
Redox-Triggered Switching of Conformational State in Triple-Decker Lanthanide Phthalocyaninates

 , ,
, ,  and
and
Abstract
1. Introduction
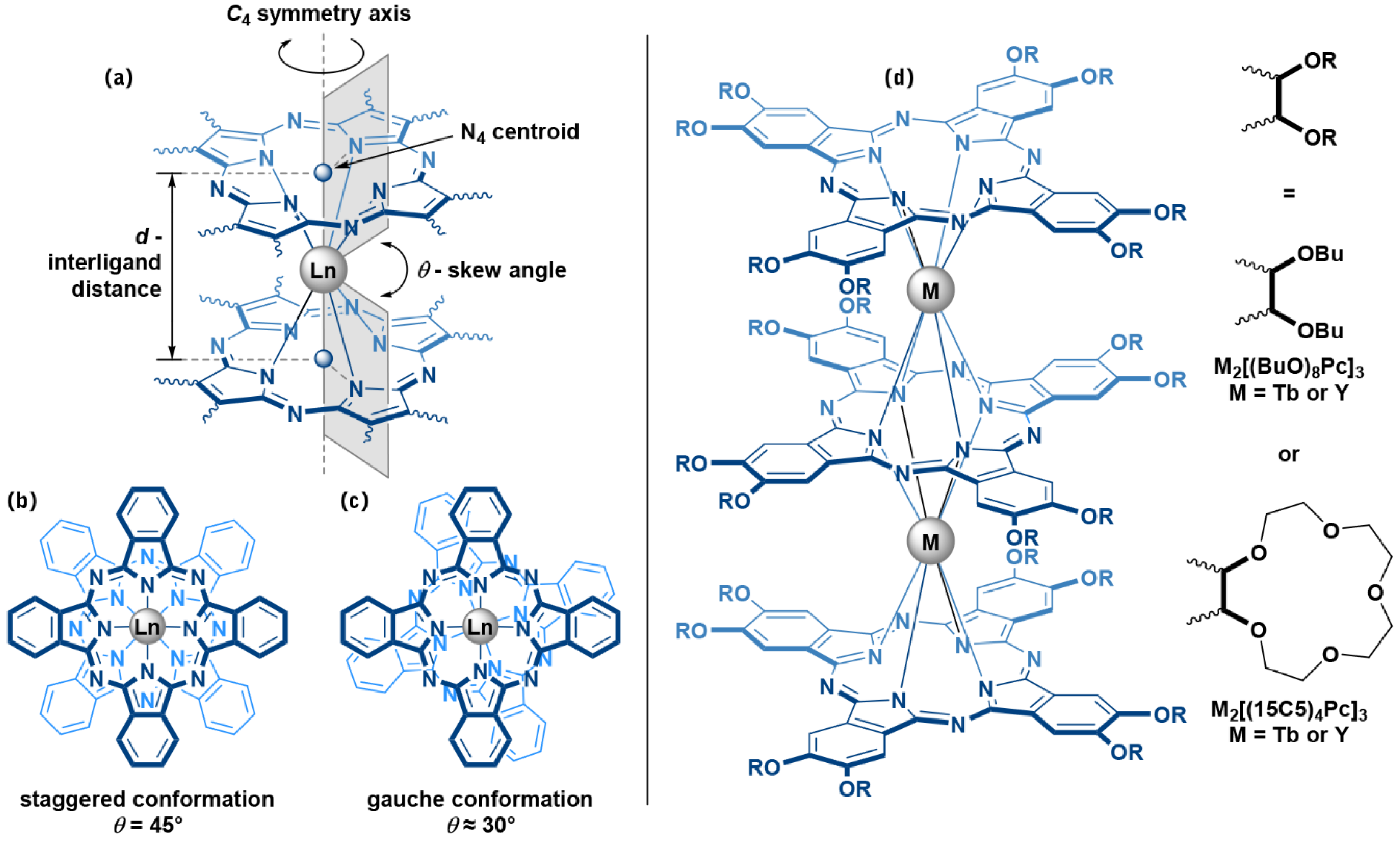
2. Results and Discussion
2.1. Synthesis of Trisphthalocyaninates
