
Experimental Examination of Solubility and Lipophilicity as Pharmaceutically Relevant Points of Novel Bioactive Hybrid Compounds



Abstract
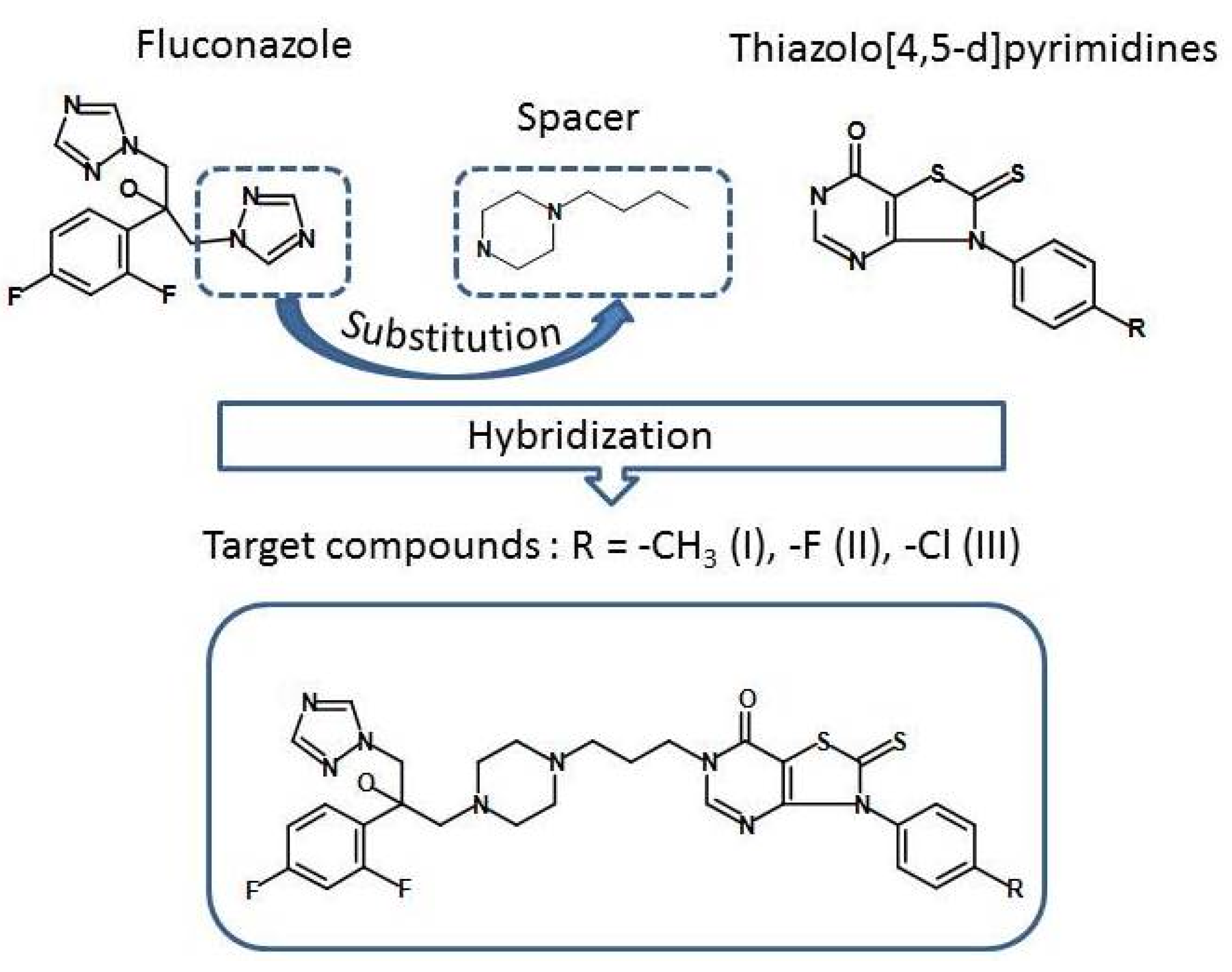
1. Introduction
- −
- Evaluation of the kinetic solubility in buffer solutions of various acidity degrees;
- −
- Determination of the temperature dependence of equilibrium solubility in buffer solutions and 1-octanol;
- −
- Measurement of partition coefficients in the two-phase system of immiscible solvents—1-octanol/buffer pH 7.4—within the temperature range of 293.15–313.15 K;
- −
- Calculation of the thermodynamic parameters of dissolution and partition processes in the studied systems and identification of their driving forces.
