
Fermentation Extract of Naringenin Increases the Expression of Estrogenic Receptor β and Modulates Genes Related to the p53 Signalling Pathway, miR-200c and miR-141 in Human Colon Cancer Cells Exposed to BPA

 , ,
, ,  , and
, and
Abstract
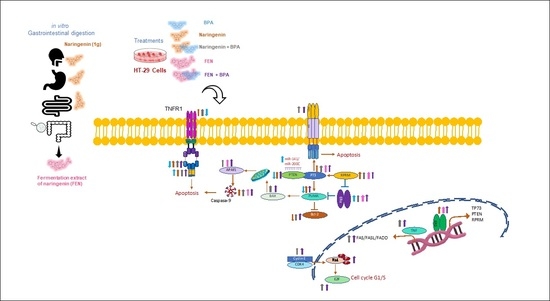
1. Introduction
2. Results
2.1. Products Generated by Microbial Metabolic Fermentation from Naringenin
Antioxidant Capacity of FEN and Naringenin
2.2. Naringenin and Its Fermented Extract Decrease Cell Viability in Co-Exposition with BPA
2.2.1. Naringenin and FEN Induce Apoptotic Cell Death and BPA Induces Necrosis in HT-29 Cells
2.2.2. Naringenin and FEN Treatments Increase SOD Activity and Decrease the Amount of GSH
2.2.3. Naringenin and FEN Increase the Expression of ERβ Activating Apoptotic Genes, whereas BPA Increases ERα Expression, Related to Survival
2.2.4. miR-200c Regulates PTEN Protein Expression
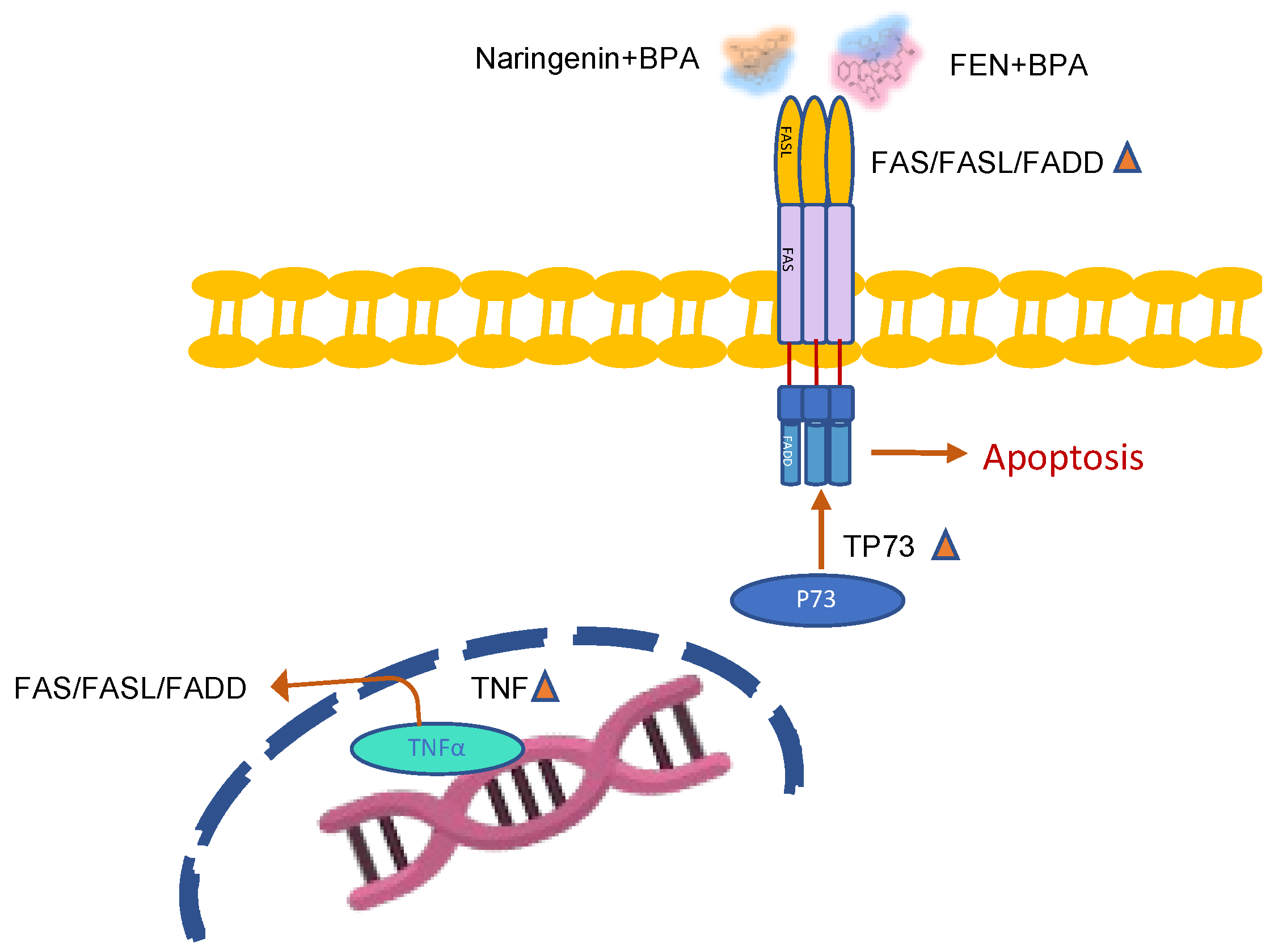
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. In Vitro Gastrointestinal Digestion and Fermentation of Naringenin
4.2.1. Identification of Metabolites
4.2.2. Antioxidant Capacity
4.3. Cell Culture
4.3.1. Treatments
4.3.2. Cell Culture and Viability Test
4.3.3. Detection of Apoptosis
4.3.4. Cellular Necrosis
4.3.5. Superoxide Dismutase (SOD) Activity
4.3.6. Quantification of Reduced Glutathione (GSH)
4.4. Gene Expression
4.5. miRNAs Expression
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Mattiuzzi, C.; Sanchis-Gomar, F.; Lippi, G. Concise Update on Colorectal Cancer Epidemiology. Ann. Transl. Med. 2019, 7, 609. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhu, Y.; Li, Q.; Wang, F.; Ge, X.; Zhou, G.; Miao, L. Association between Mediterranean Diet Adherence and Colorectal Cancer: A Dose-Response Meta-Analysis. Am. J. Clin. Nutr. 2020, 111, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Lei, L.; Zhou, Y.; Ye, F.; Zhao, G. Dietary Flavonoids and the Risk of Colorectal Cancer: An Updated Meta-Analysis of Epidemiological Studies. Nutrients 2018, 10, 950. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.U.; Rahman Mir, M.U.; Farooq, A.; Rashid, S.M.; Ahmad, B.; Bilal Ahmad, S.; Ali, R.; Hussain, I.; Masoodi, M.; Muzamil, S.; et al. Naringenin (4,5,7-Trihydroxyflavanone) Suppresses the Development of Precancerous Lesions via Controlling Hyperproliferation and Inflammation in the Colon of Wistar Rats. Environ. Toxicol. 2018, 33, 422–435. [Google Scholar] [CrossRef]
- Zhao, Y.; Fan, D.; Ru, B.; Cheng, K.-W.; Hu, S.; Zhang, J.; Li, E.T.S.; Wang, M. 6-C-(E-Phenylethenyl) Naringenin Induces Cell Growth Inhibition and Cytoprotective Autophagy in Colon Cancer Cells. Eur. J. Cancer 2016, 68, 38–50. [Google Scholar] [CrossRef]
- Song, H.M.; Park, G.H.; Eo, H.J.; Jeong, J.B. Naringenin-Mediated ATF3 Expression Contributes to Apoptosis in Human Colon Cancer. Biomol. Ther. 2016, 24, 140–146. [Google Scholar] [CrossRef]
- Li, H.; Zhu, F.; Chen, H.; Cheng, K.W.; Zykova, T.; Oi, N.; Lubet, R.A.; Bode, A.M.; Wang, M.; Dong, Z. 6-C-(E-Phenylethenyl)-Naringenin Suppresses Colorectal Cancer Growth by Inhibiting Cyclooxygenase-1. Cancer Res. 2014, 74, 243–252. [Google Scholar] [CrossRef]
- Sasso, C.V.; Santiano, F.E.; Campo Verde Arboccó, F.; Zyla, L.E.; Semino, S.N.; Guerrero-Gimenez, M.E.; Pistone Creydt, V.; López Fontana, C.M.; Carón, R.W. Estradiol and Progesterone Regulate Proliferation and Apoptosis in Colon Cancer. Endocr. Connect. 2019, 8, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Leiszter, K.; Galamb, O.; Kalmár, A.; Zsigrai, S.; Valcz, G.; Szigeti, K.A.; Barták, B.K.; Nagy, Z.B.; Dank, M.; Liposits, Z.; et al. Potential role of estrogens in colorectal tumour development. Orv. Hetil. 2020, 161, 532–543. [Google Scholar] [CrossRef]
- Luceri, C.; Femia, A.P.; Tortora, K.; D’Ambrosio, M.; Fabbri, S.; Fazi, M.; Caderni, G. Supplementation with Phytoestrogens and Insoluble Fibers Reduces Intestinal Carcinogenesis and Restores ER-β Expression in Apc-Driven Colorectal Carcinogenesis. Eur. J. Cancer Prev. 2020, 29, 27–35. [Google Scholar] [CrossRef]
- Bolli, A.; Bulzomi, P.; Galluzzo, P.; Acconcia, F.; Marino, M. Bisphenol A Impairs Estradiol-Induced Protective Effects against DLD-1 Colon Cancer Cell Growth. IUBMB Life 2010, 62, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Burns, K.A.; Arao, Y.; Luh, C.J.; Korach, K.S. Differential Estrogenic Actions of Endocrine-Disrupting Chemicals Bisphenol A, Bisphenol AF, and Zearalenone through Estrogen Receptor α and β in Vitro. Environ. Health Perspect. 2012, 120, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Puranik, N.V.; Srivastava, P.; Bhatt, G.; John Mary, D.J.S.; Limaye, A.M.; Sivaraman, J. Determination and Analysis of Agonist and Antagonist Potential of Naturally Occurring Flavonoids for Estrogen Receptor (ERα) by Various Parameters and Molecular Modelling Approach. Sci. Rep. 2019, 9, 7450. [Google Scholar] [CrossRef]
