
Fullerene-Perylenediimide (C60-PDI) Based Systems: An Overview and Synthesis of a Versatile Platform for Their Anchor Engineering



Abstract

1. Introduction
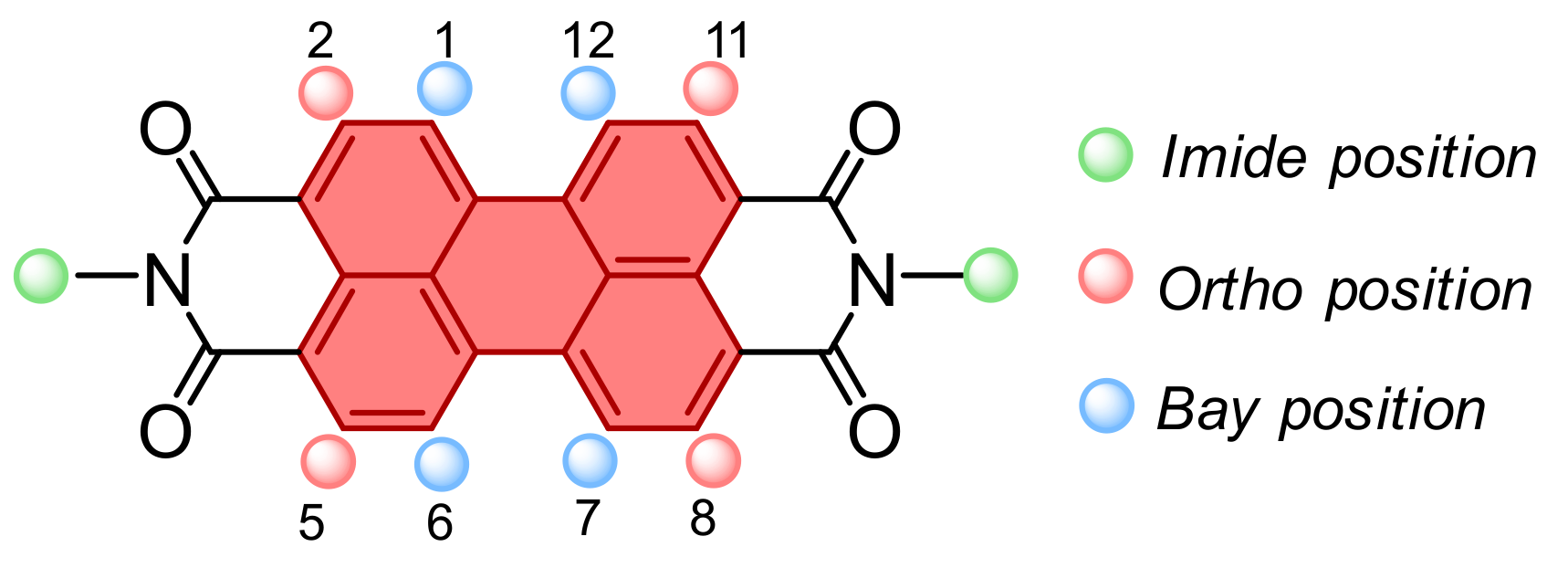
2. State-of-the-Art of Covalently Linked C60-PDI Based Systems
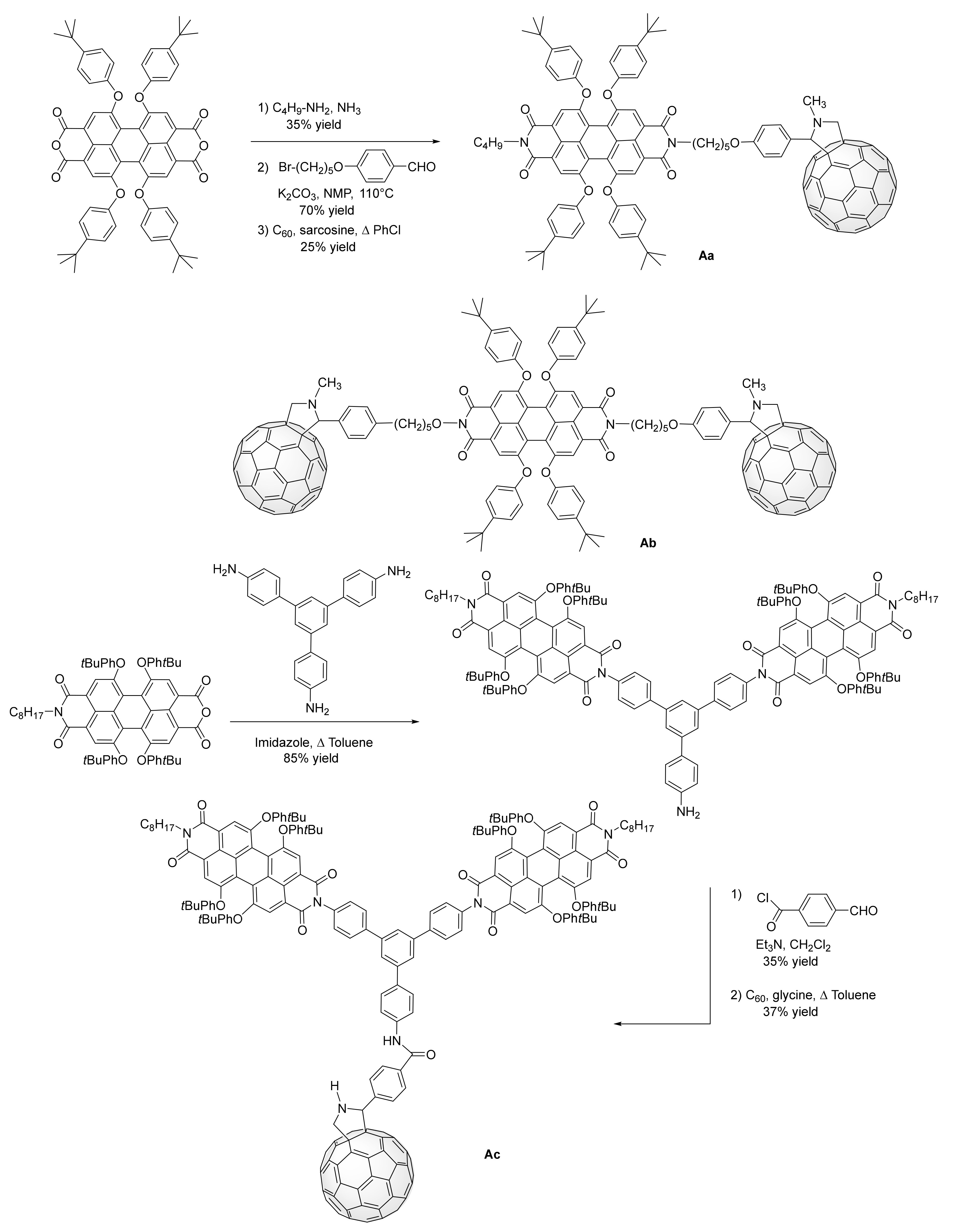
2.1. Grafting C60 at the Imide PDI Scaffold
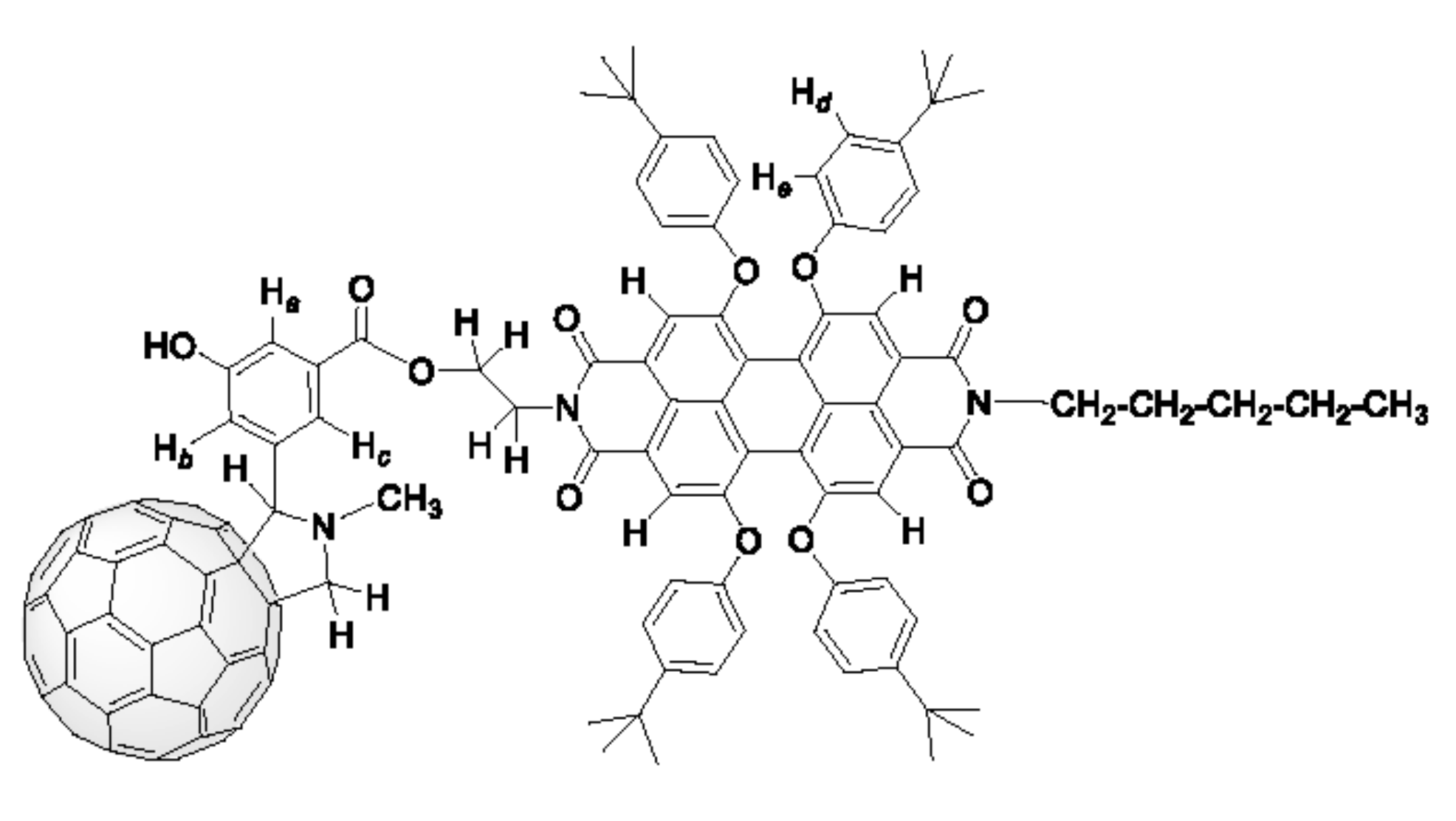
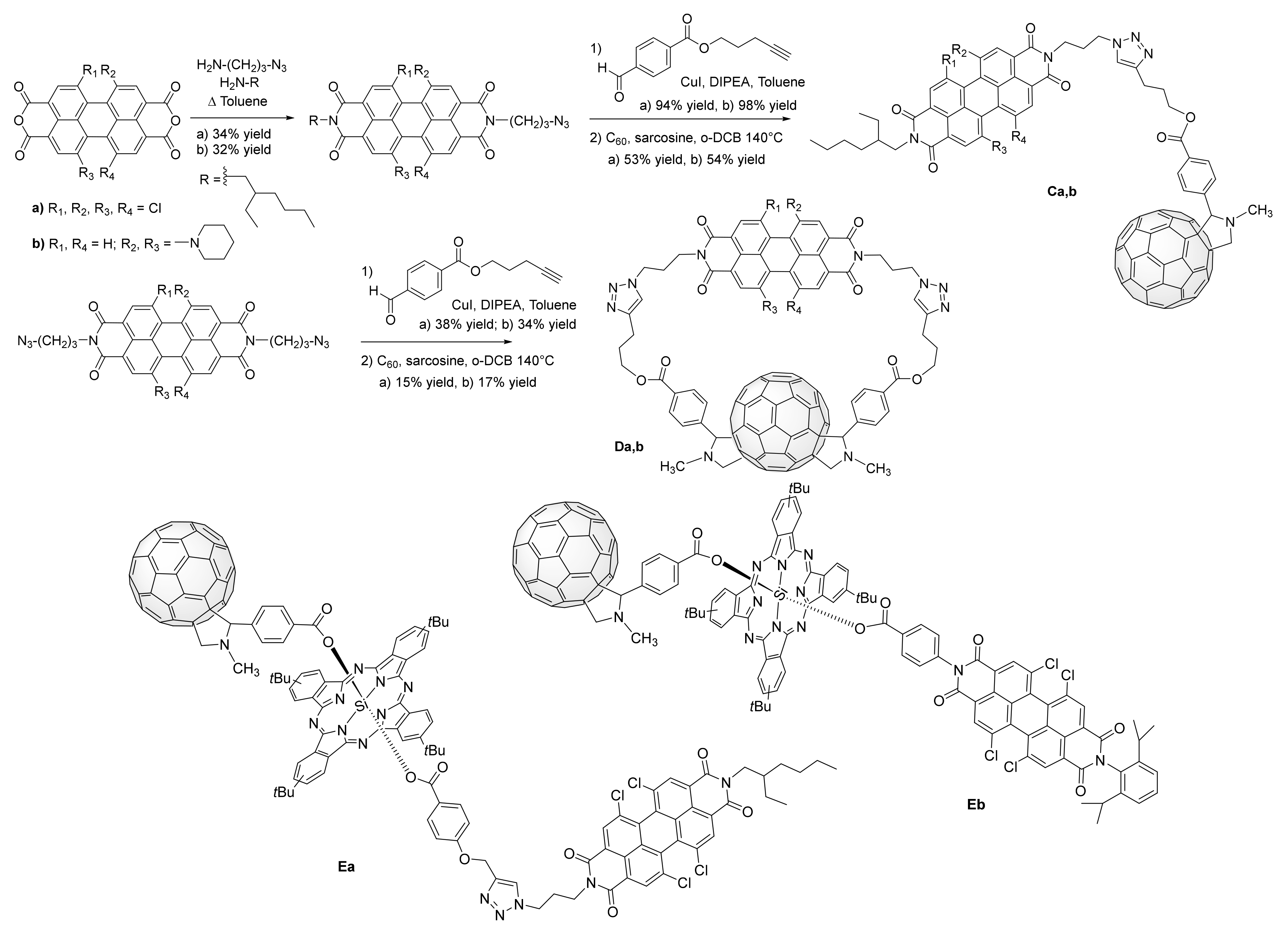
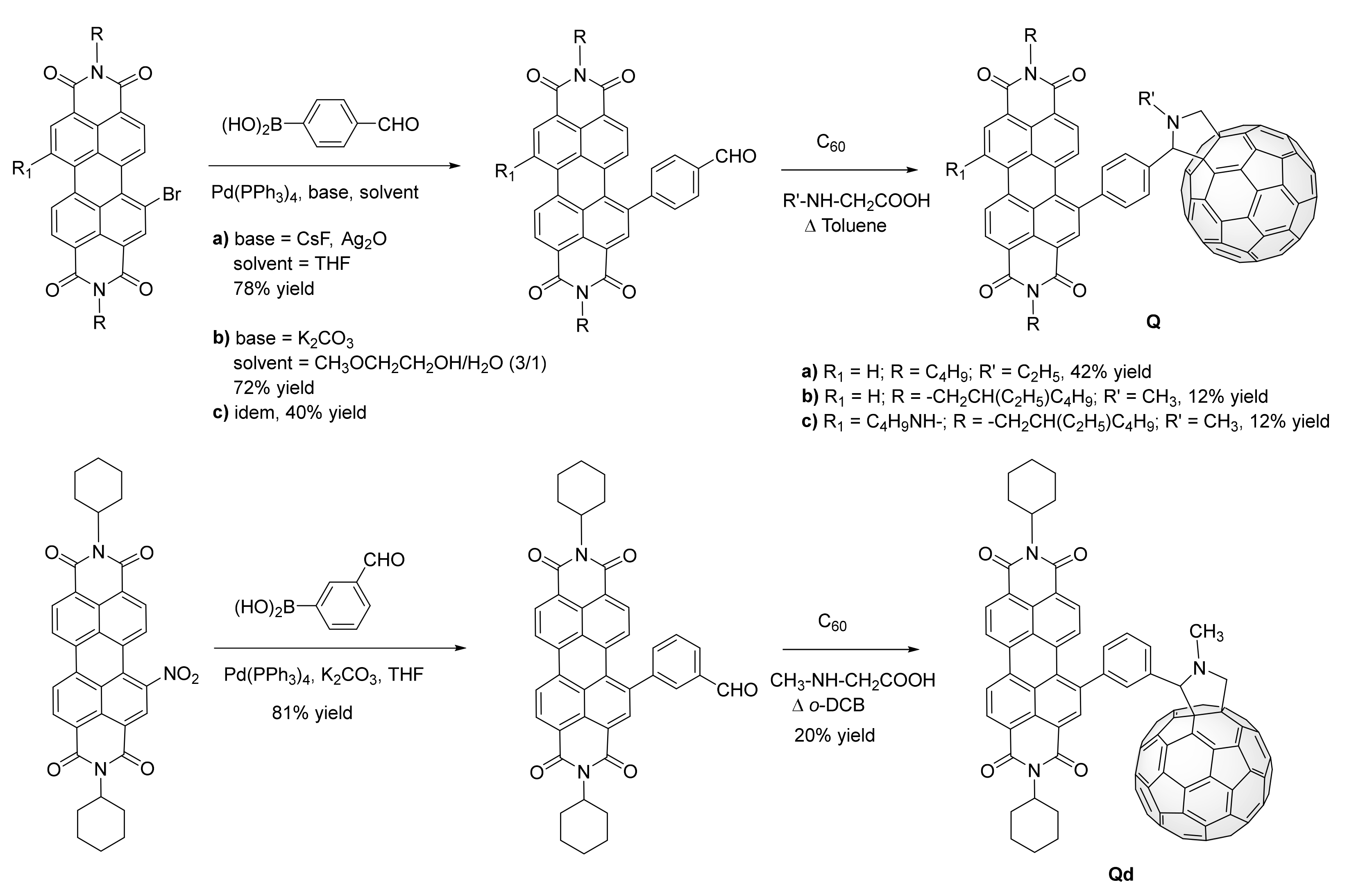
2.2. Grafting C60 onto Bay-PDI Backbone
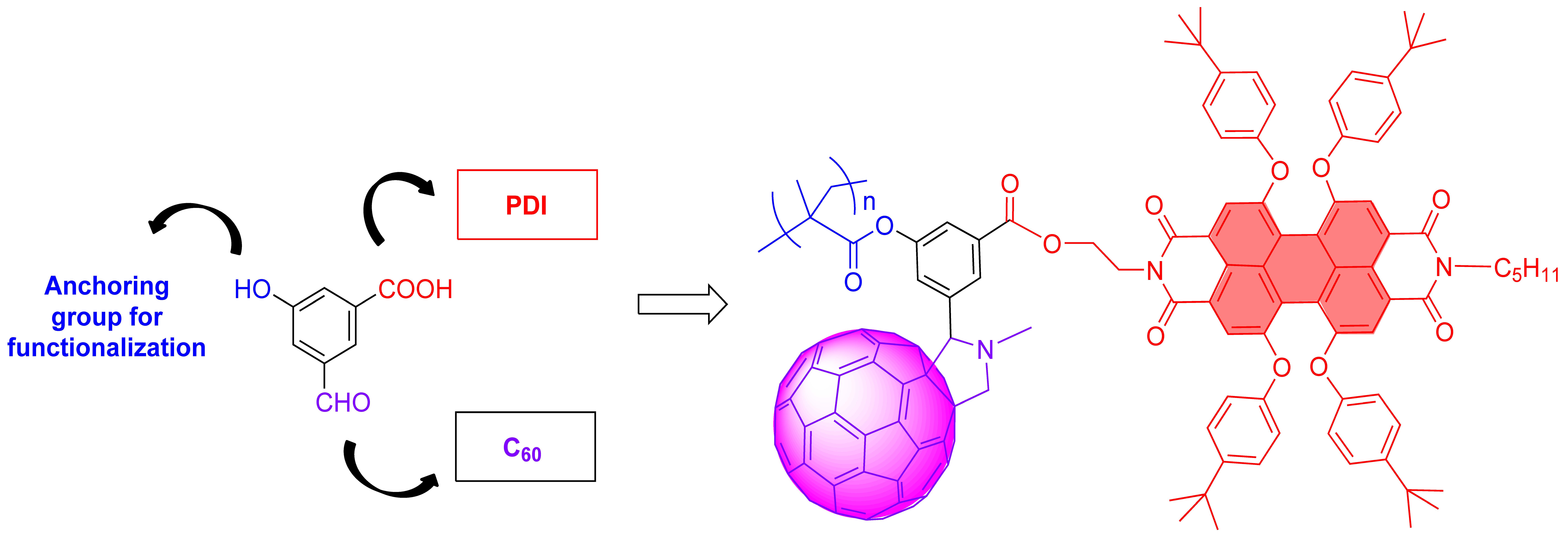
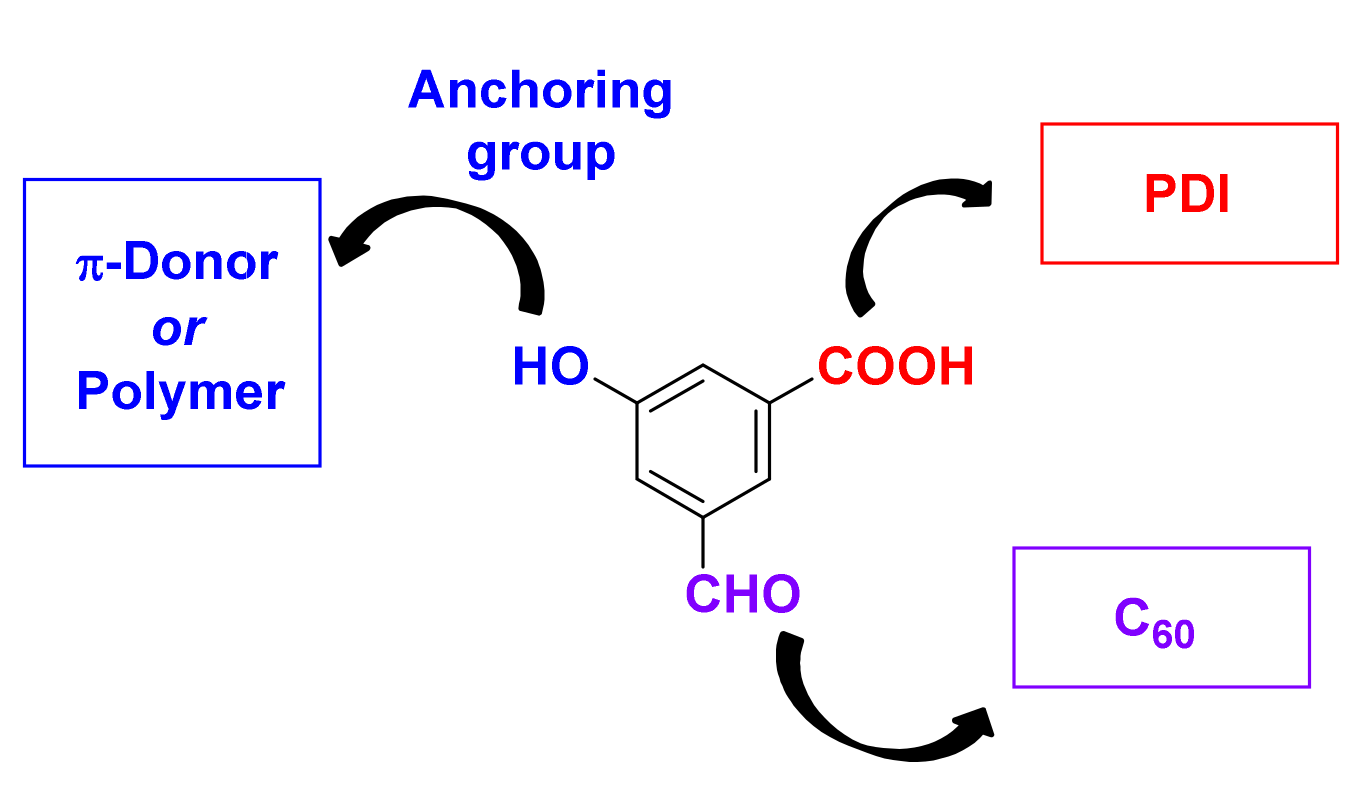
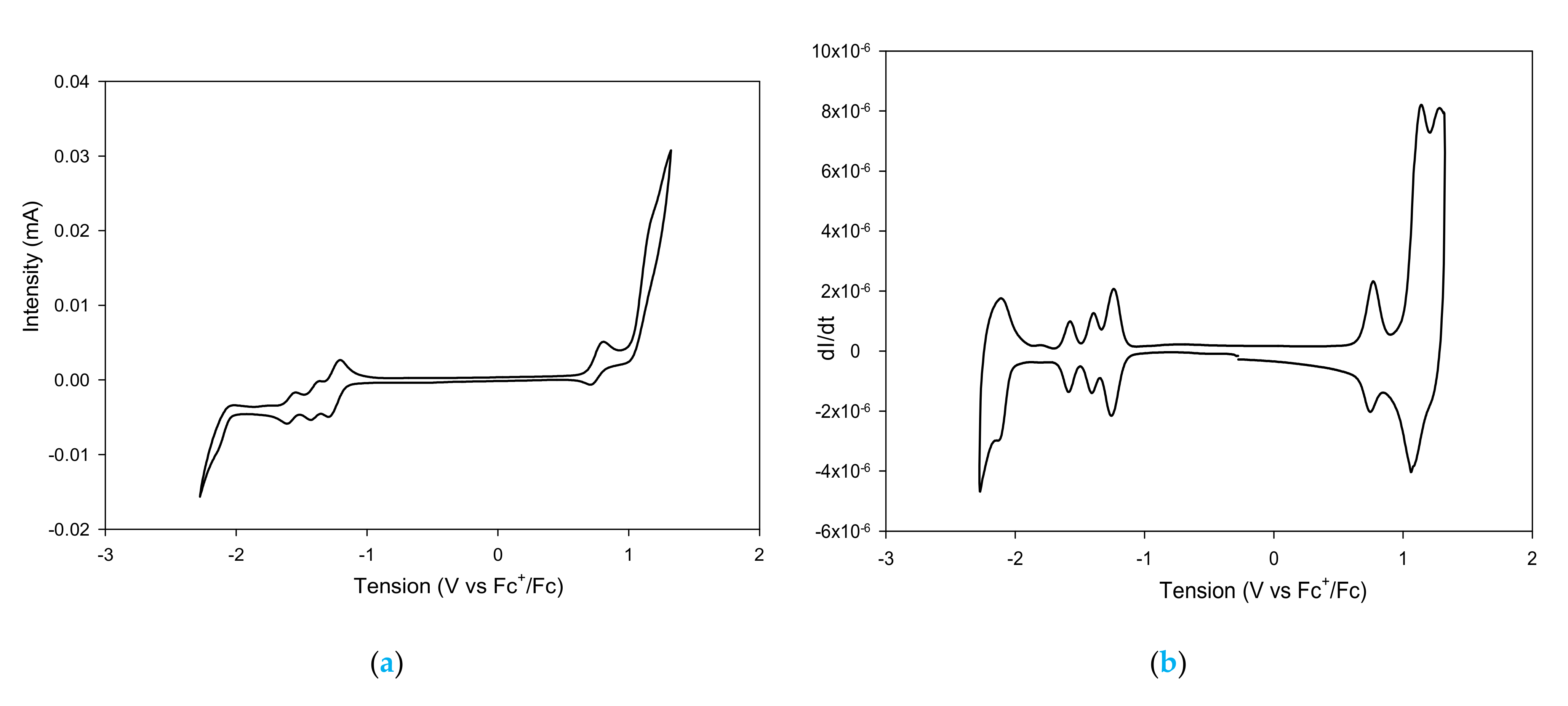
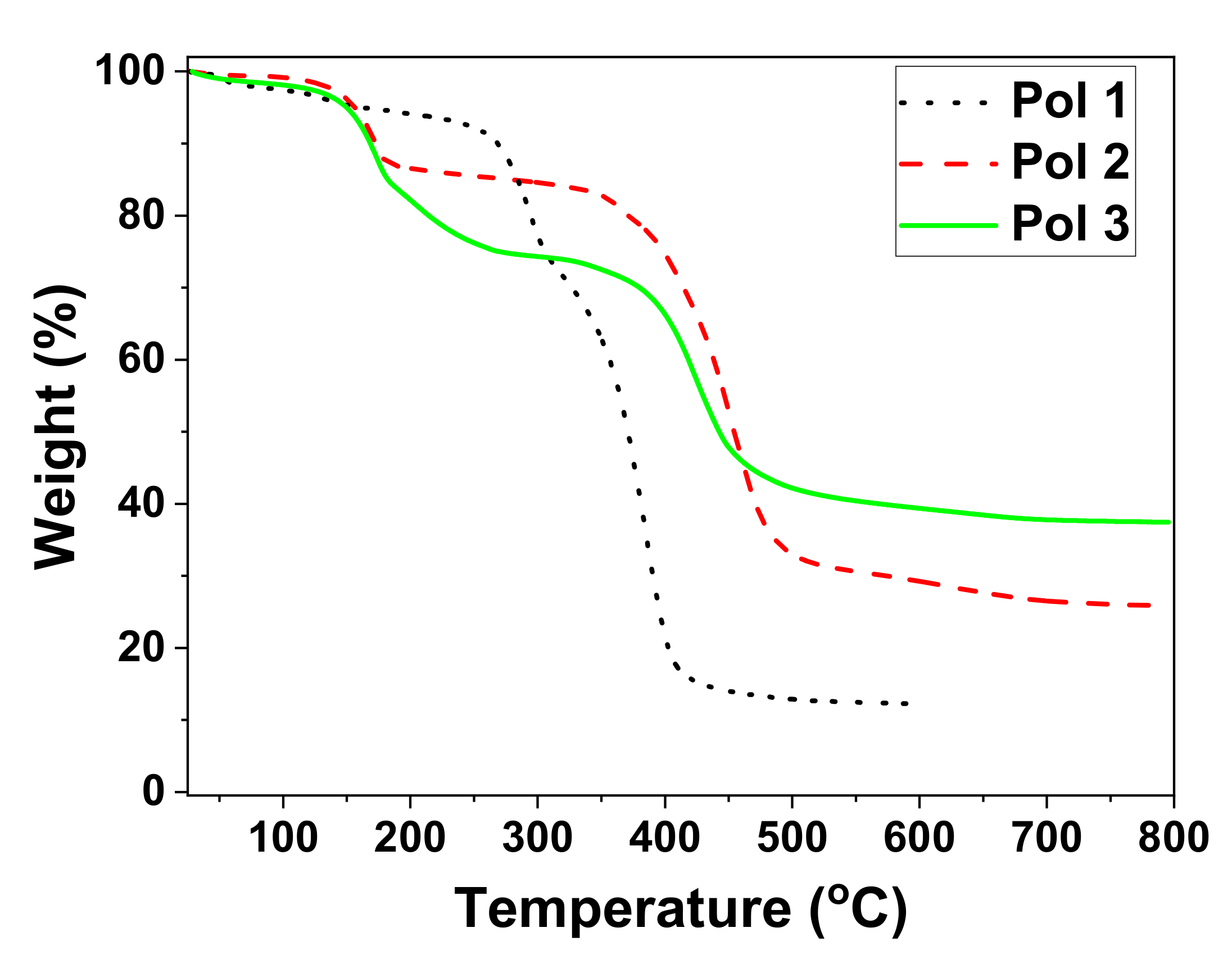
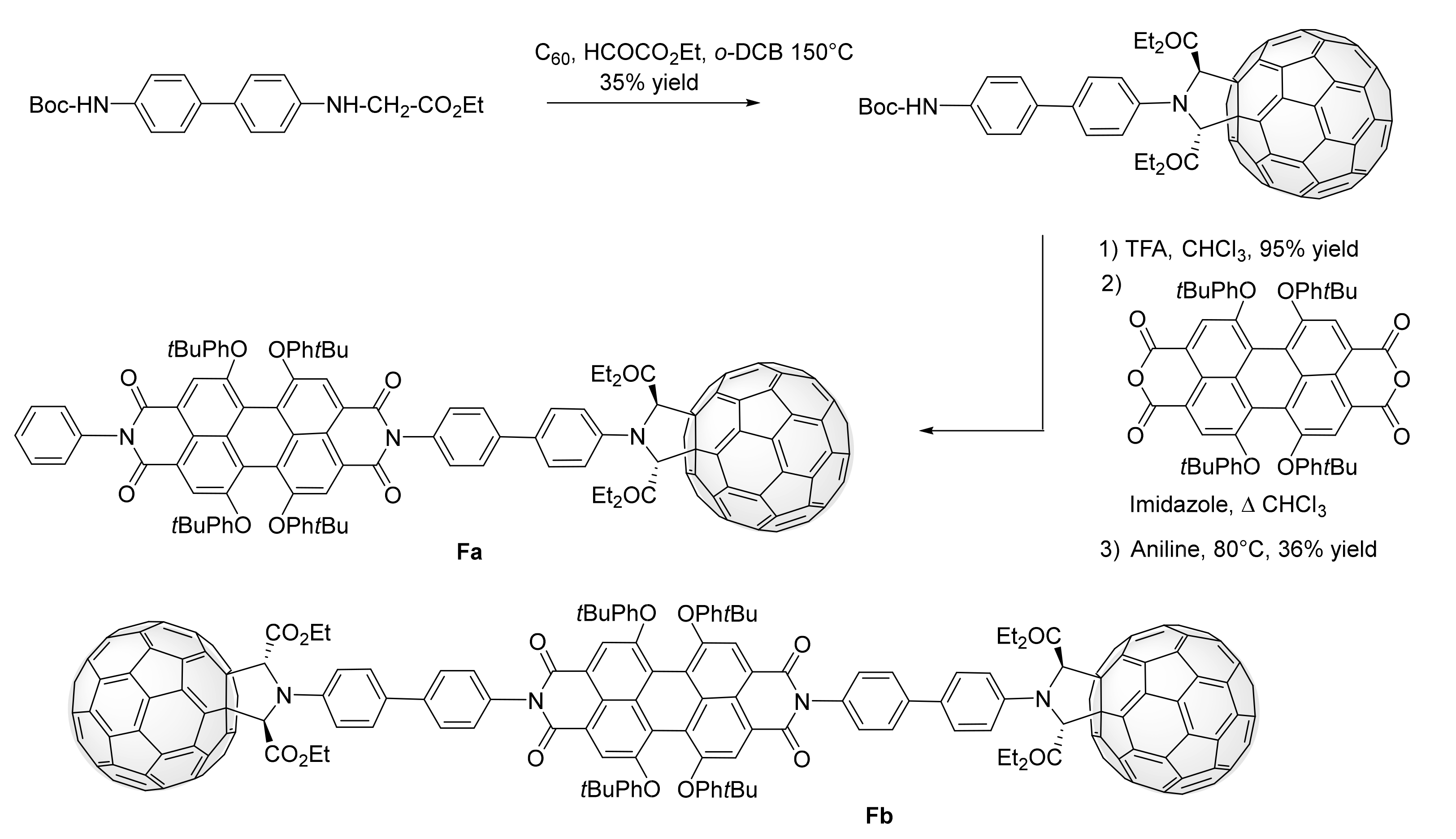
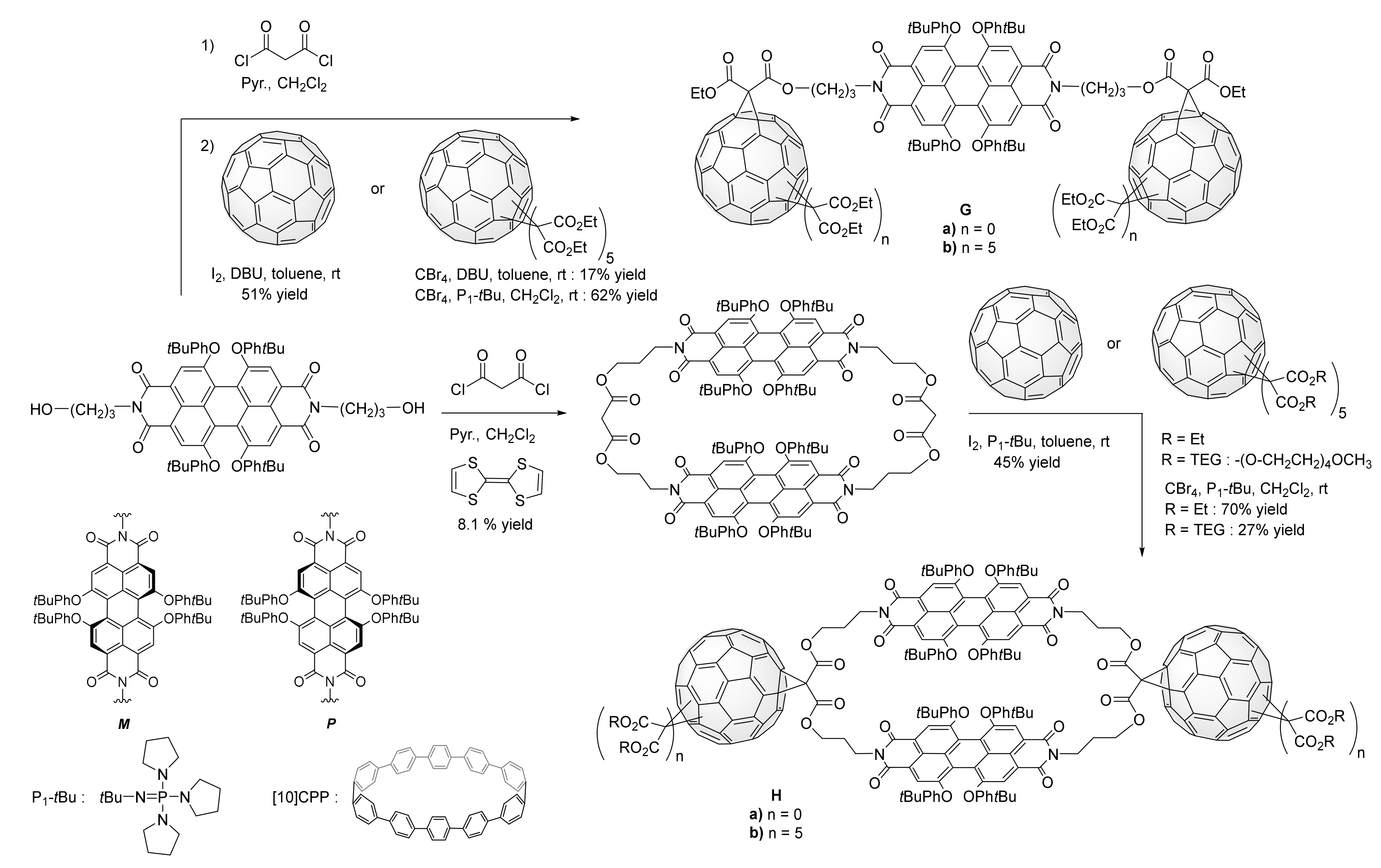
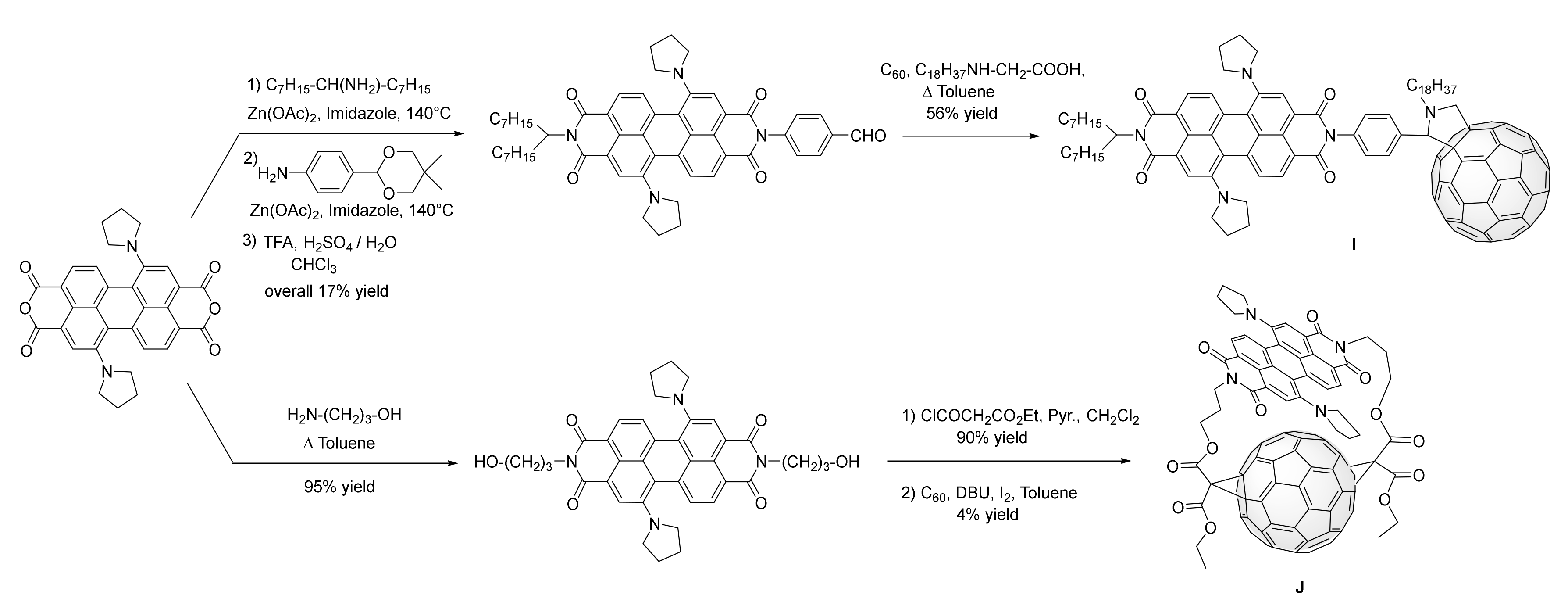
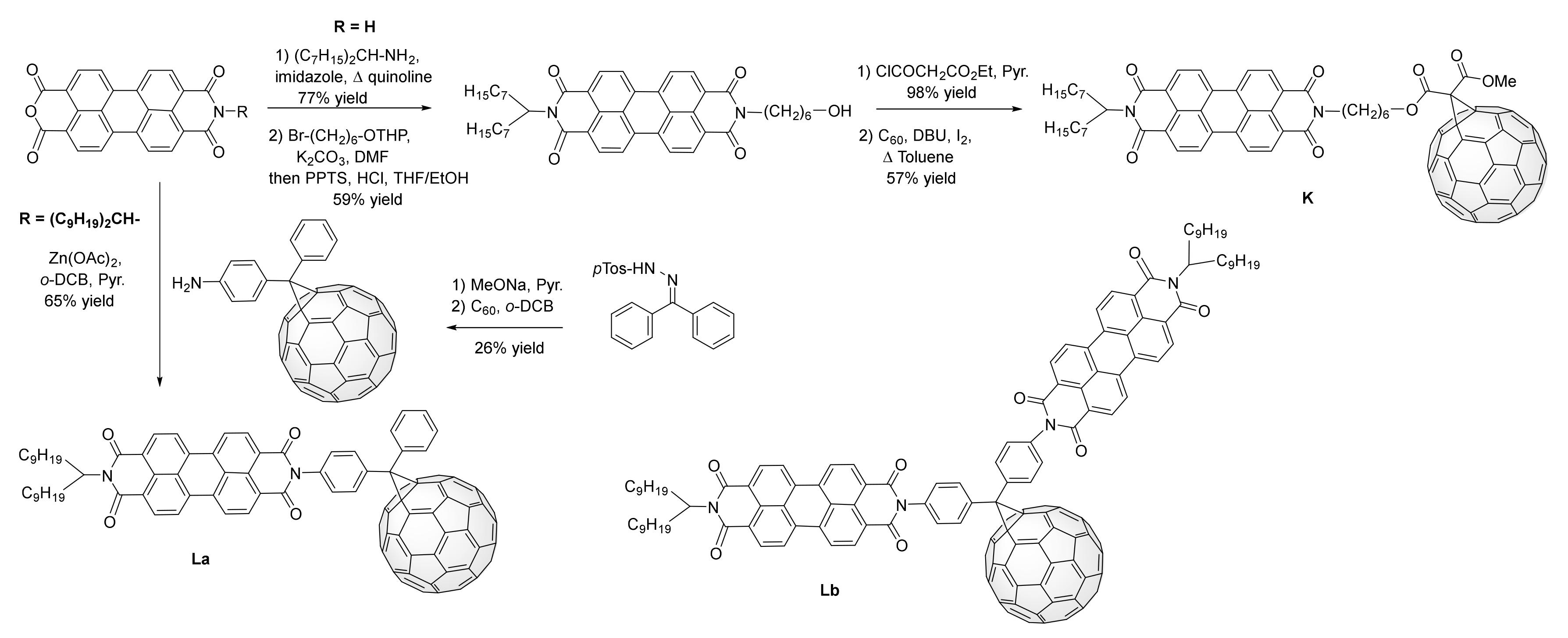
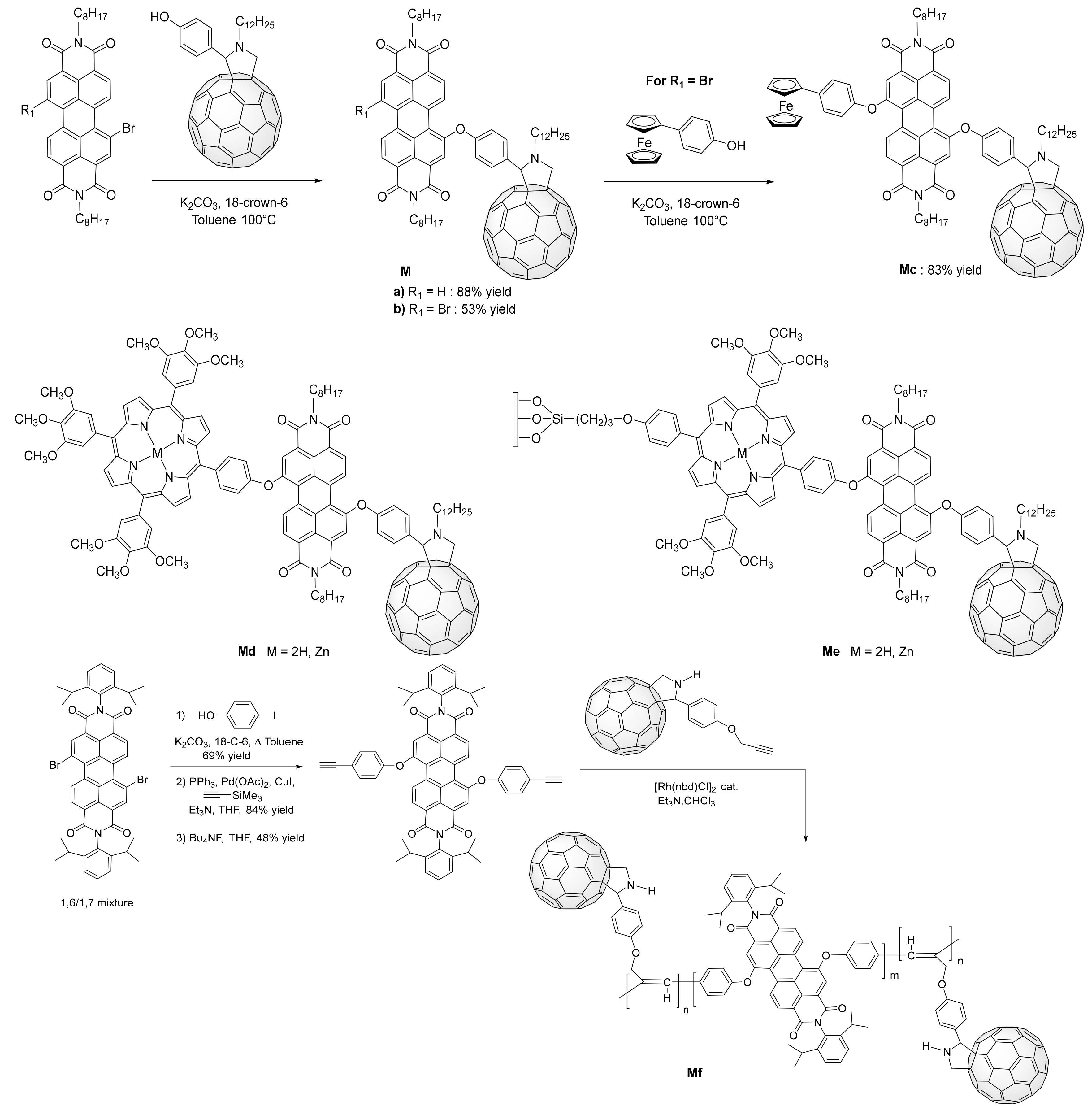
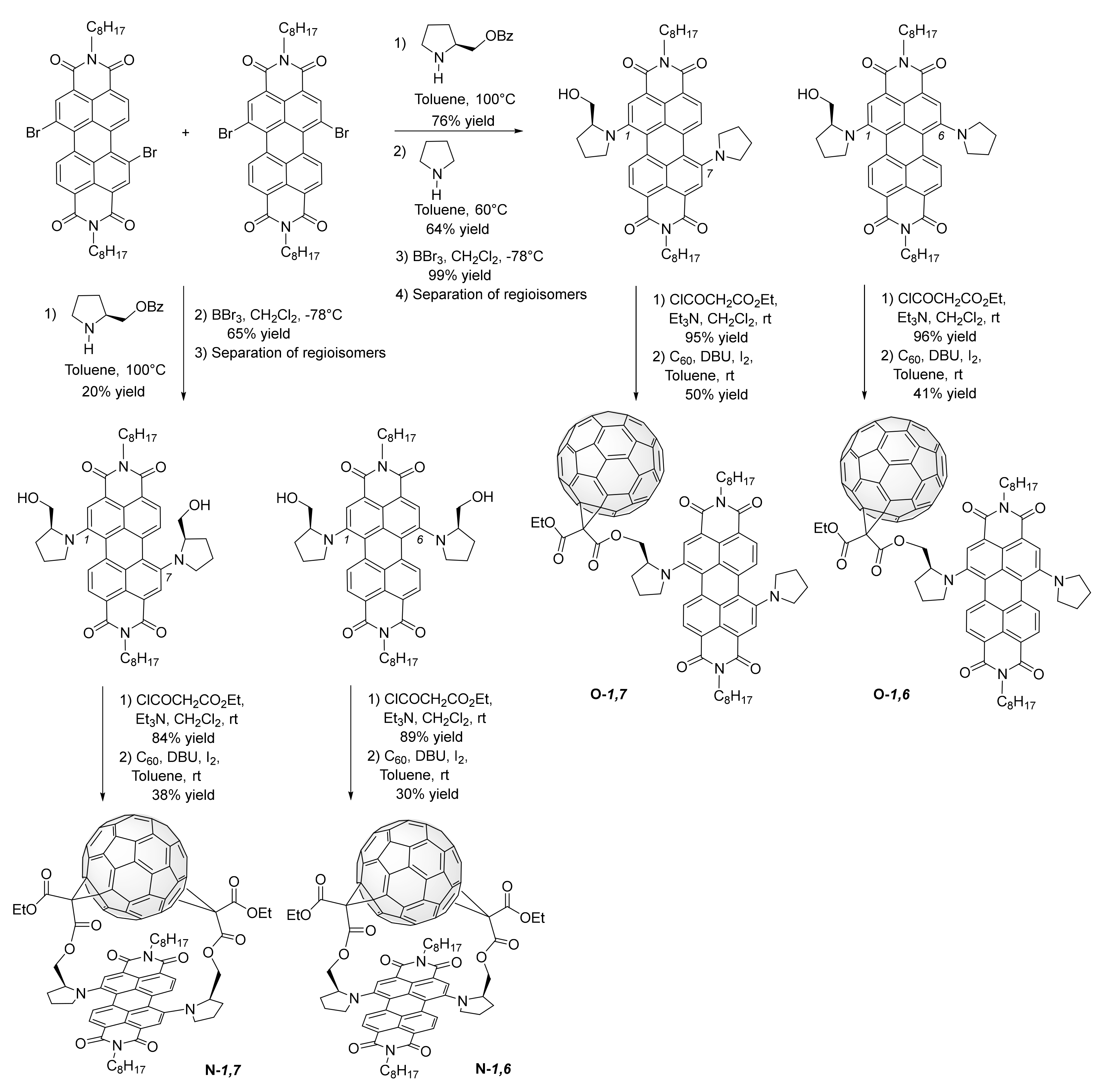
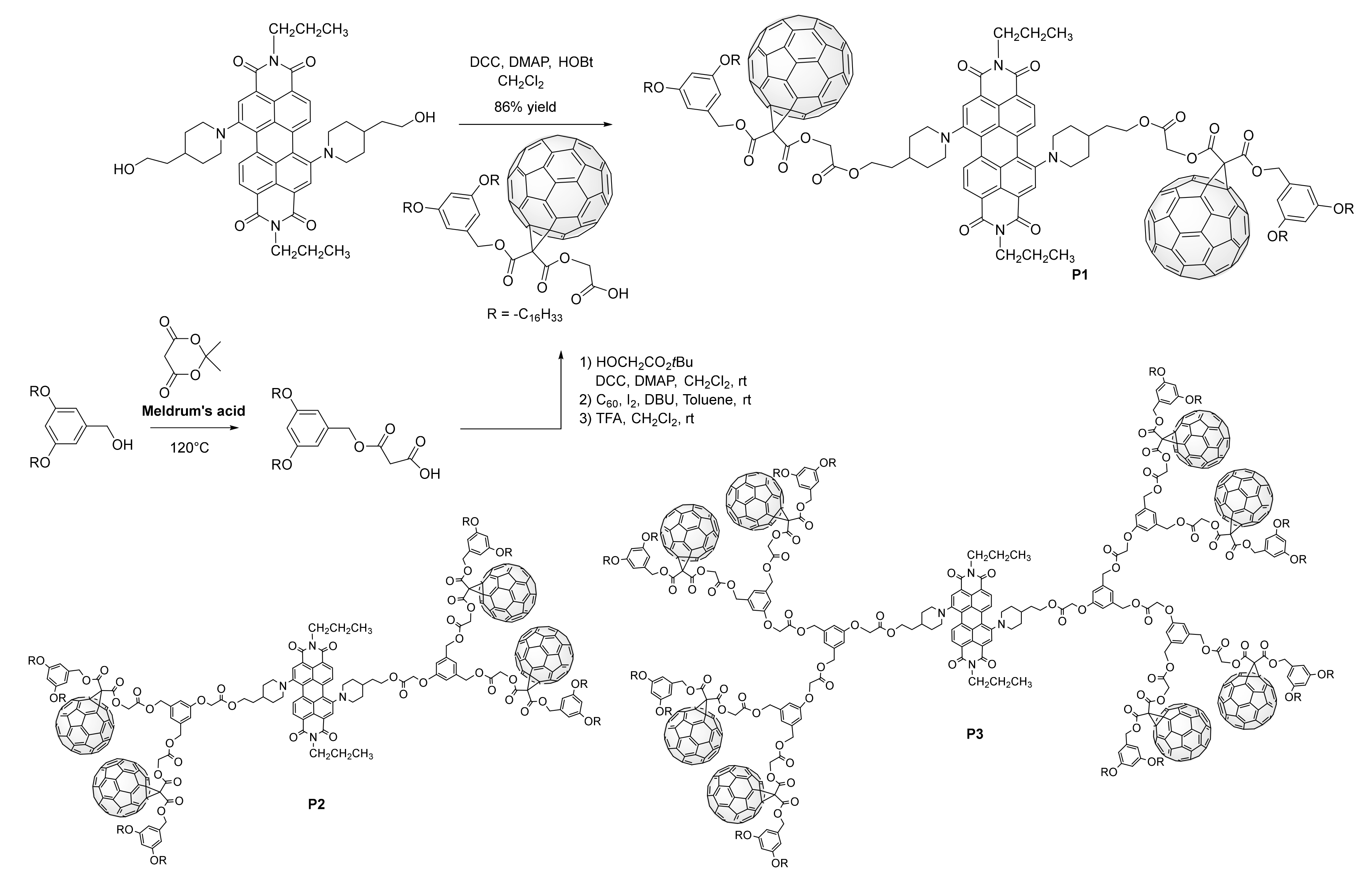
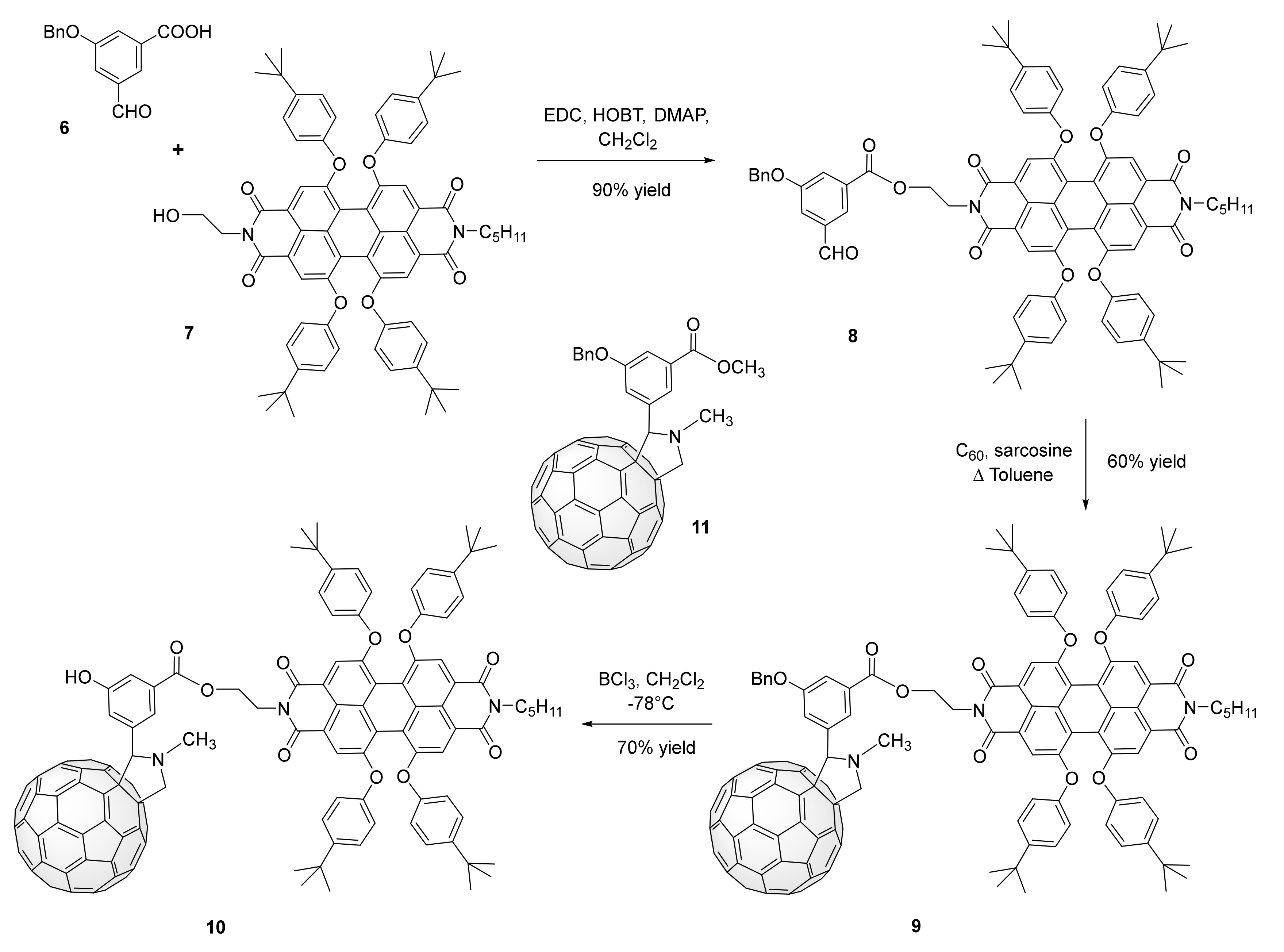
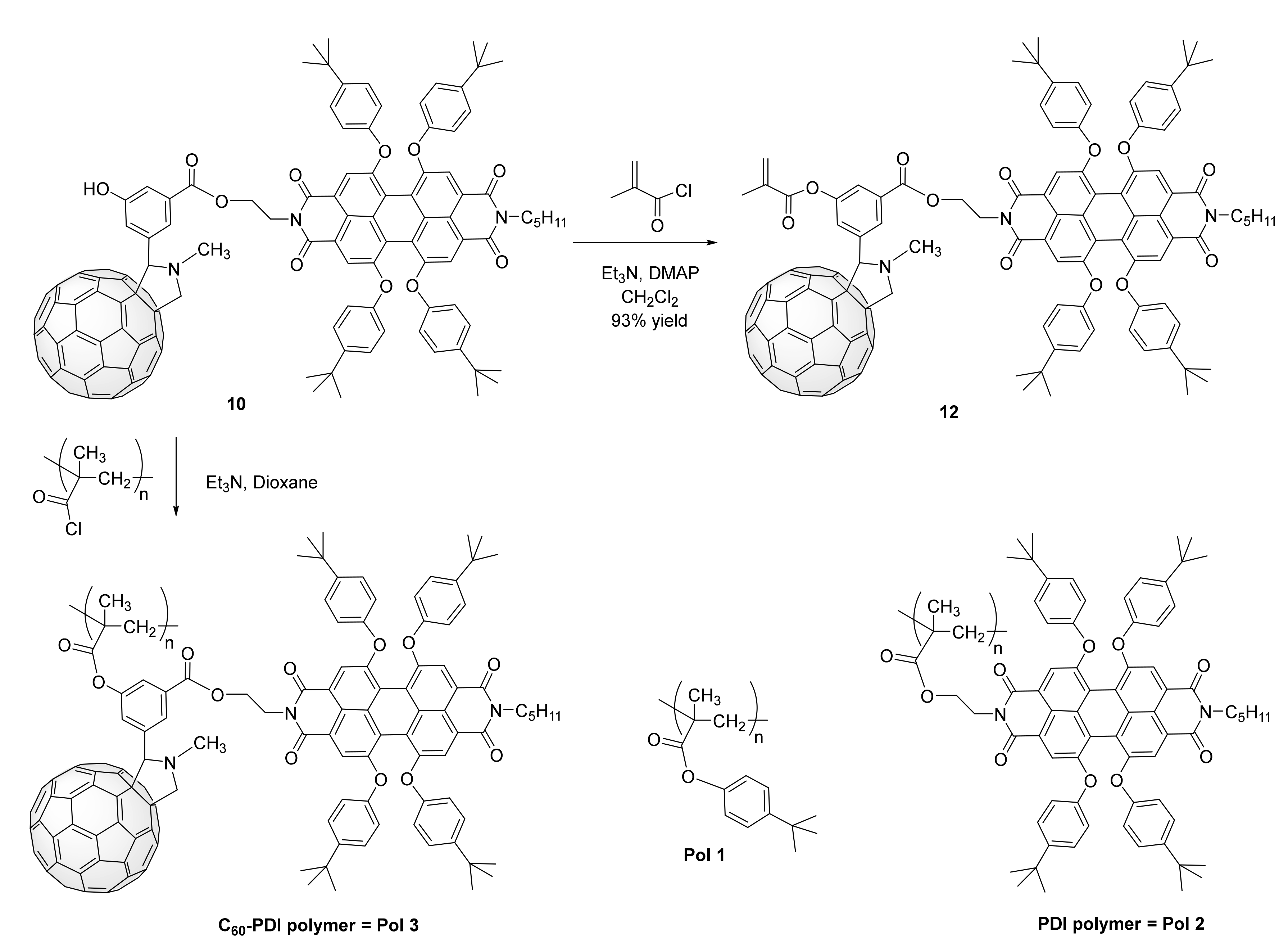
3. Synthesis of a Platform for New C60-PDI Assemblies
4. Conclusions
5. Experimental Procedures
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blankenship, R.E.; Tiede, D.M.; Barber, J.; Brudvig, G.W.; Fleming, G.; Ghirardi, M.; Gunner, M.R.; Junge, W.; Kramer, D.M.; Melis, A.; et al. Comparing photosynthetic and photovoltaic efficiencies and recognizing the potential for improvement. Science 2011, 332, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Niklas, J.; Beaupré, S.; Leclerc, M.; Xu, T.; Yu, L.; Sperlich, A.; Dyakonov, V.; Poluektov, O.G. Photoinduced dynamics of charge separation: From photosynthesis to polymer–fullerene bulk heterojunctions. J. Phys. Chem. B 2015, 119, 7407–7416. [Google Scholar] [CrossRef] [PubMed]
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Krätschmer, W.; Lamb, L.D.; Fostiropoulos, K.; Huffman, D.R. Solid C60: A new form of carbon. Nature 1990, 347, 354–358. [Google Scholar] [CrossRef]
- Kroto, H.W.; Allaf, A.W.; Balm, S.P. C60: Buckminsterfullerene. Chem. Rev. 1991, 91, 1213–1235. [Google Scholar] [CrossRef]
- Langa De La Puente, F.; Nierengarten, J.-F. Fullerenes: Principles and Applications: Edition 2; RSC Publishing: Cambridge, UK, 2011. [Google Scholar]
- Nierengarten, J.-F. Fullerenes and Other Carbon-Rich Nanostructures; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Echegoyen, L.; Echegoyen, L.E. Electrochemistry of fullerenes and their derivatives. Acc. Chem. Res. 1998, 31, 593–601. [Google Scholar] [CrossRef]
- Sariciftci, N.S.; Smilowitz, L.; Heeger, A.J.; Wudl, F. Photoinduced electron transfer from a conducting polymer to Buckminsterfullerene. Science 1992, 258, 1474–1476. [Google Scholar] [CrossRef]
- Sariciftci, N.S.; Braun, D.; Zhang, C.; Srdanov, V.I.; Heeger, A.J.; Stucky, G.; Wudl, F. Semiconducting polymer-buckminsterfullerene heterojunctions: Diodes, photodiodes, and photovoltaic cells. Appl. Phys. Lett. 1993, 62, 585–587. [Google Scholar] [CrossRef]
- Hummelen, J.C.; Knight, B.W.; LePeq, F.; Wudl, F.; Yao, J.; Wilkins, C.L. Preparation and characterization of fulleroid and methanofullerene derivatives. J. Org. Chem. 1995, 60, 532–538. [Google Scholar] [CrossRef]
- Dennler, G.; Scharber, M.C.; Brabec, C.J. Polymer-fullerene bulk-heterojunction solar cells. Adv. Mater. 2009, 21, 1323–1338. [Google Scholar] [CrossRef]
- He, Y.; Li, Y. Fullerene derivative acceptors for high performance polymer solar cells. Phys. Chem. Chem. Phys. 2011, 13, 1970–1983. [Google Scholar] [CrossRef]