2. Results
2.1. Bioactive Assay
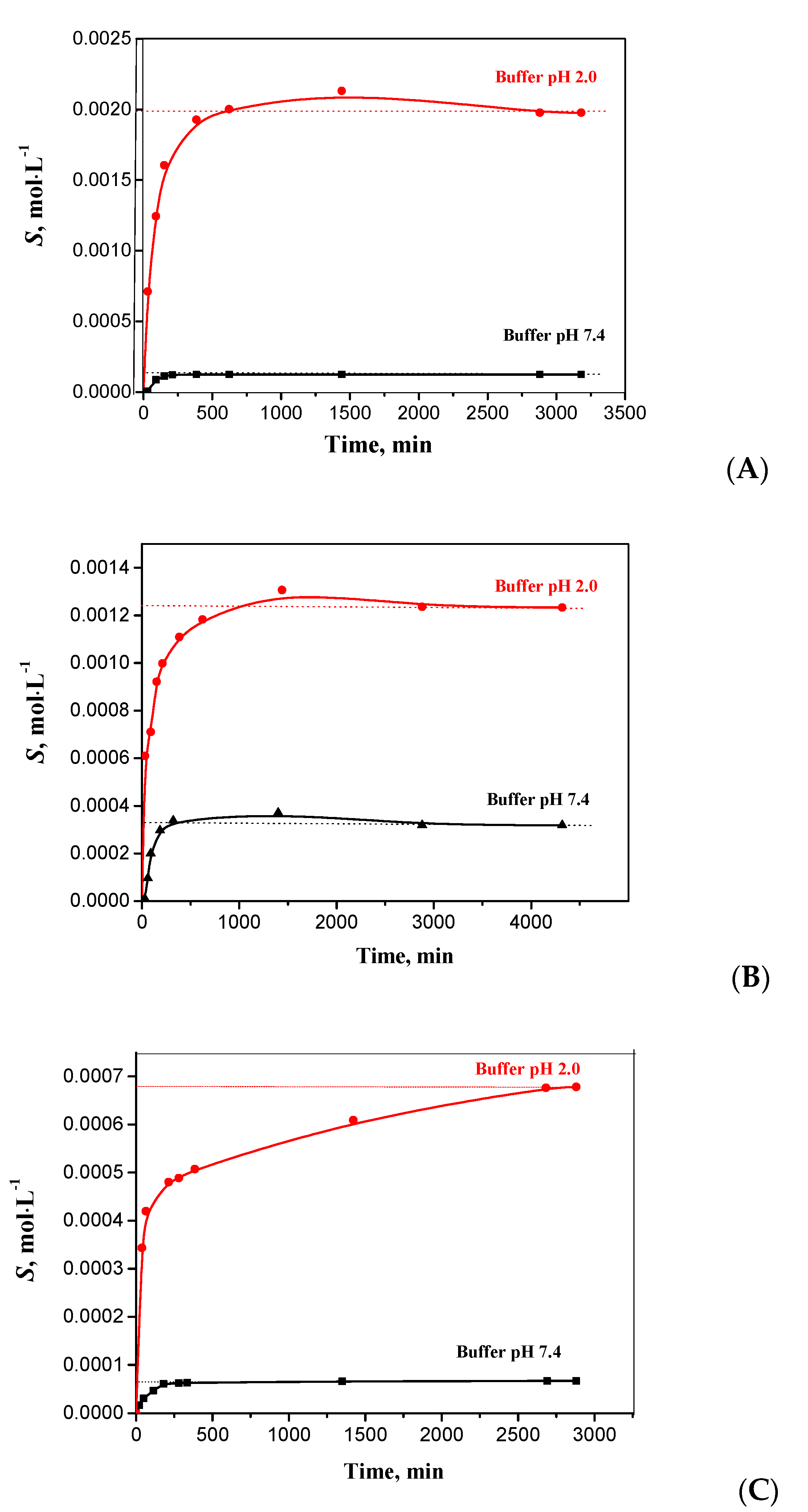
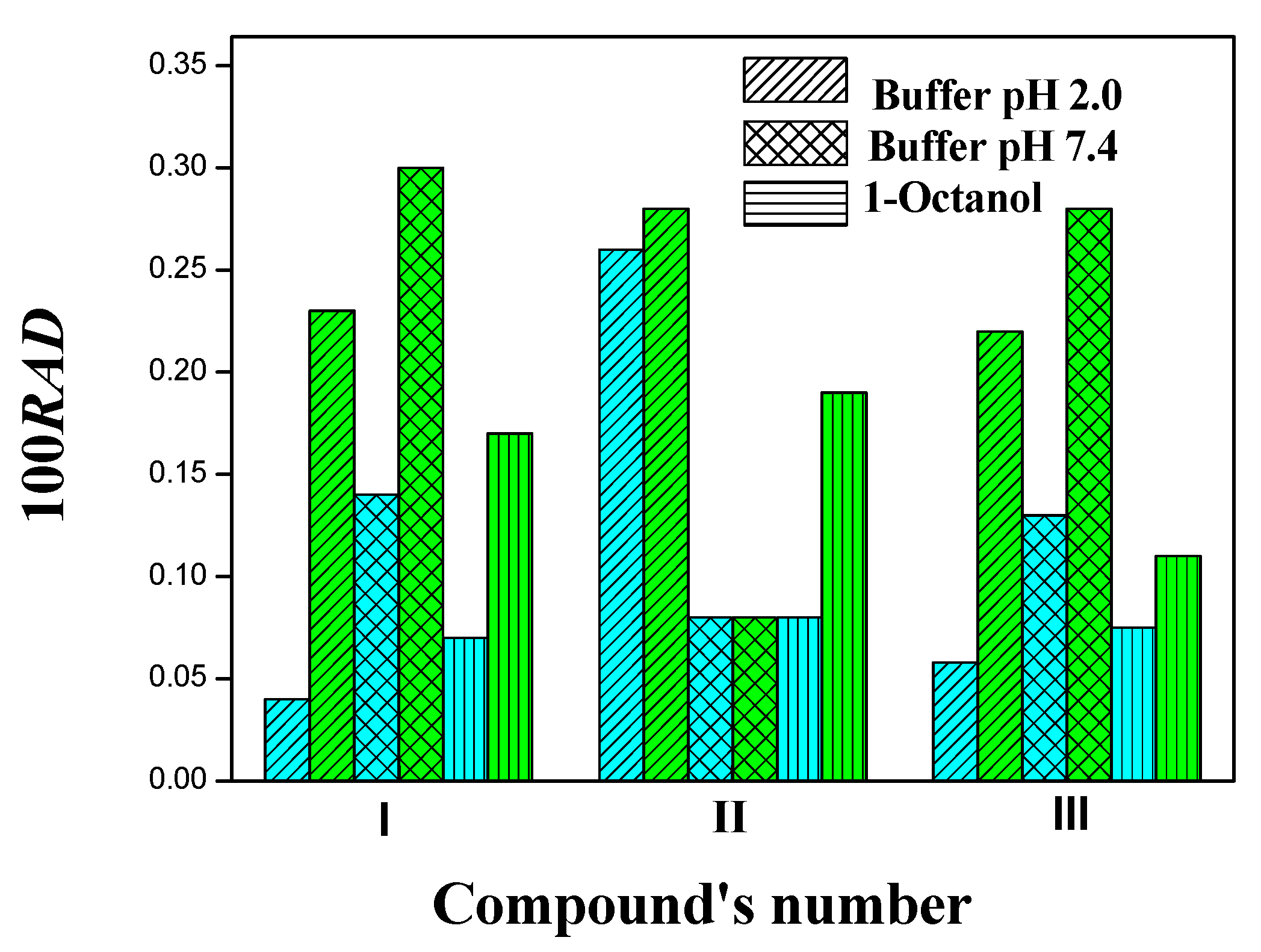
2.2. Kinetic Solubility
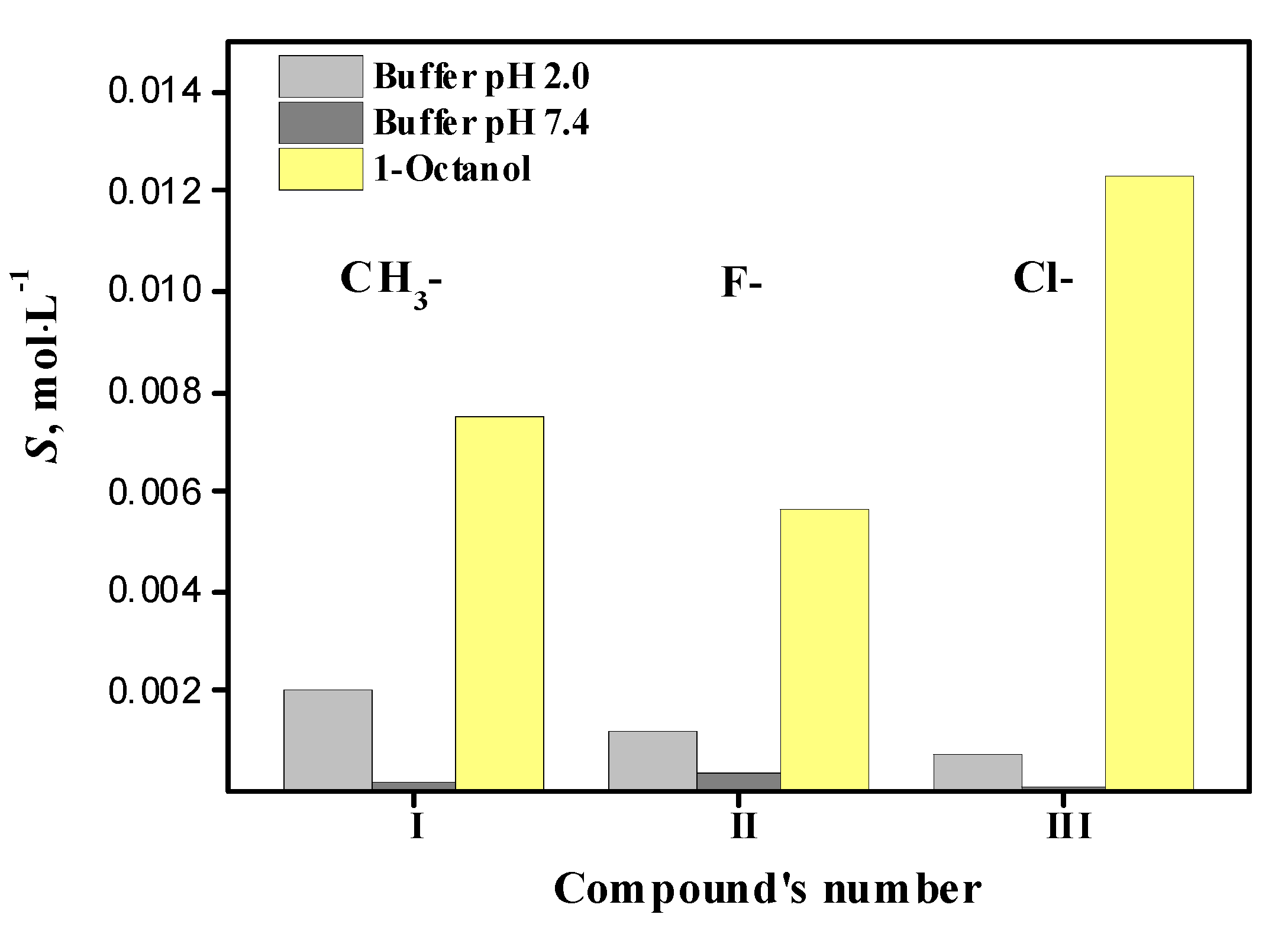
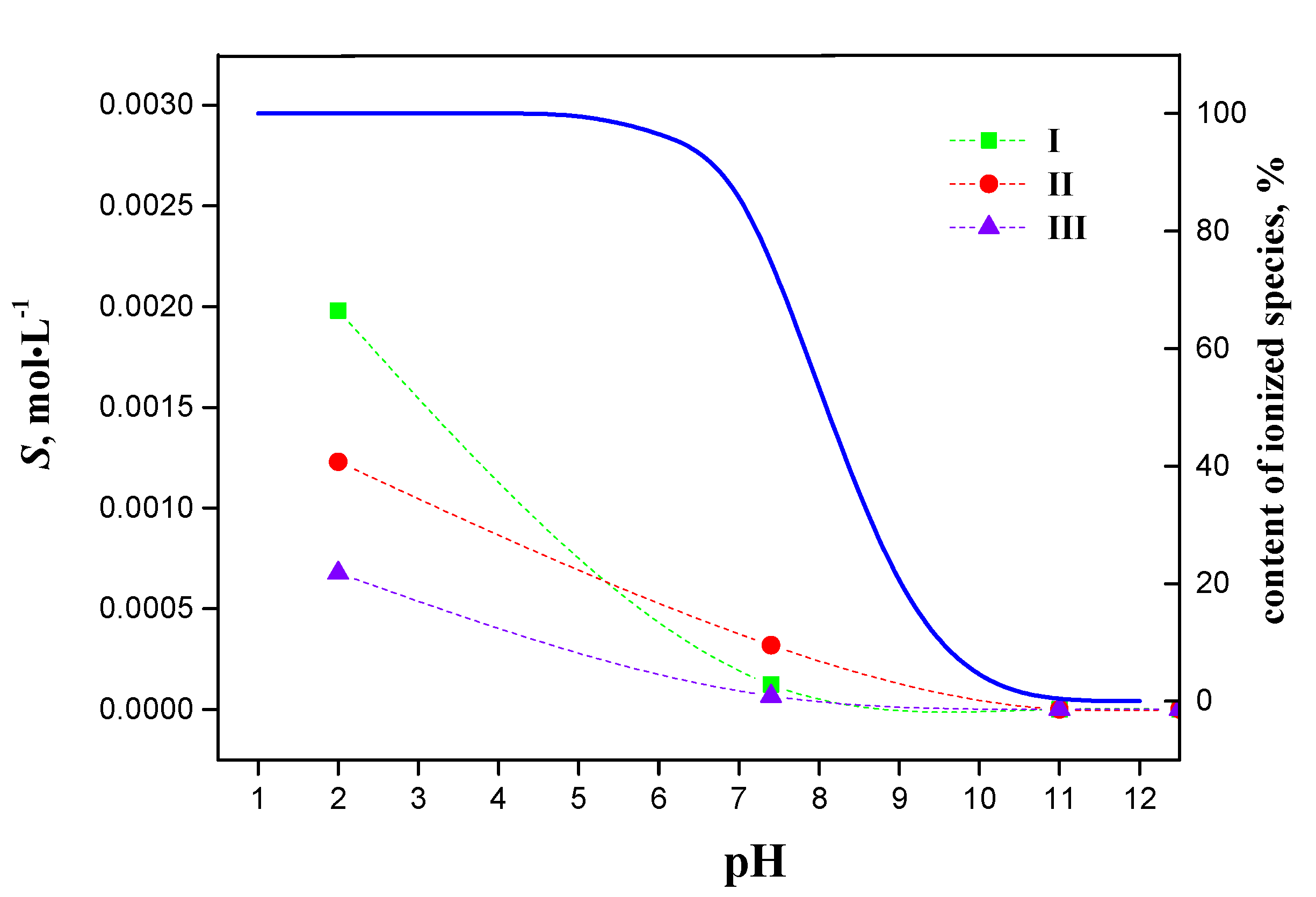
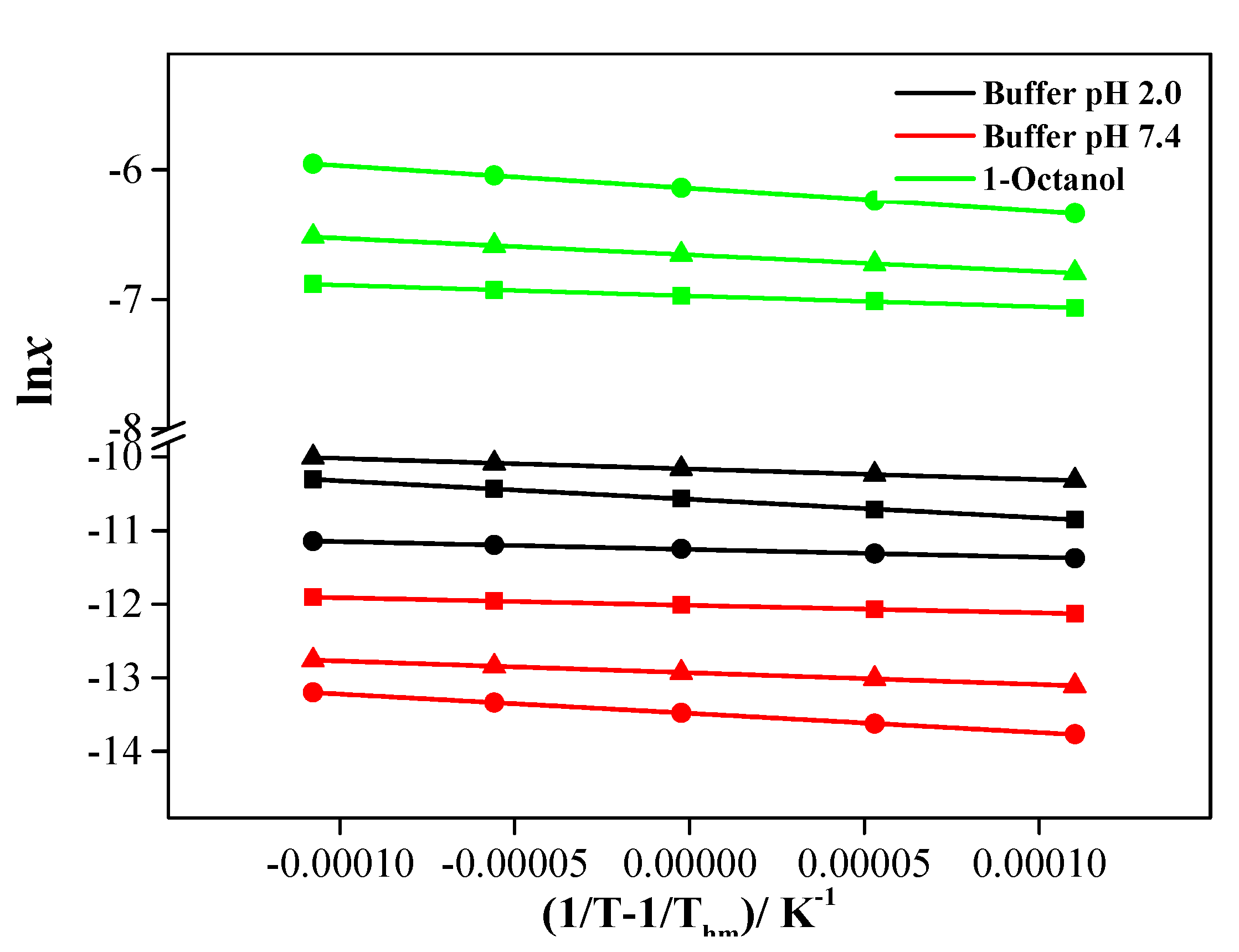
2.3. Equilibrium Solubility
2.4. Application of Hansen Solubility Parameters
2.5. Solubility Modeling
2.6. Dissolution Thermodynamics
2.7. Partition in System 1-Octanol/Buffer pH 7.4 and Transfer Thermodynamics
3. Materials and Methods
3.1. Materials
3.2. Synthesis and Characterization
3.3. Antifungal Activity Study
3.4. Kinetic Solubility
3.5. Equilibrium Solubility
3.6. Partition Experiment
3.7. Theoretical Basis
3.7.1. Van’t Hoff Equation
3.7.2. Modified Apelblat Equation