- Pang, X.; Fu, W.; Wang, J.; Kang, D.; Xu, L.; Zhao, Y.; Liu, A.-L.; Du, G.-H. Identification of Estrogen Receptor α Antagonists from Natural Products via In Vitro and In Silico Approaches. Oxid. Med. Cell. Longev. 2018, 2018, 6040149. [Google Scholar] [CrossRef]
- Feng, X.; Wang, Z.; Fillmore, R.; Xi, Y. MiR-200, a New Star MiRNA in Human Cancer. Cancer Lett. 2014, 344, 166–173. [Google Scholar] [CrossRef]
- Najmanová, I.; Vopršalová, M.; Saso, L.; Mladěnka, P. The Pharmacokinetics of Flavanones. Crit. Rev. Food Sci. Nutr. 2020, 60, 3155–3171. [Google Scholar] [CrossRef]
- Pereira-Caro, G.; Borges, G.; Ky, I.; Ribas, A.; Calani, L.; Del Rio, D.; Clifford, M.N.; Roberts, S.A.; Crozier, A. In Vitro Colonic Catabolism of Orange Juice (Poly)Phenols. Mol. Nutr. Food Res. 2015, 59, 465–475. [Google Scholar] [CrossRef]
- Márquez Campos, E.; Stehle, P.; Simon, M.-C. Microbial Metabolites of Flavan-3-Ols and Their Biological Activity. Nutrients 2019, 11, 2260. [Google Scholar] [CrossRef]
- Merlani, M.; Barbakadze, V.; Amiranashvili, L.; Gogilashvili, L.; Yannakopoulou, E.; Papadopoulos, K.; Chankvetadze, B. Enantioselective Synthesis and Antioxidant Activity of 3-(3,4-Dihydroxyphenyl)-Glyceric Acid--Basic Monomeric Moiety of a Biologically Active Polyether from Symphytum asperum and S. caucasicum. Chirality 2010, 22, 717–725. [Google Scholar]
- Kim, J.-S.; Lee, J.-H. Antioxidant and Anti-Inflammatory Activity of Phloroglucinol from Seaweeds. FASEB J. 2016, 30, 1174.12. [Google Scholar]
- Rechner, A.R.; Smith, M.A.; Kuhnle, G.; Gibson, G.R.; Debnam, E.S.; Srai, S.K.S.; Moore, K.P.; Rice-Evans, C.A. Colonic Metabolism of Dietary Polyphenols: Influence of Structure on Microbial Fermentation Products. Free Radic. Biol. Med. 2004, 36, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Su, W.; Zheng, Y.; He, Y.; He, Y.; Rao, H.; Peng, W.; Yao, H. Pharmacokinetics, Tissue Distribution, Metabolism, and Excretion of Naringin in Aged Rats. Front. Pharmacol 2019, 10, 34. [Google Scholar] [CrossRef]
- Bouchier-Hayes, L. The Role of Caspase-2 in Stress-Induced Apoptosis. J. Cell. Mol. Med. 2010, 14, 1212–1224. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.A.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef]
- Fusco, N.; Sajjadi, E.; Venetis, K.; Gaudioso, G.; Lopez, G.; Corti, C.; Guerini Rocco, E.; Criscitiello, C.; Malapelle, U.; Invernizzi, M. PTEN Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine? Genes 2020, 11, 719. [Google Scholar] [CrossRef]
- Martini, S.; Conte, A.; Tagliazucchi, D. Antiproliferative Activity and Cell Metabolism of Hydroxycinnamic Acids in Human Colon Adenocarcinoma Cell Lines. J. Agric. Food Chem. 2019, 10, 3919–3931. [Google Scholar] [CrossRef]
- Skroza, D.; Šimat, V.; Vrdoljak, L.; Jolić, N.; Skelin, A.; Čagalj, M.; Frleta, R.; Generalić-Mekinić, I. Investigation of Antioxidant Synergisms and Antagonisms among Phenolic Acids in the Model Matrices Using FRAP and ORAC Methods. Antioxidants 2022, 9, 1784. [Google Scholar] [CrossRef]
- Heim, K.E.; Tagliaferro, A.R.; Bobilya, D.J. Flavonoid antioxidants: Chemistry, metabolism and structure-activity relationships. J. Nutr. Biochem. 2002, 13, 572–584. [Google Scholar] [CrossRef]
- Kachadourian, R.; Day, B.J. Flavonoid-induced glutathione depletion: Potential implications for cancer treatment. Free Radic. Biol. Med. 2006, 41, 65–76. [Google Scholar] [CrossRef]
- Ghanbari-Movahed, M.; Jackson, G.; Farzaei, M.H.; Bishayee, A.A. Systematic Review of the Preventive and Therapeutic Effects of Naringin Against Human Malignancies. Front. Pharmacol. 2021, 12, 639840. [Google Scholar] [CrossRef]
- Frydoonfar, H.R.; McGrath, D.R.; Spigelman, A.D. The Variable Effect on Proliferation of a Colon Cancer Cell Line by the Citrus Fruit Flavonoid Naringenin. Colorectal Dis. 2003, 5, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Orrego-Lagarón, N.; Vallverdú-Queralt, A.; Martínez-Huélamo, M.; Lamuela-Raventos, R.M.; Escribano-Ferrer, E. Metabolic Profile of Naringenin in the Stomach and Colon Using Liquid Chromatography/Electrospray Ionization Linear Ion Trap Quadrupole-Orbitrap-Mass Spectrometry (LC-ESI-LTQ-Orbitrap-MS) and LC-ESI-MS/MS. J. Pharm. Biomed. Anal. 2016, 120, 38–45. [Google Scholar] [CrossRef]