- Cheng, P.; Zhan, X. Stability of organic solar cells: Challenges and strategies. Chem. Soc. Rev. 2016, 45, 2544–2582. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Q. Recent progress in non-fullerene small molecule acceptors in organic solar cells (OSCs). J. Mater. Chem. C 2017, 5, 1275–1302. [Google Scholar] [CrossRef]
- Hou, J.; Inganäs, O.; Friend, R.H.; Gao, F. Organic solar cells based on non-fullerene acceptors. Nat. Mater. 2018, 17, 119–128. [Google Scholar] [CrossRef]
- Yan, C.; Barlow, S.; Wang, Z.; Yan, H.; Jen, A.K.Y.; Marder, S.R.; Zhan, X. Non-fullerene acceptors for organic solar cells. Nat. Rev. Mater. 2018, 3, 18003. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, J.; Chow, P.C.Y.; Jiang, K.; Zhang, J.; Zhu, Z.; Zhang, J.; Huang, F.; Yan, H. Nonfullerene Acceptor Molecules for Bulk Heterojunction Organic Solar Cells. Chem. Rev. 2018, 118, 3447–3507. [Google Scholar] [CrossRef]
- Wadsworth, A.; Moser, M.; Marks, A.; Little, M.S.; Gasparini, N.; Brabec, C.J.; Baran, D.; McCulloch, I. Critical review of the molecular design progress in non-fullerene electron acceptors towards commercially viable organic solar cells. Chem. Soc. Rev. 2019, 48, 1596–1625. [Google Scholar] [CrossRef]
- Zhang, J.; Tan, H.S.; Guo, X.; Facchetti, A.; Yan, H. Material insights and challenges for non-fullerene organic solar cells based on small molecular acceptors. Nat. Energy 2018, 3, 720–731. [Google Scholar] [CrossRef]
- Huber, R. A structural basis of light energy and electron transfer in biology (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1989, 28, 848–869. [Google Scholar] [CrossRef]
- Wasielewski, M.R. Photoinduced electron transfer in supramolecular systems for artificial photosynthesis. Chem. Rev. 1992, 92, 435–461. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.; Moore, A. Mimicking photosynthetic solar energy transduction. Acc. Chem. Res. 2001, 34, 40–48. [Google Scholar] [CrossRef]
- Martín, N.; Sánchez, L.; Illescas, B.; Pérez, I. C60-Based electroactive organofullerenes. Chem. Rev. 1998, 98, 2527–2548. [Google Scholar] [CrossRef]
- Huang, C.; Barlow, S.; Marder, S.R. Perylene-3,4,9,10-tetracarboxylic Acid Diimides: Synthesis, Physical Properties, and Use in Organic Electronics. J. Org. Chem. 2011, 76, 2386–2407. [Google Scholar] [CrossRef]
- Nowak-Król, A.; Shoyama, K.; Stolte, M.; Würthner, F. Naphthalene and perylene diimides—better alternatives to fullerenes for organic electronics? Chem. Commun. 2018, 54, 13763–13772. [Google Scholar] [CrossRef]
- Li, C.; Wonneberger, H. Perylene Imides for Organic Photovoltaics: Yesterday, Today, and Tomorrow. Adv. Mater. 2012, 24, 613–636. [Google Scholar] [CrossRef]
- Fernández-Lázaro, F.; Zink-Lorre, N.; Sastre Santos, A. Perylenediimides as non-fullerene acceptors in bulk-heterojunction solar cells (BHJSCs). J. Mater. Chem. A 2016, 4, 9336–9346. [Google Scholar] [CrossRef]
- Shi, Q.; Wu, J.; Wu, X.; Peng, A.; Huang, H. Perylene Diimide-Based Conjugated Polymers for All-Polymer Solar Cells. Chem. Eur. J. 2020, 26, 12510–12522. [Google Scholar] [CrossRef]
- Sharma, V.; Koenig, J.D.B.; Welch, G.C. Perylene diimide based non-fullerene acceptors: Top performers and an emerging class featuring N-annulation. J. Mater. Chem. A 2021, 9, 6775–6789. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, Y.; Zhang, Q.; Gao, X. Non-fullerene small molecule acceptors based on perylene diimides. J. Mater. Chem. A 2016, 4, 17604–17622. [Google Scholar] [CrossRef]