3.7.3. Evaluation of Precision for Used Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Perlin, D.S.; Rautemaa-Richardson, R.; Alastruey-Izquierdo, A. The global problem of antifungal resistance: Prevalence, mechanisms, and managemen. Lancet Infect. Dis. 2017, 17, e383–e392. [Google Scholar] [CrossRef]
- Soković, M.; Liaras, K. Fungal infections—Background to specific fungal species. In Antifungal Compounds Discovery; Elsevier Inc.: Amsterdam, The Netherlands, 2021; pp. 15–48. [Google Scholar] [CrossRef]
- Shaveta; Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem 2016, 124, 500–536. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.-F.; Li, D.; Tangadanchu, V.K.R.; Gopala, L.; Gao, W.-W.; Zhou, C.-H. Novel potentially antifungal hybrids of 5-flucytosine and fluconazole: Design, synthesis and bioactive evaluation. Bioorg. Med. Chem. Lett. 2017, 27, 4964–4969. [Google Scholar] [CrossRef] [PubMed]
- Borate, H.B.; Sawargave, S.P.; Chavana, S.P.; Chandavarkar, M.A.; Iyer, R.; Tawte, A.; Rao, D.; Deore, J.V.; Kudale, A.S.; Mahajan, P.S.; et al. Novel hybrids of fluconazole and furanones: Design, synthesis and antifungal activity. Bioorg. Med. Chem. Lett. 2011, 21, 4873–4878. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, R.A.; Bakr, R.B. New advances in synthesis and clinical aspects of pyrazolo[3,4-d]pyrimidine scaffolds. Bioorg. Chem. 2018, 78, 341–357. [Google Scholar] [CrossRef] [PubMed]
- Blokhina, S.V.; Sharapova, A.V.; Ol’khovich, M.V.; Doroshenko, I.A.; Levshin, I.B.; Perlovich, G.L. Synthesis and antifungal activity of new hybrids thiazolo[4,5-d]pyrimidines with (1H-1,2,4)triazole. Bioorg. Med. Chem. Lett. 2021, 40, 127944. [Google Scholar] [CrossRef]
- Stegemann, S.; Leveiller, F.; Franchi, D.; de Jong, H.; Lindrn, H. When poor solubility becomes an issue: From early stage to proof of concept. Eur. J. Pharm. Sci. 2007, 31, 249–261. [Google Scholar] [CrossRef]
- Kerns, E.H.; Di, L. Drug-like Properties: Concepts, Structure Design and Methods; Academic Press: Cambridge, MA, USA, 2008; pp. 43–47. [Google Scholar] [CrossRef]
- Danishuddin; Khan, A.U. Descriptors and their selection methods in QSAR analysis: Paradigm for drug design. Drug Discov. Today 2016, 21, 1291–1302. [Google Scholar] [CrossRef]
- European Pharmacopoeia, Guideline on the Quality of Water for Pharmaceutical Use. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-quality-water-pharmaceutical-use_en.pdf (accessed on 1 January 2018).