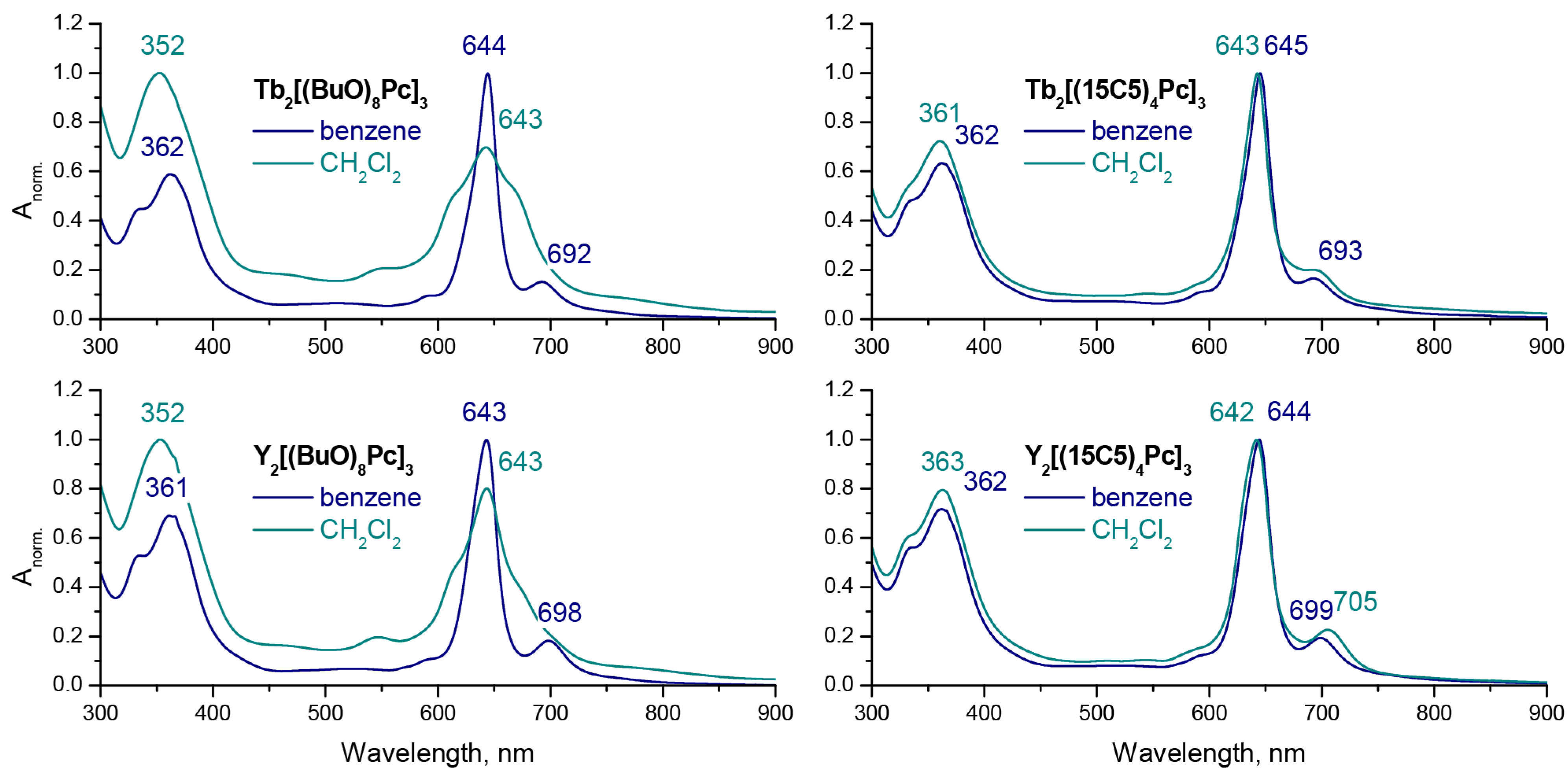
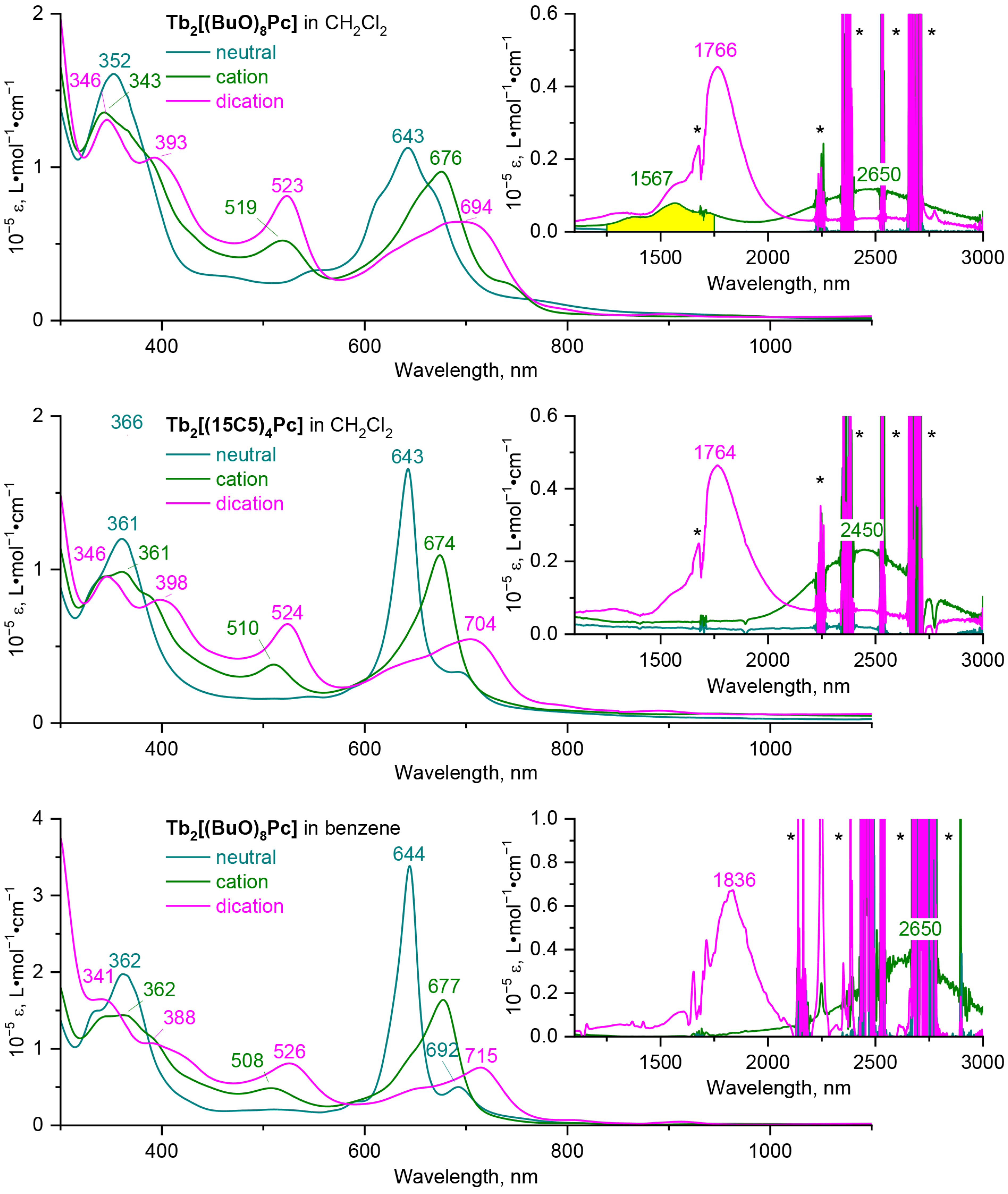
2.2. Solvatochromic Behaviour