- Kang, M.H.; Kim, I.H.; Nam, T.J.N. Phloroglucinol Induces Apoptosis via Apoptotic Signaling Pathways in HT-29 Colon Cancer Cells. Oncol. Rep. 2014, 32, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.K.; Ziegler, Y.S.; McLeod, I.X.; Yates, J.R.; Nardulli, A.M. Effects of Cu/Zn Superoxide Dismutase on Estrogen Responsiveness and Oxidative Stress in Human Breast Cancer Cells. Mol. Endocrinol. 2008, 22, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Griess, B.; Tom, E.; Domann, F.; Teoh-Fitzgerald, M. Extracellular Superoxide Dismutase and Its Role in Cancer. Free Radic. Biol. Med. 2017, 112, 464–479. [Google Scholar] [CrossRef]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as Anticancer Agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione Metabolism in Cancer Progression and Treatment Resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Chen, Z.-J.; Zhang, K.-S.; Ge, L.-C.; Liu, H.; Chen, L.-K.; Du, J.; Wang, H.-S. Signals Involved in the Effects of Bisphenol A (BPA) on Proliferation and Motility of Leydig Cells: A Comparative Proteomic Analysis. Toxicol. Res. 2016, 5, 1573–1584. [Google Scholar] [CrossRef]
- Kim, S.; Park, T.I. Naringenin: A Partial Agonist on Estrogen Receptor in T47D-KBluc Breast Cancer Cells. Int. J. Clin. Exp. Med. 2013, 6, 890–899. [Google Scholar] [PubMed]
- Totta, P.; Acconcia, F.; Leone, S.; Cardillo, I.; Marino, M. Mechanisms of Naringenin-Induced Apoptotic Cascade in Cancer Cells: Involvement of Estrogen Receptor Alpha and Beta Signalling. IUBMB Life 2004, 56, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Bulzomi, P.; Bolli, A.; Galluzzo, P.; Acconcia, F.; Ascenzi, P.; Marino, M. The Naringenin-Induced Proapoptotic Effect in Breast Cancer Cell Lines Holds out against a High Bisphenol a Background. IUBMB Life 2012, 64, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Kocyigit, A.; Koyuncu, I.; Dikilitas, M.; Bahadori, F.; Turkkan, B. Cytotoxic, Genotoxic and Apoptotic Effects of Naringenin-Oxime Relative to Naringenin on Normal and Cancer Cell Lines. Asian Pac. J. Trop. Biomed. 2016, 6, 872–880. [Google Scholar] [CrossRef]
- Gibellini, L.; Pinti, M.; Nasi, M.; De Biasi, S.; Roat, E.; Bertoncelli, L.; Cossarizza, A. Interfering with ROS Metabolism in Cancer Cells: The Potential Role of Quercetin. Cancers 2010, 2, 1288–1311. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Huang, C.; Wu, H.; Huang, J. Estrogen Receptor Beta (ERβ) Mediated-CyclinD1 Degradation via Autophagy Plays an Anti-Proliferation Role in Colon Cells. Int. J. Biol. Sci. 2019, 15, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, F.; Ryan, E.J.; Doherty, G.; Winter, D.C.; Sheahan, K. Estrogen Receptors and Their Implications in Colorectal Carcinogenesis. Front. Oncol. 2015, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Hess-Wilson, J.K.; Webb, S.L.; Daly, H.K.; Leung, Y.-K.; Boldison, J.; Comstock, C.E.S.; Sartor, M.A.; Ho, S.-M.; Knudsen, K.E. Unique Bisphenol A Transcriptome in Prostate Cancer: Novel Effects on ERβ Expression That Correspond to Androgen Receptor Mutation Status. Environ. Health Perspect. 2007, 115, 1646–1653. [Google Scholar] [CrossRef]
- Dong, S.; Terasaka, S.; Kiyama, R. Bisphenol A Induces a Rapid Activation of Erk1/2 through GPR30 in Human Breast Cancer Cells. Environ. Pollut. 2011, 159, 212–218. [Google Scholar] [CrossRef]
- Morton, M.S.; Arisaka, O.; Miyake, N.; Morgan, L.D.; Evans, B.A.J. Phytoestrogen Concentrations in Serum from Japanese Men and Women over Forty Years of Age. J. Nutr. 2002, 132, 3168–3171. [Google Scholar] [CrossRef]
- Shang, D.; Li, Z.; Zhu, Z.; Chen, H.; Zhao, L.; Wang, X.; Chen, Y. Baicalein Suppresses 17-β-Estradiol-Induced Migration, Adhesion and Invasion of Breast Cancer Cells via the G Protein-Coupled Receptor 30 Signaling Pathway. Oncol. Rep. 2015, 33, 2077–2085. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Cho, H.J.; Yu, R.; Lee, K.W.; Chun, H.S.; Park, J.H.Y. Mechanisms Underlying Apoptosis-Inducing Effects of Kaempferol in HT-29 Human Colon Cancer Cells. Int. J. Mol. Sci. 2014, 15, 2722–2737. [Google Scholar] [CrossRef] [PubMed]
- Naujokat, C.; Sezer, O.; Possinger, K. Tumor Necrosis Factor-α and Interferon-γ Induce Expression of Functional Fas Ligand on HT29 and MCF7 Adenocarcinoma Cells. Biochem. Biophys. Res. Commun. 1999, 264, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.; Guha, D.; Chakraborty, J.; Banerjee, S.; Adhikary, A.; Chakraborty, S.; Das, T.; Sa, G. Crocetin Exploits P53-Induced Death Domain (PIDD) and FAS-Associated Death Domain (FADD) Proteins to Induce Apoptosis in Colorectal Cancer. Sci. Rep. 2016, 6, 32979. [Google Scholar] [CrossRef]