- Rybtchinski, B.; Sinks, L.E.; Wasielewski, M.R. Combining Light-Harvesting and Charge Separation in a Self-Assembled Artificial Photosynthetic System Based on Perylenediimide Chromophores. J. Amer. Chem. Soc. 2004, 126, 12268–12269. [Google Scholar] [CrossRef]
- Nowak-Król, A.; Würthner, F. Progress in the synthesis of perylene bisimide dyes. Org. Chem. Front. 2019, 6, 1272–1318. [Google Scholar] [CrossRef]
- Bingel, C. Cyclopropanierung von fullerenen. Chem. Ber. 1993, 126, 1957–1959. [Google Scholar] [CrossRef]
- Maggini, M.; Scorrano, G.; Prato, M. Addition of azomethine ylides to C60: Synthesis, characterization, and functionalization of fullerene pyrrolidines. J. Am. Chem. Soc. 1993, 115, 9798–9799. [Google Scholar] [CrossRef]
- Nierengarten, J.-F.; Gramlich, V.; Cardullo, F.; Diederich, F. Regio- and diastereoselective bisfunctionalization of C60 and enantioselective synthesis of a C60 derivative with a chiral addition pattern. Angew. Chem. Int. Ed. Engl. 1996, 35, 2101–2103. [Google Scholar] [CrossRef]
- Camps, X.; Hirsch, A. Efficient cyclopropanation of C60 starting from malonates. J. Chem. Soc. Perkin Trans. 1 1997, 11, 1595–1596. [Google Scholar] [CrossRef]
- Hua, J.; Meng, F.; Ding, F.; Li, F.; Tian, H. Novel soluble and thermally-stable fullerene dyad containing perylene. J. Mater. Chem. 2004, 14, 1849–1853. [Google Scholar] [CrossRef]
- Hua, J.; Meng, F.; Ding, F.; Tian, H. Novel soluble and thermally stable perylene dye with two [60]fullerene units. Chem. Lett. 2004, 33, 432–433. [Google Scholar] [CrossRef]
- Wang, N.; Li, Y.; He, X.; Gan, H.; Li, Y.; Huang, C.; Xu, X.; Xiao, J.; Wang, S.; Liu, H.; et al. Synthesis and characterization of a novel electrical and optical-active triads containing fullerene and perylenebisimide units. Tetrahedron 2006, 62, 1216–1222. [Google Scholar] [CrossRef]
- Fortage, J.; Séverac, M.; Houarner-Rassin, C.; Pellegrin, Y.; Blart, E.; Odobel, F. Synthesis of new perylene imide dyes and their photovoltaic performances in nanocrystalline TiO2 dye-sensitized solar cells. J. Photochem. Photobiol. A 2008, 197, 156–169. [Google Scholar] [CrossRef]
- Rybtchinski, B.; Sinks, L.E.; Wasielewski, M.R. Photoinduced Electron Transfer in Self-Assembled Dimers of 3-Fold Symmetric Donor−Acceptor Molecules Based on Perylene-3,4:9,10-bis(dicarboximide). J. Phys. Chem. A 2004, 108, 7497–7505. [Google Scholar] [CrossRef]
- Leroy-Lhez, S.; Baffreau, J.; Perrin, L.; Levillain, E.; Allain, M.; Blesa, M.-J.; Hudhomme, P. Tetrathiafulvalene in a Perylene-3,4:9,10-bis(dicarboximide)-Based Dyad: A New Reversible Fluorescence-Redox Dependent Molecular System. J. Org. Chem. 2005, 70, 6313–6320. [Google Scholar] [CrossRef]
- Leroy-Lhez, S.; Perrin, L.; Baffreau, J.; Hudhomme, P. Perylenediimide derivatives in new donor–acceptor dyads. C. R. Chim. 2006, 9, 240–246. [Google Scholar] [CrossRef]
- Baffreau, J.; Perrin, L.; Leroy-Lhez, S.; Hudhomme, P. Perylene-3,4:9,10-bis(dicarboximide) linked to [60]fullerene as a light-harvesting antenna. Tet. Lett. 2005, 46, 4599–4603. [Google Scholar] [CrossRef]
- Baffreau, J.; Leroy-Lhez, S.; Anh, N.V.; Williams, R.M.; Hudhomme, P. Fullerene C60-perylene-3,4:9,10-bis(dicarboximide) light-harvesting dyads: Spacer-length and bay-substituent effects on intramolecular singlet and triplet energy transfer. Chem. Eur. J. 2008, 14, 4974–4992. [Google Scholar] [CrossRef]
- Baffreau, J.; Leroy-Lhez, S.; Gallego-Planas, N.; Hudhomme, P. Correlation of electrochemical and theoretical parameters in perylenediimide-[60]fullerene dyads. J. Mol. Struct. THEOCHEM 2007, 815, 145–150. [Google Scholar] [CrossRef]