of Trisphthalocyaninates Depending on the Nature of Substituents and Redox State
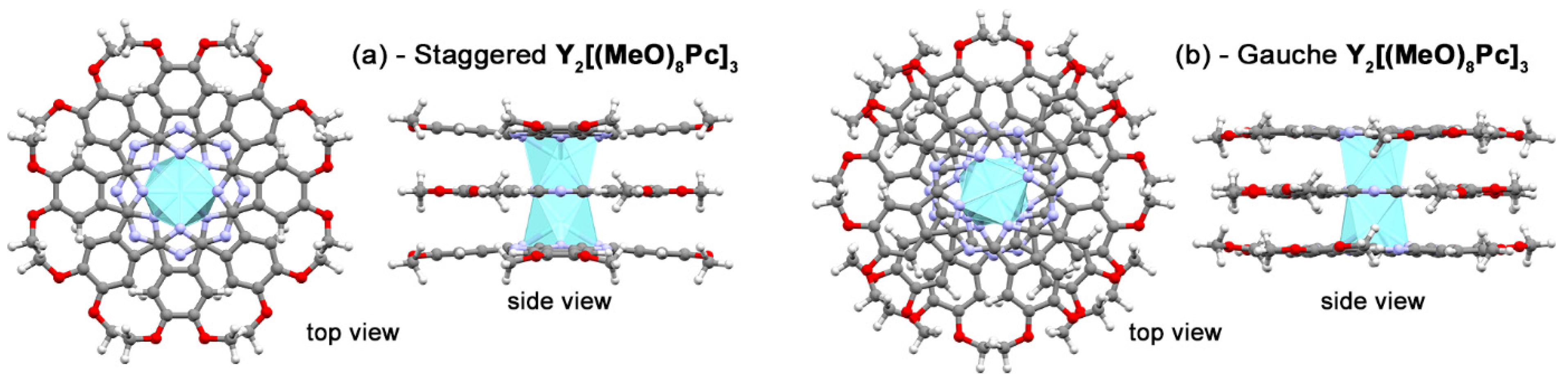