- Di Donato, N.; Jean, Y.Y.; Maga, A.M.; Krewson, B.D.; Shupp, A.B.; Avrutsky, M.I.; Roy, A.; Collins, S.; Olds, C.; Willert, R.A.; et al. Mutations in CRADD Result in Reduced Caspase-2-Mediated Neuronal Apoptosis and Cause Megalencephaly with a Rare Lissencephaly Variant. Am. J. Hum. Genet. 2016, 99, 1117–1129. [Google Scholar] [CrossRef]
- Sladky, V.C.; Villunger, A. Uncovering the PIDDosome and Caspase-2 as Regulators of Organogenesis and Cellular Differentiation. Cell Death Differ. 2020, 27, 2037–2047. [Google Scholar] [CrossRef]
- Vigneswara, V.; Ahmed, Z. The Role of Caspase-2 in Regulating Cell Fate. Cells 2020, 9, 1259. [Google Scholar] [CrossRef]
- Shangary, S.; Ding, K.; Qiu, S.; Nikolovska-Coleska, Z.; Bauer, J.A.; Liu, M.; Wang, G.; Lu, Y.; McEachern, D.; Bernard, D.; et al. Reactivation of P53 by a Specific MDM2 Antagonist (MI-43) Leads to P21-Mediated Cell Cycle Arrest and Selective Cell Death in Colon Cancer. Mol. Cancer Ther. 2008, 7, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Chen, L.; Jost, C.A.; Maya, R.; Keller, D.; Wang, X.; Kaelin, W.G.; Oren, M.; Chen, J.; Lu, H. MDM2 Suppresses P73 Function without Promoting P73 Degradation. Mol. Cell Biol. 1999, 19, 3257–3266. [Google Scholar] [CrossRef]
- Dabiri, Y.; Abu el Maaty, M.A.; Chan, H.Y.; Wölker, J.; Ott, I.; Wölfl, S.; Cheng, X. P53-Dependent Anti-Proliferative and Pro-Apoptotic Effects of a Gold(I) N-Heterocyclic Carbene (NHC) Complex in Colorectal Cancer Cells. Front. Oncol. 2019, 9, 438. [Google Scholar] [CrossRef]
- Lehman, J.A.; Waning, D.L.; Batuello, C.N.; Cipriano, R.; Kadakia, M.P.; Mayo, L.D. Induction of Apoptotic Genes by a P73-Phosphatase and Tensin Homolog (P73-PTEN) Protein Complex in Response to Genotoxic Stress. J. Biol. Chem. 2011, 286, 36631–36640. [Google Scholar] [CrossRef] [PubMed]
- Ming, L.; Sakaida, T.; Yue, W.; Jha, A.; Zhang, L.; Yu, J. Sp1 and P73 Activate PUMA Following Serum Starvation. Carcinogenesis 2008, 29, 1878–1884. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Michaille, J.-J.; Alder, H.; Volinia, S.; Delmas, D.; Latruffe, N.; Croce, C.M. Resveratrol Modulates the Levels of MicroRNAs Targeting Genes Encoding Tumor-Suppressors and Effectors of TGFβ Signaling Pathway in SW480 Cells. Biochem. Pharmacol. 2010, 80, 2057–2065. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Gu, J.; Danen, E.H.; Takino, T.; Miyamoto, S.; Yamada, K.M. PTEN Interactions with Focal Adhesion Kinase and Suppression of the Extracellular Matrix-Dependent Phosphatidylinositol 3-Kinase/Akt Cell Survival Pathway. J. Biol. Chem. 1999, 274, 20693–20703. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, M.; Liu, W.; Chen, H.; Cai, T.; Xiong, H.; Sheng, X.; Liu, S.; Peng, J.; Wang, F.; et al. Estrogen Affects the Negative Feedback Loop of PTENP1-MiR200c to Inhibit PTEN Expression in the Development of Endometrioid Endometrial Carcinoma. Cell Death Dis. 2018, 10, 4. [Google Scholar] [CrossRef]
- Mei, S.; Xin, J.; Liu, Y.; Zhang, Y.; Liang, X.; Su, X.; Yan, H.; Huang, Y.; Yang, R. MicroRNA-200c Promotes Suppressive Potential of Myeloid-Derived Suppressor Cells by Modulating PTEN and FOG2 Expression. PLoS ONE 2015, 10, e0135867. [Google Scholar] [CrossRef]
- Sasikirana, W.; Radifar, M.; Herawati, C.; Surono, A.; Astuti, I.; Aryandono, T.; Haryana, S.M. The Expression of MiR-141 and MRNA PTEN with Cisplatin Therapy on NPC. KnE Life Sci. 2019, 4, 67–75. [Google Scholar] [CrossRef]
- Haier, J.; Nicolson, G.L. PTEN Regulates Tumor Cell Adhesion of Colon Carcinoma Cells under Dynamic Conditions of Fluid Flow. Oncogene 2002, 21, 1450–1460. [Google Scholar] [CrossRef]
- Lu, J.-Y.; Jin, P.; Gao, W.; Wang, D.-Z.; Sheng, J.-Q. Estrogen Enhances Mismatch Repair by Induction of MLH1 Expression via Estrogen Receptor-β. Oncotarget 2017, 8, 38767–38779. [Google Scholar] [CrossRef]
- Jin, P.; Lu, X.; Sheng, J.; Fu, L.; Meng, X.; Wang, X.; Shi, T.; Li, S.; Rao, J. Estrogen Stimulates the Expression of Mismatch Repair Gene HMLH1 in Colonic Epithelial Cells. Cancer Prev. Res. 2010, 3, 910–916. [Google Scholar] [CrossRef]
- Groh, I.A.M.; Esselen, M. Methyleugenol and Selected Oxidative Metabolites Affect DNA-Damage Signalling Pathways and Induce Apoptosis in Human Colon Tumour HT29 Cells. Food Chemi. Toxicol. 2017, 108, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Ohki, R.; Nemoto, J.; Murasawa, H.; Oda, E.; Inazawa, J.; Tanaka, N.; Taniguchi, T. Reprimo, a New Candidate Mediator of the P53-Mediated Cell Cycle Arrest at the G2 Phase. J. Biol. Chem. 2000, 275, 22627–22630. [Google Scholar] [CrossRef]