- Hudhomme, P.; Williams, R.M. Handbook of Carbon Nano Materials; Electron transfer and applications; D’Souza, F., Kadish, D.M., Eds.; World Scientific Publishing Company: Singapore, 2011; Volume 2. [Google Scholar]
- Baffreau, J.; Leroy-Lhez, S.; Hudhomme, P.; Groeneveld, M.M.; van Stokkum, I.H.; Williams, R.M. Superabsorbing fullerenes: Spectral and kinetic characterization of photoinduced interactions in perylenediimide-fullerene-C60 dyads. J. Phys. Chem. A 2006, 110, 13123–13125. [Google Scholar] [CrossRef]
- Baffreau, J.; Leroy-Lhez, S.; Derbal, H.; Inigo, A.R.; Nunzi, J.M.; Groeneveld, M.M.; Williams, R.M.; Hudhomme, P. Light-harvesting fullerenes for organic solar cells. Eur. Phys. J. Appl. Phys. 2006, 36, 301–305. [Google Scholar] [CrossRef]
- Qu, J.; Kohl, C.; Pottek, M.; Müllen, K. Ionic Perylenetetracarboxdiimides: Highly Fluorescent and Water-Soluble Dyes for Biolabeling. Angew. Chem. Int. Ed. 2004, 43, 1528–1531. [Google Scholar] [CrossRef]
- Pla, S.; Martín-Gomis, L.; Ohkubo, K.; Fukuzumi, S.; Fernández-Lázaro, F.; Sastre Santos, A. Macrocyclic dyads based on C60 and perylenediimides connected by click chemistry. Asian J. Org. Chem. 2014, 3, 185–197. [Google Scholar] [CrossRef]
- Martín-Gomis, L.; Peralta-Ruiz, F.; Thomas, M.B.; Fernández-Lázaro, F.; D’Souza, F.; Sastre Santos, A. Multichromophoric perylenediimide-silicon phthalocyanine-C60 system as an artificial photosynthetic analogue. Chem. Eur. J. 2017, 23, 3863–3874. [Google Scholar] [CrossRef]
- Martín-Gomis, L.; Díaz-Puertas, R.; Seetharaman, S.; Karr, P.A.; Fernández-Lázaro, F.; D’Souza, F.; Sastre Santos, Á. Distance Matters: Effect of the Spacer Length on the Photophysical Properties of Multimodular Perylenediimide–Silicon Phthalocyanine–Fullerene Triads. Chem. Eur. J. 2020, 26, 4822–4832. [Google Scholar] [CrossRef]
- Zhu, S.-E.; Liu, K.-Q.; Wang, X.-F.; Xia, A.-D.; Wang, G.-W. Synthesis and properties of axially symmetrical rigid visible light-harvesting systems containing [60]fullerene and perylenebisimide. J. Org. Chem. 2016, 81, 12223–12231. [Google Scholar] [CrossRef]
- Solymosi, I.; Krishna, S.; Nuin, E.; Maid, H.; Scholz, B.; Guldi, D.M.; Pérez-Ojeda, M.E.; Hirsch, A. Diastereoselective formation of homochiral flexible perylene bisimide cyclophanes and their hybrids with fullerenes. Chem. Sci. 2021, 12, 15491–15502. [Google Scholar] [CrossRef]
- Solymosi, I.; Sabin, J.; Maid, H.; Friedrich, L.; Nuin, E.; Perez-Ojeda, M.E.; Hirsch, A. Bis-pseudorotaxane Formation of Perylene Bisimide-Linked [60]Fullerene Dumbbell-Like Molecules with [10]Cycloparaphenylene. Org. Mater. 2022, 4, 73–85. [Google Scholar] [CrossRef]
- Pérez-Ojeda, M.E.; Wabra, I.; Böttcher, C.; Hirsch, A. Fullerene Building Blocks with Tailor-Made Solubility and New Insights into Their Hierarchical Self-Assembly. Chem. Eur. J. 2018, 24, 14088–14100. [Google Scholar] [CrossRef]
- Maxouti, K.L.; Hirsch, A. Sequential Tether-Directed Synthesis of Pentakis-Adducts of C60 with a Mixed [3:2] Octahedral Addition Pattern. Eur. J. Org. Chem. 2018, 2018, 2579–2586. [Google Scholar] [CrossRef]
- Würthner, F. Bay-substituted perylene bisimides: Twisted fluorophores for supramolecular chemistry. Pure Appl. Chem. 2006, 78, 2341–2349. [Google Scholar] [CrossRef]
- Schönamsgruber, J.; Hirsch, A. Benz-Bisimidazole-Bridged Perylenes—Linearly Expanded Chromophores. Eur. J. Org. Chem. 2015, 2015, 2167–2174. [Google Scholar] [CrossRef]