- Jinno, J.; Oh, D.; Crison, J.R.; Amidon, G.L. Dissolution of ionizable water-insoluble drugs: The combined effect of pH and surfactant. J. Pharm. Sci. 2000, 89, 268–274. [Google Scholar] [CrossRef]
- Alantary, D.; Yalkowsky, S. Calculating the Solubilities of Drugs and Drug-Like Compounds in Octanol. J. Pharm. Sci. 2016, 105, 2770–2773. [Google Scholar] [CrossRef]
- Blokhina, S.V.; Sharapova, A.V.; Ol’khovich, M.V.; Levshin, I.B.; Perlovich, G.L. Study of dissolution and transfer processes of new bioactive thiazolo[4,5-d]pyrimidine derivatives in modeling biological systems. J. Mol. Liq. 2021, 337, 116395. [Google Scholar] [CrossRef]
- Blokhina, S.V.; Sharapova, A.V.; Ol’khovich, M.V.; Levshin, I.B.; Perlovich, G.L. Solid–liquid phase equilibrium and thermodynamic analysis of novel thiazolidine-2,4-dione derivative in different solvents. J. Mol. Liq. 2021, 326, 115273. [Google Scholar] [CrossRef]
- Saal, C.; Petereit, A.C. Optimizing solubility: Kinetic versus thermodynamic solubility temptations and risks. Eur. J. Pharm. Sci. 2012, 47, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A. Suggested improvements for measurement of equilibrium solubility-pH of ionizable drugs. ADMET DMPK 2015, 3, 84–109. [Google Scholar] [CrossRef]
- Avdeef, A. Anomalous Solubility Behavior of Several Acidic Drugs. ADMET DMPK 2014, 2, 33–42. [Google Scholar] [CrossRef]
- Nair, A.; Saal, C. Solubility in Pharmaceutical Chemistry; De Gruyter: Berlin, Germany, 2020. [Google Scholar] [CrossRef]
- Petrikova, E.; Patera, J.; Gorlova, O. Influence of active pharmaceutical ingredient structures on Hansen solubility parameters. Eur. J. Pharm. Sci. 2021, 167, 106016. [Google Scholar] [CrossRef]
- Hansen, C.M. Hansen Solubility Parameters: A User’s Handbook; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Just, S.; Sievert, F.; Thommes, M.; Breitkreutz, J. Improved Group Contribution Parameter Set for the Application of Solubility Parameters to Melt Extrusion. Eur. J. Pharm. Biopharm. 2013, 85, 1191–1199. [Google Scholar] [CrossRef]
- Van Krevelen, D.W. Properties of Polymers, Their Estimation and Correlation with Chemical Structure; Elsevier: Amsterdam, The Netherlands, 1976. [Google Scholar]
- Greenhalgh, D.J.; Williams, A.C.; Timmins, P.; York, P. Solubility parameters as predictors of miscibility in solid dispersions. J. Pharm. Sci. 1999, 88, 1182–1190. [Google Scholar] [CrossRef]
- Ruelle, P.; Kesselring, U.W. The Hydrophobic Effect. 2. Relative Importance of the Hydrophobic Effect on the Solubility of Hydrophobes and Pharmaceuticals in H-Bonded Solvents. J. Pharm. Sci. 1998, 87, 998–1014. [Google Scholar] [CrossRef]
- Frank, H.S.; Evans, M.W. Free volume and entropy in condensed systems. III. Entropy in binary liquid mixtures; Partial molal entropy in dilute solutions; Structure and thermodynamics in aqueous electrolytes. J. Chem. Phys. 1945, 13, 507–532. [Google Scholar] [CrossRef]
- Huyskens, P.L.; Haulait-Pirson, M.C.; Siegel, G.G.; Kapuku, F. Gibbs energy of some binary systems containing hydrogen bonds. J. Phys. Chem. 1988, 92, 6841–6847. [Google Scholar] [CrossRef]
- Abraham, M.H.; Grellier, P.L.; Mcgill, R.A. A quantitative measure of solvent solvophobic effect. J. Chem. Soc. Perkin Trans. 1988, 2, 339–345. [Google Scholar] [CrossRef]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Comer, J.E. High throughput measurement of logD and pKa. In Methods and Principles in Medicinal Chemistry; Artursson, P., Lennernas, H., van de Waterbeemd, H., Eds.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Ruelle, P. The n-octanol and n-hexane/water partition coefficient of environmentally relevant chemicals predicted from the mobile order and disorder (MOD) thermodynamics. Chemosphere 2000, 40, 457–512. [Google Scholar] [CrossRef]
- Buffers. A Guide for the Preparation and Use of Buffers in Biological Systems; Mohan, C., Ed.; EMD Bioscience: Darmstadt, Germany, 2006. [Google Scholar]
- Ombrato, R. New Antibacterial Agents. International Application No. PCT/EP2017/0636012017211760, 14 December 2017. [Google Scholar]
- Clinical Laboratory Standards Institute (CLSI). Reference Method for Broth Dilution Testing of Yeasts, 3rd ed.; CLSI: Wayne, PA, USA, 2008. [Google Scholar]