- Saavedra, K.; Valbuena, J.; Olivares, W.; Marchant, M.J.; Rodríguez, A.; Torres-Estay, V.; Carrasco-Avino, G.; Guzmán, L.; Aguayo, F.; Roa, J.C.; et al. Loss of Expression of Reprimo, a P53-Induced Cell Cycle Arrest Gene, Correlates with Invasive Stage of Tumor Progression and P73 Expression in Gastric Cancer. PLoS ONE 2015, 10, e0125834. [Google Scholar] [CrossRef] [PubMed]
- Buchegger, K.; Riquelme, I.; Viscarra, T.; Ili, C.; Brebi, P.; Huang, T.H.-M.; Roa, J.C. Reprimo, a Potential P53-Dependent Tumor Suppressor Gene, Is Frequently Hypermethylated in Estrogen Receptor α-Positive Breast Cancer. Int. J. Mol. Sci. 2017, 18, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.P.; Sato, F.; Jin, Z.; Greenwald, B.D.; Ito, T.; Mori, Y.; Paun, B.C.; Kan, T.; Cheng, Y.; Wang, S.; et al. Reprimo Methylation Is a Potential Biomarker of Barrett’s-Associated Esophageal Neoplastic Progression. Clin. Cancer Res. 2006, 12, 6637–6642. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Fukushima, N.; Matsubayashi, H.; Iacobuzio-Donahue, C.A.; Yeo, C.J.; Goggins, M. Aberrant Methylation of Reprimo Correlates with Genetic Instability and Predicts Poor Prognosis in Pancreatic Ductal Adenocarcinoma. Cancer 2006, 107, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.-B.; Cheng, Y.-K.; Zhang, L.; Wang, X.-P.; Wang, L.; Lan, P. Prognostic Value of Estrogen Receptor-α and Progesterone Receptor in Curatively Resected Colorectal Cancer: A Retrospective Analysis with Independent Validations. BMC Cancer 2019, 19, 933. [Google Scholar] [CrossRef]
- Nüssler, N.C.; Reinbacher, K.; Shanny, N.; Schirmeier, A.; Glanemann, M.; Neuhaus, P.; Nussler, A.K.; Kirschner, M. Sex-Specific Differences in the Expression Levels of Estrogen Receptor Subtypes in Colorectal Cancer. Gend. Med. 2008, 5, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Wu, J.; Su, X.; Yan, H.; Sun, Z. Effects of Low Dose of Bisphenol A on the Proliferation and Mechanism of Primary Cultured Prostate Epithelial Cells in Rodents. Oncol. Lett. 2017, 14, 2635–2642. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Shaw, I.C. Food Flavonoid Ligand Structure/Estrogen Receptor-α Affinity Relationships—Toxicity or Food Functionality? Food Chem. Toxicol. 2019, 129, 328–336. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Kominea, A.; Vandoros, G.; Sykiotis, G.P.; Andricopoulos, P.; Varakis, I.; Sotiropoulou-Bonikou, G.; Papavassiliou, A.G. Oestrogen Receptor Beta (ERbeta) Is Abundantly Expressed in Normal Colonic Mucosa, but Declines in Colon Adenocarcinoma Paralleling the Tumour’s Dedifferentiation. Eur. J. Cancer 2003, 39, 1251–1258. [Google Scholar] [CrossRef]
- Pinton, G.; Nilsson, S.; Moro, L. Targeting Estrogen Receptor Beta (ERβ) for Treatment of Ovarian Cancer: Importance of KDM6B and SIRT1 for ERβ Expression and Functionality. Oncogenesis 2018, 7, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-N.; Lin, V.C.-H.; Rau, K.-M.; Shieh, P.-C.; Kuo, D.-H.; Shieh, J.-C.; Chen, W.-J.; Tsai, S.-C.; Way, T.-D. Resveratrol Modulates Tumor Cell Proliferation and Protein Translation via SIRT1-Dependent AMPK Activation. J. Agric. Food Chem. 2010, 58, 1584–1592. [Google Scholar] [CrossRef]
- Moore, R.L.; Dai, Y.; Faller, D.V. Sirtuin 1 (SIRT1) and Steroid Hormone Receptor Activity in Cancer. J. Endocrinol. 2012, 213, 37–48. [Google Scholar] [CrossRef]
- Sun, H.; Tian, A.; Zhang, J.; Liao, X.; Zhang, N. Epithelial-Mesenchymal Transition Induced by MyoD Inhibits Growth of High Metastatic Colorectal Cancer. Med. Hypotheses 2019, 130, 109285. [Google Scholar] [CrossRef]
- Campos-Vega, R.; Vázquez-Sánchez, K.; López-Barrera, D.; Loarca-Piña, G.; Mendoza-Díaz, S.; Oomah, B.D. Simulated Gastrointestinal Digestion and in Vitro Colonic Fermentation of Spent Coffee (Coffea arabica L.): Bioaccessibility and Intestinal Permeability. Food Res. Int. 2015, 77, 156–161. [Google Scholar] [CrossRef]
- Campos-Vega, R.; Reynoso-Camacho, R.; Pedraza-Aboytes, G.; Acosta-Gallegos, J.A.; Guzman-Maldonado, S.H.; Paredes-Lopez, O.; Oomah, B.D.; Loarca-Piña, G. Chemical Composition and in Vitro Polysaccharide Fermentation of Different Beans (Phaseolus vulgaris L.). J. Food Sci. 2009, 74, T59–T65. [Google Scholar] [CrossRef] [PubMed]