- Kim, W.; Nowak-Król, A.; Hong, Y.; Schlosser, F.; Würthner, F.; Kim, D. Solvent-Modulated Charge-Transfer Resonance Enhancement in the Excimer State of a Bay-Substituted Perylene Bisimide Cyclophane. J. Phys. Chem. Lett. 2019, 10, 1919–1927. [Google Scholar] [CrossRef]
- Shibano, Y.; Umeyama, T.; Matano, Y.; Tkachenko, N.V.; Lemmetyinen, H.; Imahori, H. Synthesis and photophysical properties of electron-rich perylenediimide-fullerene dyad. Org. Lett. 2006, 8, 4425–4428. [Google Scholar] [CrossRef]
- Shibano, Y.; Umeyama, T.; Matano, Y.; Tkachenko, N.V.; Lemmetyinen, H.; Araki, Y.; Ito, O.; Imahori, H. Large reorganization energy of pyrrolidine-substituted perylenediimide in electron transfer. J. Phys. Chem. C 2007, 111, 6133–6142. [Google Scholar] [CrossRef]
- Pla, S.; Martín-Gomis, L.; Fernández-Lázaro, F.; Sastre Santos, A.; Niemi, M.; Lemmetyinen, H.; Tkachenko, N.V. Charge separation and charge recombination photophysical studies in a series of perylene-C60 linear and cyclic dyads. Phys. Chem. Chem. Phys. 2016, 18, 3598–3605. [Google Scholar] [CrossRef] [PubMed]
- Langhals, H. Cyclic carboxylic imide structures as structure elements of high stability. Novel developments in perylene dye chemistry. Heterocycles 1995, 40, 477–500. [Google Scholar] [CrossRef]
- Hofmann, C.C.; Lindner, S.M.; Ruppert, M.; Hirsch, A.; Haque, S.A.; Thelakkat, M.; Köhler, J. Mutual interplay of light harvesting and triplet sensitizing in a perylene bisimide antenna−fullerene dyad. J. Phys. Chem. B 2010, 114, 9148–9156. [Google Scholar] [CrossRef] [PubMed]
- Gómez, R.; Segura, J.L.; Martín, N. Highly efficient light-harvesting organofullerenes. Org. Lett. 2005, 7, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Bamford, W.R.; Stevens, T.S. The decomposition of toluene-p-sulphonylhydrazones by alkali. J. Chem. Soc. 1952, 28, 4735–4740. [Google Scholar] [CrossRef]
- Wienk, M.M.; Kroon, J.M.; Verhees, W.J.H.; Knol, J.; Hummelen, J.C.; van Hal, P.A.; Janssen, R.A.J. Efficient methano[70]fullerene/MDMO-PPV bulk heterojunction photovoltaic cells. Angew. Chem. Int. Ed. 2003, 42, 3371–3375. [Google Scholar] [CrossRef]
- Xiao, S.; Li, Y.; Li, Y.; Zhuang, J.; Wang, N.; Liu, H.; Ning, B.; Liu, Y.; Lu, F.; Fan, L.; et al. [60]Fullerene-based molecular triads with expanded absorptions in the visible region: Synthesis and photovoltaic properties. J. Phys. Chem. B 2004, 108, 16677–16685. [Google Scholar] [CrossRef]
- Li, Y.; Wang, N.; He, X.; Wang, S.; Liu, H.; Li, Y.; Li, X.; Zhuang, J.; Zhu, D. Synthesis and characterization of ferrocene-perylenetetracarboxylic diimide–fullerene triad. Tetrahedron 2005, 61, 1563–1569. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Li, H.; Shi, Q.; Fu, H.; Yao, J. N-type cascade electron transfer along an oxidative gradient. Chem. Commun. 2009, 45, 6955–6957. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Liu, H.; Wang, S.; Wang, N.; Zhuang, J.; Li, X.; He, X.; Zhu, D. Self-assembled monolayers of porphyrin–perylenetetracarboxylic diimide–[60] fullerene on indium tin oxide electrodes: Enhancement of light harvesting in the visible light region. Nanotechnology 2005, 16, 1899–1904. [Google Scholar] [CrossRef]
- Würthner, F.; Stepanenko, V.; Chen, Z.; Saha-Möller, C.R.; Kocher, N.; Stalke, D. Preparation and characterization of regioisomerically pure 1,7-disubstituted perylene bisimide dyes. J. Org. Chem. 2004, 69, 7933–7939. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, N.; Li, Y.; Liu, H.; Li, Y.; Xiao, J.; Xu, X.; Huang, C.; Cui, S.; Zhu, D. A new class of conjugated polyacetylenes having perylene bisimide units and pendant fullerene or porphyrin groups. Macromolecules 2005, 38, 4880–4887. [Google Scholar] [CrossRef]