- Test No. 107: Partition Coefficient (n-octanol/water): Shake Flask Method. In OECD Guidelines for the Testing of Chemicals; OECD Publishing: Paris, France, 1995. [CrossRef]
- Zarghampour, A.; Moradi, M.; Martinez, F.; Hemmati, S.; Rahimpour, E.; Jouyban, A. Solubility study of mesalazine in the aqueous mixtures of a deep-eutectic solvent at different temperatures. J. Mol. Liq. 2021, 336, 116300. [Google Scholar] [CrossRef]
- Liu, J.; Li, Z.; Wang, M.; Wan, X.; Jia, S.; Cao, Y.; Gao, Z. Solid-liquid phase equilibrium of N, N’-diphenyl thiourea (DPTU) in twelve pure solvents: Solubility determination, correlation, molecular simulation and thermodynamic analysis. J. Chem. Thermodyn. 2021, 163, 106605. [Google Scholar] [CrossRef]
- Grant, D.J.W.; Mehdizadeh, M.; Chow, A.H.-L.; Fairbrother, J.E. Non-linear van’t Hoff solubility-temperature plots and their pharmaceutical interpretation. Int. J. Pharm. 1984, 18, 25–38. [Google Scholar] [CrossRef]
- Atkins, P.; De Paula, J. Physical Chemistry; W.H. Freeman and Company: New York, NY, USA, 2006. [Google Scholar]
- Borra, A.; Kosuru, R.K.; Bharadwaj, R.; Satyavathi, B. Measurement and modeling of solubility of furan 2-carboxylic acid in mono and binary systems. J. Mol. Liq. 2021, 326, 115271. [Google Scholar] [CrossRef]
- Li, M.; Liu, Y.; Li, M.; Shang, Z.; Liu, M.; Han, D. Measurement and Correlation of the Solubility of Kaempferol Monohydrate in Pure and Binary Solvents. Fluid Phase Equilib. 2021, 539, 113027. [Google Scholar] [CrossRef]
- Blokhina, S.; Sharapova, A.; Ol’khovich, M.; Perlovich, G. A thermodynamic study of sublimation, dissolution and distribution processes of anti-inflammatory drug Clonixin. J. Chem. Thermodyn. 2019, 132, 281–288. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MIC (μg/mL) | ||
|---|---|---|---|
| C. parapsilosis ATCC 22019 | C. utilis 84 | C. glabrata 61L | |
| I | 0.5 | 4 | 2 |
| II | 0.1 | 1 | 4 |
| III | 0.25 | 2 | 2 |
| Fluconazole | 2 | 2 | 8 |
| T/K | I | II | III | ||||||
|---|---|---|---|---|---|---|---|---|---|
| a Buffer pH 2.0 | b Buffer pH 7.4 | 1-Octanol | Buffer pH 2.0 | Buffer pH 7.4 | 1-Octanol | Buffer pH 2.0 | Buffer pH 7.4 | 1-Octanol | |
| x × 105 (S × 103) | x × 106 (S × 104) | x × 103 (S × 103) | x × 105 (S × 103) | x × 106 (S × 104) | x × 104 (S × 103) | x × 105 (S × 104) | x × 106 (S × 105) | x × 103 (S × 102) | |
| 293.15 | 3.3040 (1.8307) | 2.0541 (1.1295) | 1.1199 (7.0781) | 1.9402 (1.0755) | 5.4794 (3.0126) | 8.5358 (5.3994) | 1.1474 (6.3622) | 1.0651 (5.8572) | 1.7767 (1.1196) |
| 298.15 | 3.5732 (1.9773) | 2.2653 (1.2441) | 1.1981 (7.5389) | 2.2272 (1.2330) | 5.8122 (3.1917) | 9.0027 (5.6703) | 1.2247 (6.7826) | 1.2262 (6.7347) | 1.9566 (1.2269) |
| 303.15 | 3.8570 (2.1311) | 2.4636 (1.3510) | 1.2903 (8.0826) | 2.5822 (1.4273) | 6.1623 (3.3788) | 9.3813 (5.8834) | 1.3013 (7.1958) | 1.4201 (7.7877) | 2.1577 (1.3464) |
| 308.15 | 4.1749 (2.3027) | 2.6820 (1.4682) | 1.3835 (8.6264) | 2.9451 (1.6250) | 6.4929 (3.5539) | 9.8001 (6.1187) | 1.3731 (7.5805) | 1.6349 (8.9503) | 2.3729 (1.4730) |
| 313.15 | 4.5144 (2.4851) | 2.9135 (1.5920) | 1.4834 (9.2070) | 3.3404 (1.8394) | 6.8581 (3.7469) | 10.2644 (6.3807) | 1.4468 (7.9724) | 1.8788 (10.2665) | 2.5979 (1.6045) |
| Compound | V, cm3·mol−1 | δd MPa0.5 | δp MPa0.5 | δh MPa0.5 | δt MPa0.5 | Δδt MPa0.5 | ∆δ MPa0.5 |
|---|---|---|---|---|---|---|---|
| I | 540.3 | 20.8 | 3.6 | 8.0 | 22.6 | - | - |
| Buffer solutions | 18.0 | 15.5 | 16.0 | 42.3 | 47.8 | 25.2 | 36.9 |
| 1-Octanol | 157.7 | 17.0 | 3.3 | 11.9 | 21.0 | 1.6 | 5.4 |
| II | 524.8 | 21.0 | 4.3 | 8.9 | 23.2 | - | - |
| Buffer solutions | 18.0 | 15.5 | 16.0 | 42.3 | 47.8 | 24.6 | 35.8 |
| 1-Octanol | 157.7 | 17.0 | 3.3 | 11.9 | 21.0 | 2.2 | 5.0 |
| III | 532.8 | 21.1 | 4.6 | 8.6 | 23.2 | - | - |
| Buffer solutions | 18.0 | 15.5 | 16.0 | 42.3 | 47.8 | 24.6 | 36.0 |
| 1-Octanol | 157.7 | 17.0 | 3.3 | 11.9 | 21.0 | 2.2 | 5.4 |
| Compound | ||||
|---|---|---|---|---|
| Buffer pH 2.0 | ||||
| I | 25.4 ± 0.5 | 11.9 ± 0.1 | −13.5 | −45.2 ± 2.4 |
| II | 26.6 ± 0.5 | 20.9 ± 0.2 | −5.7 | −19.1 ± 1.1 |
| III | 28.0 ± 0.4 | 8.8 ± 0.1 | −19.2 | −64.4 ± 3.8 |
| Buffer pH 7.4 | ||||
| I | 32.2 ± 0.6 | 13.2 ± 0.1 | −19.0 | −63.6 ± 4.1 |
| II | 29.9 ± 0.6 | 8.5 ± 0.1 | −21.3 | −71.4 ± 4.2 |
| III | 33.7 ± 0.7 | 21.7 ± 0.2 | −12.0 | −40.3 ± 2.3 |