- Abu-Reidah, I.M.; Arráez-Román, D.; Warad, I.; Fernández-Gutiérrez, A.; Segura-Carretero, A. UHPLC/MS2-Based Approach for the Comprehensive Metabolite Profiling of Bean (Vicia faba L.) by-Products: A Promising Source of Bioactive Constituents. Food Res. Int. 2017, 93, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Cardador-Martínez, A.; Loarca-Piña, G.; Oomah, B.D. Antioxidant Activity in Common Beans (Phaseolus vulgaris L.). J. Agric. Food Chem. 2002, 50, 6975–6980. [Google Scholar] [CrossRef] [PubMed]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and Validation of an Improved Oxygen Radical Absorbance Capacity Assay Using Fluorescein as the Fluorescent Probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Delgado, M.; Ribeiro-Varandas, E. Bisphenol A at the Reference Level Counteracts Doxorubicin Transcriptional Effects on Cancer Related Genes in HT29 Cells. Toxicol. Vitro 2015, 29, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Zazueta, M.S.; Luzardo-Ocampo, I.; García-Romo, J.S.; Noguera-Artiaga, L.; Carbonell-Barrachina, Á.A.; Taboada-Antelo, P.; Campos-Vega, R.; Rosas-Burgos, E.C.; Burboa-Zazueta, M.G.; Ezquerra-Brauer, J.M.; et al. Bioactive Compounds from Octopus Vulgaris Ink Extracts Exerted Anti-Proliferative and Anti-Inflammatory Effects in Vitro. Food Chem. Toxicol. 2021, 151, 112119. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UPLC-MS | Antioxidant Capacity | ||||
|---|---|---|---|---|---|
| Treatment | Compounds | Retention Time | Molar Mass g/mol | DPPH µM eq. Trolox | ORAC µM eq. Trolox |
| Blank | Phenylacetic acid | 6.50 ± 0.00 | 135.04 | 35.27 ± 0.70 a | 2.02 ± 0.10 a |
| Secoisolariciresinol | 11.00 ± 0.00 | 361.16 | |||
| FEN | Naringenin | 8.63 ± 0.01 | 271.06 | 52.97 ± 1.40 b | 22.12 ± 6.70 b |
| 3-HPPA * | 4.93 ± 0.03 | 167.03 | |||
| Apigenin | 8.63 ± 0.01 | 271.06 | |||
| Secoisolariciresinol | 11.60 ± 0.00 | 361.16 | |||
| Phenylacetic acid | 6.43 ± 0.01 | 135.94 | |||
| Naringenin | Naringenin | 8.63 ± 0.00 | 271.06 | 92.37 ± 1.10 c | 22.31 ± 6.80 b |
| GSH nmol GSH/mL | SOD % Inhibition of O2 | |
|---|---|---|
| Control | 160.22 ± 2.4 d | 1.62 ± 1.1 a |
| Naringenin | 110.75 ± 1.5 a | 3.05 ± 0.3 a |
| BPA | 140.03 ± 4.1 c | 6.03 ± 1.1 a |
| Naringenin + BPA | 129.19 ± 0.9 b | 5.2 ± 0.8 a |
| FEN | 144.15 ± 5.6 d | 13.18 ± 2 b |
| FEN + BPA | 157.99 ± 1.5 c | 4.8 ± 1.0 a |
| Blank | 152.67 ± 0.4 d | 3.2 ± 0.4 a |
| Symbol | Description | Naringenin | BPA | Naringenin + BPA | FEN | FEN + BPA |
|---|---|---|---|---|---|---|
| APAF1 | Apoptotic peptidase activating factor 1 (Other apoptosis genes) | 1.11 | 1.52 | 21.10 | 2.62 | 27.86 |
| ATM | Ataxia telangiectasia mutated (Negative regulation of the cell cycle) | 1.00 | 1.02 | 3.13 | 1.12 | 8.66 |
| ATR | Ataxia telangiectasia and Rad3-related (Cell cycle checkpoint) | −1.20 | −2.16 | 3.28 | −1.07 | 5.63 |
| ADGRB1 | Brain-specific angiogenesis inhibitor 1 (Other genes related to inhibition proliferation) | −1.11 | −1.04 | 16.54 | 2.39 | 16.04 |
| BAX | BCL2-associated X protein (Induction of apoptosis) | −1.27 | −1.41 | 1.80 | −1.45 | 5.06 |
| BBC3 | BCL2 binding component 3 (Induction of apoptosis) | 1.46 | −4.96 | 13.67 | 1.76 | 6.87 |
| BCL2 | B-cell CLL/lymphoma 2 (Anti-apoptosis) | −2.17 | −1.21 | 40.54 | 1.70 | 146.89 |
| BCL2A1 | BCL2-related protein A1 (Anti-apoptosis) | 1.01 | −1.21 | 19.29 | 3.76 | 43.36 |
| BID | BH3 interacting domain death agonist (Induction of apoptosis) | 1.52 | −1.16 | 1.79 | 1.11 | 1.17 |
| BIRC5 | Baculoviral IAP repeat containing 5 (Anti-apoptosis) | 1.36 | 1.41 | 10.53 | 3.12 | 21.64 |
| BRCA1 | Breast cancer 1, early onset (Cell cycle checkpoint) | 1.11 | 1.15 | 4.04 | 1.27 | 4.29 |
| BRCA2 | Breast cancer 2, early onset (Regulation of the cell cycle) | 3.28 | −1.34 | 62.42 | 1.92 | 19.02 |
| BTG2 | BTG family, member 2 (Other genes related to inhibition proliferation) | −1.24 | −1.56 | 15.30 | −2.66 | 2.88 |
| CASP2 | Caspase 2, apoptosis-related cysteine peptidase (Anti-apoptosis, apoptosis) | 2.14 | 8.11 | 2.99 | −1.58 | 4.30 |
| CASP9 | Caspase 9, apoptosis-related cysteine peptidase (Other apoptosis genes) | 3.99 | −1.06 | 2.36 | 2.03 | 12.27 |
| CCNB1 | Cyclin B1 (Other cell cycle genes) | 1.32 | 1.30 | 1.47 | 1.46 | 2.12 |
| CCNE1 | Cyclin E1 (Cell cycle checkpoint) | 1.11 | 1.13 | 3.96 | 1.88 | 8.23 |
| CCNG1 | Cyclin G1 (Cell cycle checkpoint) | 1.02 | 1.02 | −3.41 | −1.49 | 1.22 |
| CCNH | Cyclin H (Regulation of the cell cycle) | 1.31 | −1.04 | 1.76 | 1.69 | 9.19 |
| CDC25A | Cell division cycle 25 homolog A (S. pombe) (Regulation of the cell cycle) | 1.27 | −1.11 | 18.49 | 2.02 | 12.59 |
| CDC25C | Cell division cycle 25 homolog C (S. pombe) (Cell proliferation) | 1.45 | 1.14 | 4.31 | 1.73 | 7.16 |
| CDK1 | Cyclin-dependent kinase 1 (Regulation of the cell cycle) | 1.17 | 1.30 | −2.86 | 1.34 | 3.73 |