- Lu, F.; Xiao, S.; Li, Y.; Liu, H.; Li, H.; Zhuang, J.; Liu, Y.; Wang, N.; He, X.; Li, X.; et al. Synthesis and chemical properties of conjugated polyacetylenes having pendant fullerene and/or porphyrin units. Macromolecules 2004, 37, 7444–7450. [Google Scholar] [CrossRef]
- Dubey, R.K.; Niemi, M.; Kaunisto, K.; Efimov, A.; Tkachenko, N.V.; Lemmetyinen, H. Direct evidence of significantly different chemical behavior and excited-state dynamics of 1,7- and 1,6-regioisomers of pyrrolidinyl-substituted perylene diimide. Chem. Eur. J. 2013, 19, 6791–6806. [Google Scholar] [CrossRef]
- Kaunisto, K.M.; Vivo, P.; Dubey, R.K.; Chukharev, V.I.; Efimov, A.; Tkachenko, N.V.; Lemmetyinen, H.J. Charge-transfer dynamics in poly(3-hexylthiophene):perylenediimide-C60 blend films studied by ultrafast transient absorption. J. Phys. Chem. C 2014, 118, 10625–10630. [Google Scholar] [CrossRef]
- Hahn, U.; Nierengarten, J.-F.; Delavaux-Nicot, B.; Monti, F.; Chiorboli, C.; Armaroli, N. Fullerodendrimers with a perylenediimide core. New J. Chem. 2011, 35, 2234–2244. [Google Scholar] [CrossRef]
- Serin, J.M.; Brousmiche, D.W.; Fréchet, J.M.J. A FRET-based ultraviolet to near-infrared frequency converter. J. Amer. Chem. Soc. 2002, 124, 11848–11849. [Google Scholar] [CrossRef]
- Felder, D.; Nierengarten, H.; Gisselbrecht, J.-P.; Boudon, C.; Leize, E.; Nicoud, J.-F.; Gross, M.; Van Dorsselaer, A.; Nierengarten, J.-F. Fullerodendrons: Synthesis, electrochemistry and reduction in the electrospray source for mass spectrometry analysis. New J. Chem. 2000, 24, 687–695. [Google Scholar] [CrossRef]
- Hahn, U.; Hosomizu, K.; Imahori, H.; Nierengarten, J.-F. Synthesis of dendritic branches with peripheral fullerene subunits. Eur. J. Org. Chem. 2006, 2006, 85–91. [Google Scholar] [CrossRef]
- Chamberlain, T.W.; Davies, E.S.; Khlobystov, A.N.; Champness, N.R. Multi-electron-acceptor dyad and triad systems based on perylene bisimides and fullerenes. Chem. Eur. J. 2011, 17, 3759–3767. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J. Visible light-harvesting perylenebisimide–fullerene (C60) dyads with bidirectional “ping-pong” energy transfer as triplet photosensitizers for photooxidation of 1,5-dihydroxynaphthalene. Chem. Commun. 2012, 48, 3751–3753. [Google Scholar] [CrossRef] [PubMed]
- Rocard, L.; Hudhomme, P. Recent Developments in the Suzuki–Miyaura Reaction Using Nitroarenes as Electrophilic Coupling Reagents. Catalysts 2019, 9, 213. [Google Scholar] [CrossRef]
- Rocard, L.; Hatych, D.; Chartier, T.; Cauchy, T.; Hudhomme, P. Original Suzuki–Miyaura Coupling Using Nitro Derivatives for the Synthesis of Perylenediimide-Based Multimers. Eur. J. Org. Chem. 2019, 2019, 7635–7643. [Google Scholar] [CrossRef]
- Rocard, L.; Goujon, A.; Hudhomme, P. Nitro-Perylenediimide: An Emerging Building Block for the Synthesis of Functional Organic Materials. Molecules 2020, 25, 1402. [Google Scholar] [CrossRef] [PubMed]
- Hruzd, M.; Rocard, L.; Goujon, A.; Allain, M.; Cauchy, T.; Hudhomme, P. Desymmetrization of Perylenediimide Bay Regions Using Selective Suzuki–Miyaura Reactions from Dinitro Substituted Derivatives. Chem. Eur. J. 2020, 26, 15881–15891. [Google Scholar] [CrossRef] [PubMed]
- El-Berjawi, R.; Hudhomme, P. Synthesis of a perylenediimide-fullerene C60 dyad: A simple use of a nitro leaving group for a Suzuki-Miyaura coupling reaction. Dye. Pigm. 2018, 159, 551–556. [Google Scholar] [CrossRef]
