878853646) | |
| Gly13 | BARD1 | 3 | 4 | G505fs | Hereditary cancer-predisposing syndrome; Familial cancer of breast |
| CDKN2A | 4 | 7 | G122R | CMM2 | |
| CDKN2A | 4 | 7 | G122S | Biliary tract tumor | |
| CDKN2A | 1 | 7 | G23D | Pancreas tumor; melanoma; loss of CDK4 binding | |
| CDKN2A | 3 | 7 | G89D | CMM2 | |
| CDKN2A | 3 | 7 | G89S | CMM2 | |
| TRPC6 | 1 | 10 | G109S | Focal segmental glomerulosclerosis 2 (FSGS2); increases calcium ion transport | |
| Leu21 | CDKN2A | 3 | 7 | L97R | CMM2; loss of CDK4 binding |
| Leu22 | CDKN2A | 1 | 7 | L32P | Hereditary cancer-predisposing syndrome; Hereditary melanoma |
| Gly25 | ANK2 | 20 | 1 | G685E | breast cancer |
| ANKK1 | 3 | 1 | G451R | G -> R (in dbSNP:rs34983219) | |
| ANKHD1 | 1 | 3 | G228C | G -> C (in dbSNP:rs17850572) | |
| CDKN2A | 3 | 7 | G101W | Hereditary melanoma; Cutaneous malignant melanoma 2; Melanoma-pancreatic cancer syndrome; Hereditary cancer-predisposing syndrome | |
| CDKN2A | 1 | 7 | G35A | CMM2; biliary tract tumor; uveal melanoma; partial loss of CDK4 binding | |
| CDKN2A | 1 | 7 | G35E | CMM2 | |
| CDKN2A | 1 | 7 | G35V | CMM2; loss of CDK4 binding | |
| Ala26 | ANKRD16 | 3 | 6 | A128G | A -> G (in dbSNP:rs2296136) |
| CDKN2A | 3 | 7 | A102E | Seminoma; medulloblastoma tissues from Li-Fraumeni syndrome patients carrying a mutation in TP53; | |
| CDKN2A | 3 | 7 | A102T | A -> T (in dbSNP:rs35741010) | |
| CDKN2A | 1 | 7 | A36fs | Hereditary melanoma; Hereditary cancer-predisposing syndrome |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakrabarty, B.; Parekh, N. Sequence and Structure-Based Analyses of Human Ankyrin Repeats. Molecules 2022, 27, 423. https://doi.org/10.3390/molecules27020423
Chakrabarty B, Parekh N. Sequence and Structure-Based Analyses of Human Ankyrin Repeats. Molecules. 2022; 27(2):423. https://doi.org/10.3390/molecules27020423
Chicago/Turabian StyleChakrabarty, Broto, and Nita Parekh. 2022. "Sequence and Structure-Based Analyses of Human Ankyrin Repeats" Molecules 27, no. 2: 423. https://doi.org/10.3390/molecules27020423
APA StyleChakrabarty, B., & Parekh, N. (2022). Sequence and Structure-Based Analyses of Human Ankyrin Repeats. Molecules, 27(2), 423. https://doi.org/10.3390/molecules27020423