| CDK4 | Cyclin-dependent kinase 4 (Regulation of the cell cycle) | 1.13 | −1.39 | 1.84 | −1.14 | 1.23 |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) (Cell cycle arrest) | 1.36 | −1.13 | 3.37 | 5.85 | 48.32 |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A (melanoma, p16, inhibits CDK4) (Cell cycle arrest) | 1.38 | 1.11 | 2.18 | 1.44 | 1.62 |
| CHEK1 | CHK1 checkpoint homolog (S. pombe) (Cell cycle arrest) | 1.30 | 1.27 | 2.31 | 1.24 | 4.75 |
| CHEK2 | CHK2 checkpoint homolog (S. pombe) (Cell cycle arrest) | 1.54 | 1.18 | 2.74 | 1.74 | 7.30 |
| CRADD | CASP2 and RIPK1 domain containing adaptor with death domain (Induction of apoptosis) | 2.96 | −1.48 | 56.18 | 1.38 | 10.58 |
| DNMT1 | DNA (cytosine-5-)-methyltransferase 1 (DNA repair genes) | −1.05 | 1.01 | 2.20 | 1.21 | 2.07 |
| E2F1 | E2F transcription factor 1 (Regulation of the cell cycle) | 1.29 | 1.03 | 3.59 | 1.39 | 6.35 |
| E2F3 | E2F transcription factor 3 (Regulation of the cell cycle) | 1.18 | −1.01 | 6.17 | −1.44 | 5.39 |
| EGFR | Epidermal growth factor receptor (Other genes related to cell growth, proliferation, and differentiation) | 1.11 | −1.05 | 4.07 | 1.04 | 8.01 |
| EGR1 | Early growth response 1 (Other genes related to cell growth, proliferation, and differentiation) | 1.44 | −1.06 | 22.88 | −1.00 | 15.41 |
| EI24 | Etoposide induced 2.4 mRNA (Induction of apoptosis) | 1.17 | −1.12 | −1.44 | 1.06 | 1.62 |
| ESR1 | Estrogen receptor 1 (Cell growth and differentiation) | 1.11 | 3.63 | 63.17 | 5.11 | 33.01 |
| FADD | Fas (TNFRSF6)-associated via death domain (Induction of apoptosis) | 1.41 | −1.07 | 1.19 | −1.05 | 2.05 |
| FAS | Fas (TNF receptor superfamily, member 6) (Induction of apoptosis) | 1.01 | 1.43 | 12.09 | 2.13 | 15.30 |
| FASLG | Fas ligand (TNF superfamily, member 6) (Induction of apoptosis) | 1.86 | −1.31 | 35.45 | 4.63 | 52.37 |
| FOXO3 | Forkhead box O3 (Other genes related to cell growth, proliferation and differentiation) | 1.29 | 1.01 | 1.69 | 1.15 | 4.45 |
| GADD45A | Growth arrest and DNA-damage-inducible, alpha (Other apoptosis genes) | −1.16 | −1.13 | 4.97 | 2.92 | 4.79 |
| GML | Glycosylphosphatidylinositol anchored molecule like (Other genes related to cell growth, proliferation and differentiation) protein (Other apoptosis genes) | 1.95 | 1.05 | 37.20 | 4.98 | 60.37 |
| HDAC1 | Histone deacetylase 1 (Anti-apoptosis) | 1.49 | −1.01 | −1.57 | −1.05 | 1.64 |
| HK2 | Hexokinase 2 (Regulation of the cell cycle) | 1.24 | 1.62 | 3.55 | 2.79 | 9.41 |
| IGF1R | Insulin-like growth factor 1 receptor (Regulation of the cell cycle) | 1.27 | 1.17 | 3.50 | −1.05 | 5.58 |
| IL6 | Interleukin 6 (interferon, beta 2) (Positive regulation of the cell proliferation) | −1.40 | −1.01 | 13.63 | 3.45 | 30.56 |
| JUN | Jun proto-oncogene (Other genes related to cell growth, proliferation and differentiation) | −2.06 | −1.72 | 1.57 | −3.60 | 1.59 |
| KAT2B | K(lysine) acetyltransferase 2B (Other genes related to cell growth, proliferation and differentiation) | 1.18 | 1.04 | 22.40 | 2.51 | 16.65 |
| KRAS | V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (Other genes related to cell growth, proliferation) | −1.10 | −1.01 | −1.39 | −1.42 | 2.19 |
| MCL1 | Myeloid cell leukemia sequence 1 (BCL2-related) (Anti-apoptotic) | 1.12 | −1.17 | 2.52 | 1.47 | 4.03 |
| MDM2 | Mdm2 p53 binding protein homolog (mouse) (Negative regulation of cell proliferation) | 1.14 | 1.46 | 21.69 | 2.85 | 36.22 |
| MDM4 | Mdm4 p53 binding protein homolog (mouse) (Negative regulation of cell proliferation) | −1.39 | 1.66 | 3.80 | −1.28 | 7.25 |
| MLH1 | MutL homolog 1, colon cancer, nonpolyposis type 2 (E. coli) (Other cell cycle genes) | 4.48 | −7.94 | 2.15 | 1.25 | 5.32 |
| MSH2 | MutS homolog 2, colon cancer, nonpolyposis type 1 (E. coli) (DNA repair genes) | 1.39 | 1.07 | −1.24 | 1.30 | 4.25 |
| MYC | V-myc myelocytomatosis viral oncogene homolog (avian) (Cell cycle arrest) | −1.02 | −1.21 | 1.25 | −1.10 | 1.79 |
| MYOD1 | Myogenic differentiation 1 (Cell growth and differentiation) | 3.99 | −1.10 | 75.89 | 5.20 | 26.82 |
| NF1 | Neurofibromin 1 (Negative regulation of the cell cycle) | 1.16 | 1.09 | 4.35 | 1.56 | 11.20 |
| NFKB1 | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 (Anti-apoptosis) | −1.22 | −1.08 | 6.63 | −1.12 | 5.06 |
| PCNA | Proliferating cell nuclear antigen (Cell proliferation) | 1.14 | −1.21 | 2.15 | 1.01 | 2.18 |
| PIDD1 | P53-induced death domain protein (Apoptosis) | 1.09 | 1.04 | 12.74 | 1.87 | 10.41 |
| PPM1D | Protein phosphatase, Mg2+/Mn2+ dependent, 1D (Negative regulation of the cell proliferation) | −1.35 | 1.14 | 10.99 | 1.31 | 6.36 |