- Uno, M.; Ban, H.S.; Nabeyama, W.; Nakamura, H. de novo Design and synthesis of N-benzylanilines as new candidates for VEGFR tyrosinekinase inhibitors. Org. Biomol. Chem. 2008, 6, 979–981. [Google Scholar] [CrossRef]
- Coco, S.; Cordovilla, C.; Donnio, B.; Espinet, P.; García-Casas, M.J.; Guillon, D. Self-organization of dendritic supermolecules, based on isocyanide–gold(I), –copper(I), –palladium(II), and –platinum(II) complexes, into micellar cubic mesophases. Chem. Eur. J. 2008, 14, 3544–3552. [Google Scholar] [CrossRef]
- Zhao, H.; Thurkauf, A. A practical and convenient synthesis of methyl 5-formyl-3-methoxybenzoate. Synth. Commun. 2001, 31, 1921–1926. [Google Scholar] [CrossRef]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. Int. Ed. Engl. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Diacon, A.; Derue, L.; Lecourtier, C.; Dautel, O.; Wantz, G.; Hudhomme, P. Cross-linkable azido C60-fullerene derivatives for efficient thermal stabilization of polymer bulk-heterojunction solar cells. J. Mater. Chem. C 2014, 2, 7163–7167. [Google Scholar] [CrossRef]
- Boovanahalli, S.K.; Kim, D.W.; Chi, D.Y. Application of ionic liquid halide nucleophilicity for the cleavage of ethers: A green protocol for the regeneration of phenols from ethers. J. Org. Chem. 2004, 69, 3340–3344. [Google Scholar] [CrossRef]
- Bieliauskas, A.V.; Weerasinghe, S.V.W.; Pflum, M.K.H. Structural requirements of HDAC inhibitors: SAHA analogs functionalized adjacent to the hydroxamic acid. Bioorg. Med. Chem. Lett. 2007, 17, 2216–2219. [Google Scholar] [CrossRef]
- Haberkorn, N.; Kim, S.; Kim, K.-S.; Sommer, M.; Thelakkat, M.; Sohn, B.-H.; Theato, P. Template-Assisted Fabrication of Highly Ordered Interpenetrating Polymeric Donor/Acceptor Nanostructures for Photovoltaic Applications. Macromol. Chem. Phys. 2011, 212, 2142–2150. [Google Scholar] [CrossRef]
- Lindner, S.M.; Thelakkat, M. Nanostructures of n-Type Organic Semiconductor in a p-Type Matrix via Self-Assembly of Block Copolymers. Macromolecules 2004, 37, 8832–8835. [Google Scholar] [CrossRef]
- Sommer, M.; Thelakkat, M. Synthesis, characterization and application of donor-acceptor block copolymers in nanostructured bulk heterojunction solar cells. Eur. Phys. J. Appl. Phys. 2006, 36, 245–249. [Google Scholar] [CrossRef]
- Sommer, M.; Lindner, S.M.; Thelakkat, M. Microphase-Separated Donor–Acceptor Diblock Copolymers: Influence of HOMO Energy Levels and Morphology on Polymer Solar Cells. Adv. Funct. Mater. 2007, 17, 1493–1500. [Google Scholar] [CrossRef]
- Sommer, M.; Lang, A.S.; Thelakkat, M. Crystalline-crystalline donor-acceptor block copolymers. Angew. Chem. Int. Ed. Engl. 2008, 47, 7901–7904. [Google Scholar] [CrossRef]
- Hüttner, S.; Sommer, M.; Thelakkat, M. n-type organic field effect transistors from perylene bisimide block copolymers and homopolymers. Appl. Phys. Lett. 2008, 92, 093302. [Google Scholar] [CrossRef]
- Huettner, S.; Sommer, M.; Hodgkiss, J.; Kohn, P.; Thurn-Albrecht, T.; Friend, R.H.; Steiner, U.; Thelakkat, M. Tunable Charge Transport Using Supramolecular Self-Assembly of Nanostructured Crystalline Block Copolymers. ACS Nano 2011, 5, 3506–3515. [Google Scholar] [CrossRef] [PubMed]
- Yuen, J.D.; Pozdin, V.A.; Young, A.T.; Turner, B.L.; Giles, I.D.; Naciri, J.; Trammell, S.A.; Charles, P.T.; Stenger, D.A.; Daniele, M.A. Perylene-diimide-based n-type semiconductors with enhanced air and temperature stable photoconductor and transistor properties. Dye. Pigm. 2020, 174, 108014. [Google Scholar] [CrossRef]