| 1-Octanol | ||||
| I | 16.8 ± 0.2 | 10.6 ± 0.2 | −6.2 | −20.8 ± 1.1 |
| II | 17.4 ± 0.3 | 6.9 ± 0.1 | −10.4 | −35.1 ± 1.8 |
| III | 15.4 ± 0.3 | 14.5 ± 0.1 | −0.9 | −3.1 ± 0.2 |
| T/K | I | II | ||||||||||
| sb × 106 | so × 103 | logPo/b | xb × 107 | xo × 104 | logP*o/b | sb × 105 | so × 103 | logPo/b | xb × 107 | xo × 104 | logP*o/b | |
| 293.15 | 10.2 | 2.88 | 2.45 | 1.82 | 4.54 | 3.39 | 1.47 | 3.39 | 2.36 | 2.67 | 5.34 | 3.30 |
| 298.15 | 9.11 | 2.88 | 2.50 | 1.63 | 4.57 | 3.44 | 1.35 | 3.39 | 2.40 | 2.45 | 5.37 | 3.34 |
| 303.15 | 7.95 | 2.88 | 2.56 | 1.43 | 4.59 | 3.50 | 1.25 | 3.39 | 2.43 | 2.38 | 5.39 | 3.37 |
| 308.15 | 7.16 | 2.88 | 2.60 | 1.29 | 4.61 | 3.55 | 1.15 | 3.39 | 2.47 | 2.08 | 5.42 | 3.41 |
| 313.15 | 6.38 | 2.88 | 2.65 | 1.15 | 4.63 | 3.60 | 1.06 | 3.39 | 2.50 | 1.94 | 5.44 | 3.45 |
| A a | 6.71 ± 0.05 | 5.61 ± 0.05 | ||||||||||
| B a | 972 ± 16 | 678 ± 16 | ||||||||||
| R b | 0.9996 | 0.9983 | ||||||||||
| σ c | 0.7 × 10−4 | 0.2 × 10−4 | ||||||||||
| T/K | III | |||||||||||
| sb × 106 | so × 104 | logPo/b | xb × 107 | xo × 105 | logP*o/b | |||||||
| 293.15 | 9.29 | 3.89 | 1.62 | 1.69 | 6.14 | 2.56 | ||||||
| 298.15 | 8.77 | 3.90 | 1.65 | 1.60 | 6.17 | 2.59 | ||||||
| 303.15 | 8.29 | 3.91 | 1.67 | 1.51 | 6.21 | 2.61 | ||||||
| 308.15 | 7.80 | 3.91 | 1.70 | 1.42 | 6.24 | 2.64 | ||||||
| 313.15 | 7.36 | 3.92 | 1.73 | 1.35 | 6.28 | 2.69 | ||||||
| A a | 4.25 ± 0.02 | |||||||||||
| B a | 495 ± 21 | |||||||||||
| R b | 0.9972 | |||||||||||
| σ c | 0.3 × 10−4 | |||||||||||
| Compound | ∆trGo kJ·mol−1 | ∆trHo kJ·mol−1 | T∆trSo kJ·mol−1 | ∆trSo J·mol−1·K−1 |
|---|---|---|---|---|
| I II III | −19.6 ± 0.5 −19.1 ± 0.3 −14.8 ± 0.2 | 18.3 ± 0.3 12.8 ± 0.2 9.5 ± 0.1 | 38.0 ± 0.9 31.9 ± 0.8 24.3 ± 1.0 | 127.4 ± 4.7 106.9 ± 2.9 81.5 ± 1.5 |
| Chemical Name | CAS Register No. | Formula | M/g mol−1 | Source | Mass Fraction Purity |
|---|---|---|---|---|---|
| 6-[3-[4-[2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl]-1-piperazinyl]propyl]-2,3-dihydro-3-(4-methylphenyl)-2-thioxo-thiazolo[4,5-d]pyrimidin-7(6H)-one (I) | 2637523-56-7 | C30H32F2N8O2S2 | 638.75 | Synthesis | ≥0.96 |
| 6-[3-[4-[2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl]-1-piperazinyl]propyl]-2,3-dihydro-3-(4-fluorophenyl)-2-thioxo-thiazolo[4,5-d]pyrimidin-7(6H)-one (II) | 2637523-57-8 | C29H29F3N8O2S2 | 642.72 | Synthesis | ≥0.96 |
| 6-[3-[4-[2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl]-1-piperazinyl]propyl]-2,3-dihydro-3-(4-chlorophenyl)-2-thioxo-thiazolo[4,5-d]pyrimidin-7(6H)-one (III) | 2637523-58-9 | C29 H29ClF2N8O2S2 | 659.17 | Synthesis | ≥0.96 |
| 1-Octanol | 111-87-5 | C8H18O | 130.20 | Sigma- Aldrich | ≥0.99 a |
| Potassium dihydrogen phosphate | 7778-77-0 | KH2PO4 | 136.08 | Merck | ≥0.99 a |
| Disodium hydrogen phosphate dodecahydrate | 10039-32-4 | Na2HPO4·12H2O | 358.14 | Merck | ≥0.99 a |
| Potassium chloride | 7447-40-7 | KCl | 74.55 | Sigma- Aldrich | ≥0.99 a |
| Hydrochloric acid 0.1 mol/dm3 fixanal | 7647-01-0 | HCl | 36.46 | Sigma- Aldrich | ≥0.99 a |
| Fluconazole | 86386-73-4 | C13H12F2N6O | 306.27 | Quimica Sintetica | ≥0.99 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharapova, A.; Ol’khovich, M.; Blokhina, S.; Perlovich, G.L. Experimental Examination of Solubility and Lipophilicity as Pharmaceutically Relevant Points of Novel Bioactive Hybrid Compounds. Molecules 2022, 27, 6504. https://doi.org/10.3390/molecules27196504
Sharapova A, Ol’khovich M, Blokhina S, Perlovich GL. Experimental Examination of Solubility and Lipophilicity as Pharmaceutically Relevant Points of Novel Bioactive Hybrid Compounds. Molecules. 2022; 27(19):6504. https://doi.org/10.3390/molecules27196504
Chicago/Turabian StyleSharapova, Angelica, Marina Ol’khovich, Svetlana Blokhina, and German L. Perlovich. 2022. "Experimental Examination of Solubility and Lipophilicity as Pharmaceutically Relevant Points of Novel Bioactive Hybrid Compounds" Molecules 27, no. 19: 6504. https://doi.org/10.3390/molecules27196504
APA StyleSharapova, A., Ol’khovich, M., Blokhina, S., & Perlovich, G. L. (2022). Experimental Examination of Solubility and Lipophilicity as Pharmaceutically Relevant Points of Novel Bioactive Hybrid Compounds. Molecules, 27(19), 6504. https://doi.org/10.3390/molecules27196504