| PRC1 | Protein regulator of cytokinesis 1 (Other cell cycle genes) | 1.12 | −1.49 | 1.41 | −1.17 | 1.59 |
| PRKCA | Protein kinase C, alpha (Cell proliferation) | 1.75 | −1.10 | 4.29 | 1.71 | 8.28 |
| PTEN | Phosphatase and tensin homolog (Negative regulation of the cell cycle) | 2.70 | −3.46 | 1.20 | 1.72 | 6.98 |
| PTTG1 | Pituitary tumor-transforming 1 (DNA repair genes) | 1.68 | −1.38 | 1.74 | 2.72 | 6.99 |
| RB1 | Retinoblastoma 1 (Cell cycle checkpoint) | −1.01 | −3.32 | 1.04 | −1.08 | 3.02 |
| RELA | V-rel reticuloendotheliosis viral oncogene homolog A (avian) (Cell proliferation) | 1.49 | −1.19 | 1.18 | −1.19 | 1.87 |
| RPRM | Reprimo, TP53 dependent G2 arrest mediator candidate (Cell cycle arrest) | 3.15 | −1.39 | 59.96 | 5.71 | 42.08 |
| SESN2 | Sestrin 2 (Negative regulation of the cell proliferation) | −1.70 | −1.01 | 2.86 | 1.69 | 4.24 |
| SIAH1 | Seven in absentia homolog 1 (Drosophila) (Other apoptosis genes) | 1.61 | −1.42 | 1.21 | 1.39 | 4.26 |
| SIRT1 | Sirtuin 1 (Other apoptosis genes) | −1.00 | −3.18 | 2.05 | −1.09 | 2.70 |
| STAT1 | Signal transducer and activator of transcription 1, 91kDa (Regulation of the cell cycle) | −1.08 | −4.06 | 1.19 | −1.21 | −9.20 |
| TADA3 | Transcriptional adaptor 3 (Regulation of the cell cycle) | 1.39 | −2.66 | 1.67 | −1.17 | −9.68 |
| TNF | Tumor necrosis factor (Anti-apoptosis) | 1.71 | −1.62 | 32.52 | 3.89 | 47.68 |
| TNFRSF10B | Tumor necrosis factor receptor superfamily, member 10b (Induction of apoptosis) | 1.08 | 1.23 | 3.59 | 1.75 | 3.66 |
| TNFRSF10D | Tumor necrosis factor receptor superfamily, member 10d, decoy with truncated death domain (Anti-apoptosis) | 4.98 | −1.13 | 14.79 | 1.74 | 9.71 |
| p53 | Tumor protein p53 (Induction of apoptosis, negative regulation of the cell cycle, DNA repair genes) | 1.15 | 1.41 | −1.00 | −1.80 | 1.19 |
| TP53AIP1 | Tumor protein p53 regulated apoptosis inducing protein 1(Other apoptosis genes) | 3.99 | −1.10 | 75.89 | 3.62 | 27.66 |
| TP53BP2 | Tumor protein p53 binding protein, 2 (Other apoptosis genes) | 1.35 | −4.06 | 1.46 | 1.58 | 6.08 |
| TP63 | Tumor protein p63 (Induction of apoptosis) | 2.11 | −1.24 | 39.99 | 6.29 | 66.82 |
| TP73 | Tumor protein p73 (Induction of apoptosis) | 3.99 | 1.12 | 75.89 | 2.65 | 101.66 |
| TRAF2 | TNF receptor-associated factor 2 (Cell proliferation) | 1.21 | −1.20 | 2.49 | 1.36 | 4.12 |
| TSC1 | Tuberous sclerosis 1 (Negative regulation of the cell cycle) | −1.19 | 1.18 | 5.12 | 1.42 | −1.04 |
| WT1 | Wilms tumor 1 (Negative regulation of the cell cycle) | 1.39 | 1.02 | 75.89 | 3.21 | 14.29 |
| XRCC5 | X-ray repair complementing defective repair in Chinese hamster cells 5 (double-strand-break rejoining) (DNA repair genes) | 1.21 | −1.31 | −1.84 | −1.73 | −30.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lozano-Herrera, S.J.; Luna-Bárcenas, G.; Guevara-González, R.G.; Campos-Vega, R.; Solís-Sáinz, J.C.; Hernández-Puga, A.G.; Vergara-Castañeda, H.A. Fermentation Extract of Naringenin Increases the Expression of Estrogenic Receptor β and Modulates Genes Related to the p53 Signalling Pathway, miR-200c and miR-141 in Human Colon Cancer Cells Exposed to BPA. Molecules 2022, 27, 6588. https://doi.org/10.3390/molecules27196588
Lozano-Herrera SJ, Luna-Bárcenas G, Guevara-González RG, Campos-Vega R, Solís-Sáinz JC, Hernández-Puga AG, Vergara-Castañeda HA. Fermentation Extract of Naringenin Increases the Expression of Estrogenic Receptor β and Modulates Genes Related to the p53 Signalling Pathway, miR-200c and miR-141 in Human Colon Cancer Cells Exposed to BPA. Molecules. 2022; 27(19):6588. https://doi.org/10.3390/molecules27196588
Chicago/Turabian StyleLozano-Herrera, Sara Julietta, Gabriel Luna-Bárcenas, Ramón Gerardo Guevara-González, Rocio Campos-Vega, Juan Carlos Solís-Sáinz, Ana Gabriela Hernández-Puga, and Haydé Azeneth Vergara-Castañeda. 2022. "Fermentation Extract of Naringenin Increases the Expression of Estrogenic Receptor β and Modulates Genes Related to the p53 Signalling Pathway, miR-200c and miR-141 in Human Colon Cancer Cells Exposed to BPA" Molecules 27, no. 19: 6588. https://doi.org/10.3390/molecules27196588
APA StyleLozano-Herrera, S. J., Luna-Bárcenas, G., Guevara-González, R. G., Campos-Vega, R., Solís-Sáinz, J. C., Hernández-Puga, A. G., & Vergara-Castañeda, H. A. (2022). Fermentation Extract of Naringenin Increases the Expression of Estrogenic Receptor β and Modulates Genes Related to the p53 Signalling Pathway, miR-200c and miR-141 in Human Colon Cancer Cells Exposed to BPA. Molecules, 27(19), 6588. https://doi.org/10.3390/molecules27196588