- Aoki, H.; Takahashi, T.; Tamai, Y.; Sekine, R.; Aoki, S.; Tani, K.; Ito, S. Poly(methacrylate)s labeled by perylene diimide: Synthesis and applications in single chain detection studies. Polym. J. 2009, 41, 778–783. [Google Scholar] [CrossRef]
- Davis, N.J.L.K.; MacQueen, R.W.; Roberts, D.A.; Danos, A.; Dehn, S.; Perrier, S.; Schmidt, T.W. Energy transfer in pendant perylene diimide copolymers. J. Mater. Chem. C 2016, 4, 8270–8275. [Google Scholar] [CrossRef]
- Sato, H.; Matsuda, D.; Ogino, K. Synthesis and polymerization of methacrylate having fullerene. Polym. J. 1998, 30, 904–909. [Google Scholar] [CrossRef][Green Version]
- Zheng, J.; Goh, S.H.; Lee, S.Y. Synthesis and thermal properties of fullerene-containing polymethacrylates. Polym. Bull. 1997, 39, 79–84. [Google Scholar] [CrossRef]
- Kurmaz, S.V.; Ozhiganov, V.V. Fullerene-containing branched polymethacrylates and polymer networks: Synthesis, structure, and properties. Polym. Sci. Ser. A 2011, 53, 232–245. [Google Scholar] [CrossRef]
- Lopatin, M.A.; Evlampieva, N.P.; Lopatina, T.I.; Kuznetsova, Y.L.; Lavrenko, P.N. Methyl methacrylate polymerization in the presence of C60 (C70) and molecular characteristics of fullerene-containing poly(methyl methacrylate). Russ. J. Gen. Chem. 2008, 78, 1545–1552. [Google Scholar] [CrossRef]
- Lin, Q.; Xu Xu, R.H.J.; Yang, N.; Karim, A.A.; Loh, X.J.; Zhang, K. UV Protection and Antioxidant Activity of Nanodiamonds and Fullerenes for Sunscreen Formulations. ACS Appl. Nano Mater. 2019, 2, 7604–7616. [Google Scholar] [CrossRef]
- Heuts, J.P.A.; Gilbert, R.G.; Maxwell, I.A. Penultimate Unit Effect in Free-Radical Copolymerization. Macromolecules 1997, 30, 726–736. [Google Scholar] [CrossRef]
- Günther, H.; Guenther, H. NMR Spectroscopy: An Introduction; Wiley Publishing: New York, NY, USA, 1980. [Google Scholar]
- Du, H.; Fuh, R.-C.A.; Li, J.; Corkan, L.A.; Lindsey, J.S. Photochemistry and Photobiology; Wiley Publishing: New York, NY, USA, 1998; Volume 68, pp. 141–142. [Google Scholar]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | E0red3 | E0red2 | E0red1 | E0ox1 |
|---|---|---|---|---|
| PDI 8 | - | −1.40 | −1.24 | +0.77 |
| Dyad 9 | −1.55 | −1.38 | −1.22 * | +0.81 * |
| FP 11 | −1.78 | −1.55 | −1.18 | +0.98 ** |
| Compound | λabs (nm) (CH2Cl2) | λabs (nm) (toluene) | λem (nm) (CH2Cl2) | λem (nm) (toluene) | φF (CH2Cl2) | φF (Toluene) |
|---|---|---|---|---|---|---|
| PDI 7 | 452, 540, 580 | 448, 535, 575 | 607 | 601 | ~1 | ~1 |
| Dyad 9 | 229, 452, 543, 579 | 283, 451, 535, 573 | 610 | 603 | 2.8 × 10−3 | 1.1 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diacon, A.; Krupka, O.; Hudhomme, P. Fullerene-Perylenediimide (C60-PDI) Based Systems: An Overview and Synthesis of a Versatile Platform for Their Anchor Engineering. Molecules 2022, 27, 6522. https://doi.org/10.3390/molecules27196522
Diacon A, Krupka O, Hudhomme P. Fullerene-Perylenediimide (C60-PDI) Based Systems: An Overview and Synthesis of a Versatile Platform for Their Anchor Engineering. Molecules. 2022; 27(19):6522. https://doi.org/10.3390/molecules27196522
Chicago/Turabian StyleDiacon, Aurel, Oksana Krupka, and Piétrick Hudhomme. 2022. "Fullerene-Perylenediimide (C60-PDI) Based Systems: An Overview and Synthesis of a Versatile Platform for Their Anchor Engineering" Molecules 27, no. 19: 6522. https://doi.org/10.3390/molecules27196522
APA StyleDiacon, A., Krupka, O., & Hudhomme, P. (2022). Fullerene-Perylenediimide (C60-PDI) Based Systems: An Overview and Synthesis of a Versatile Platform for Their Anchor Engineering. Molecules, 27(19), 6522. https://doi.org/10.3390/molecules27196522