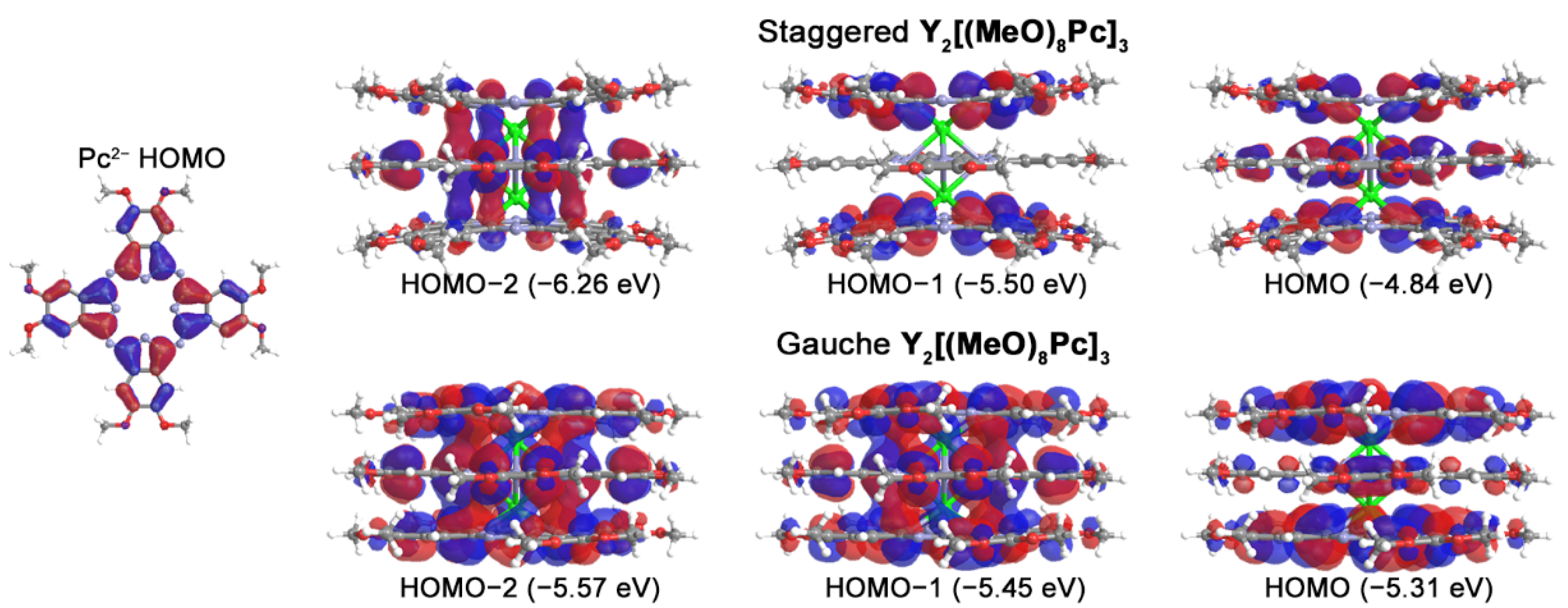
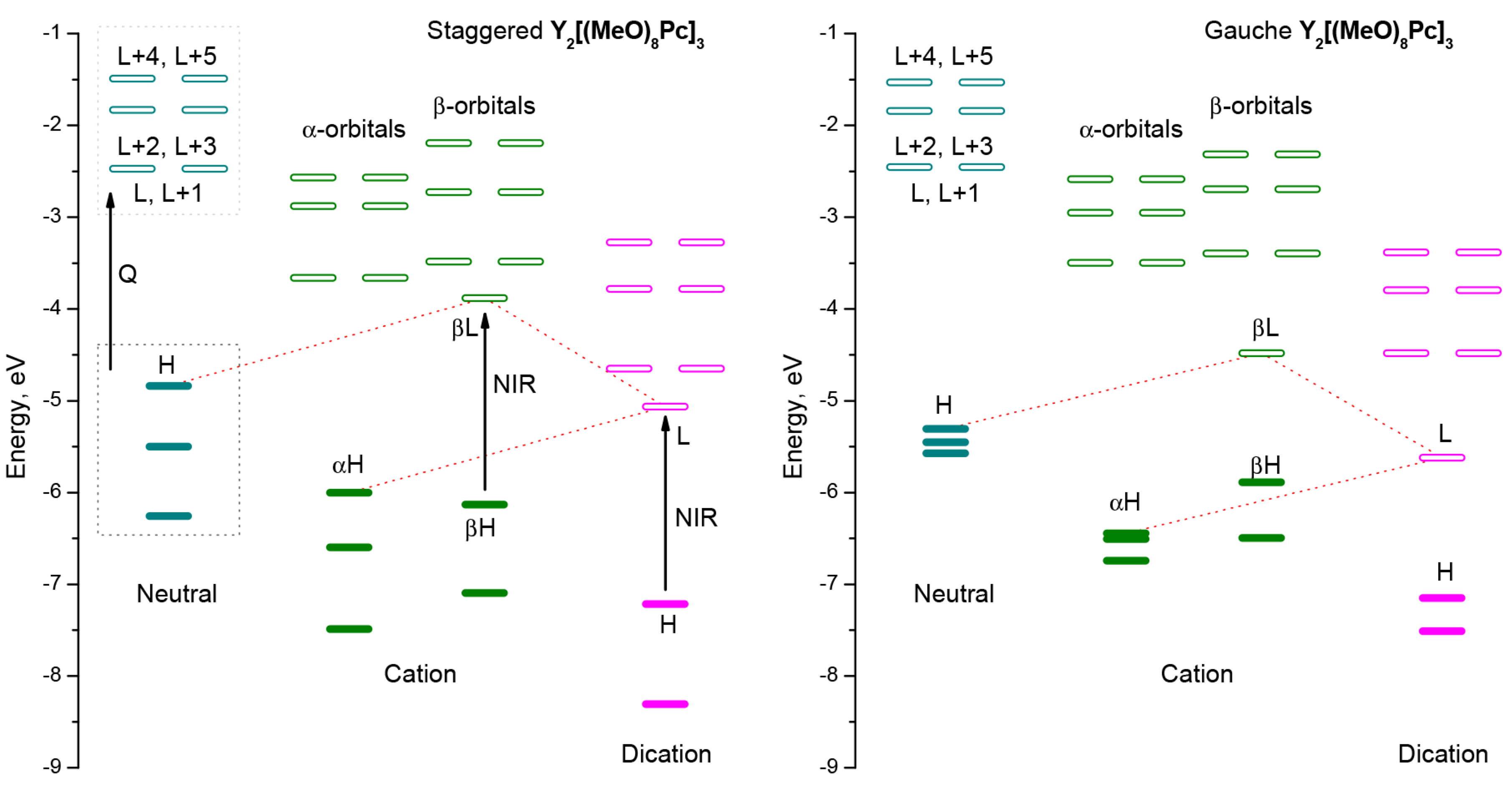
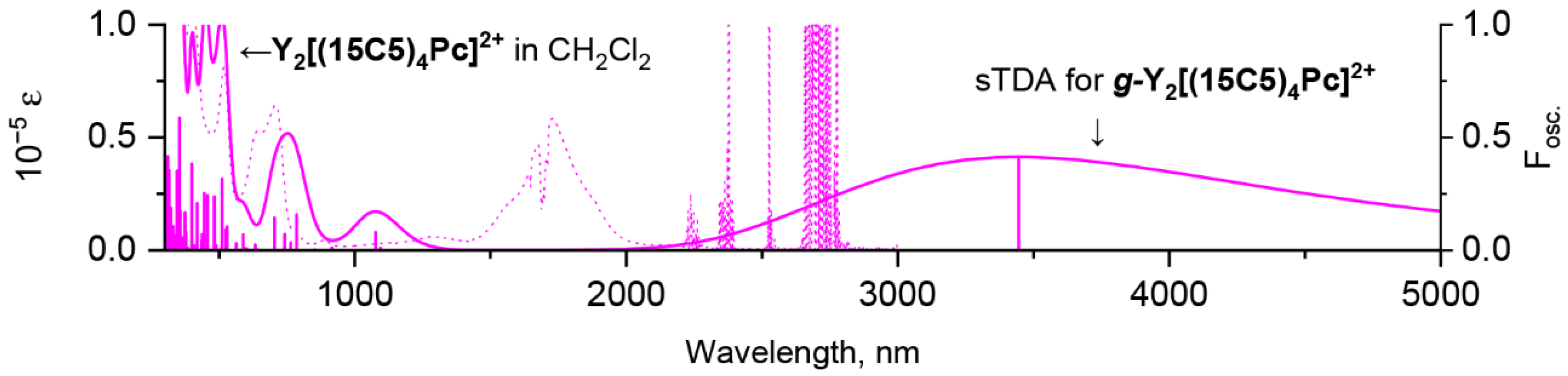
2.3. Quantum-Chemical Modelling of Conformation- and Redox-Dependent UV-vis-NIR Spectra of Trisphthalocyaninates
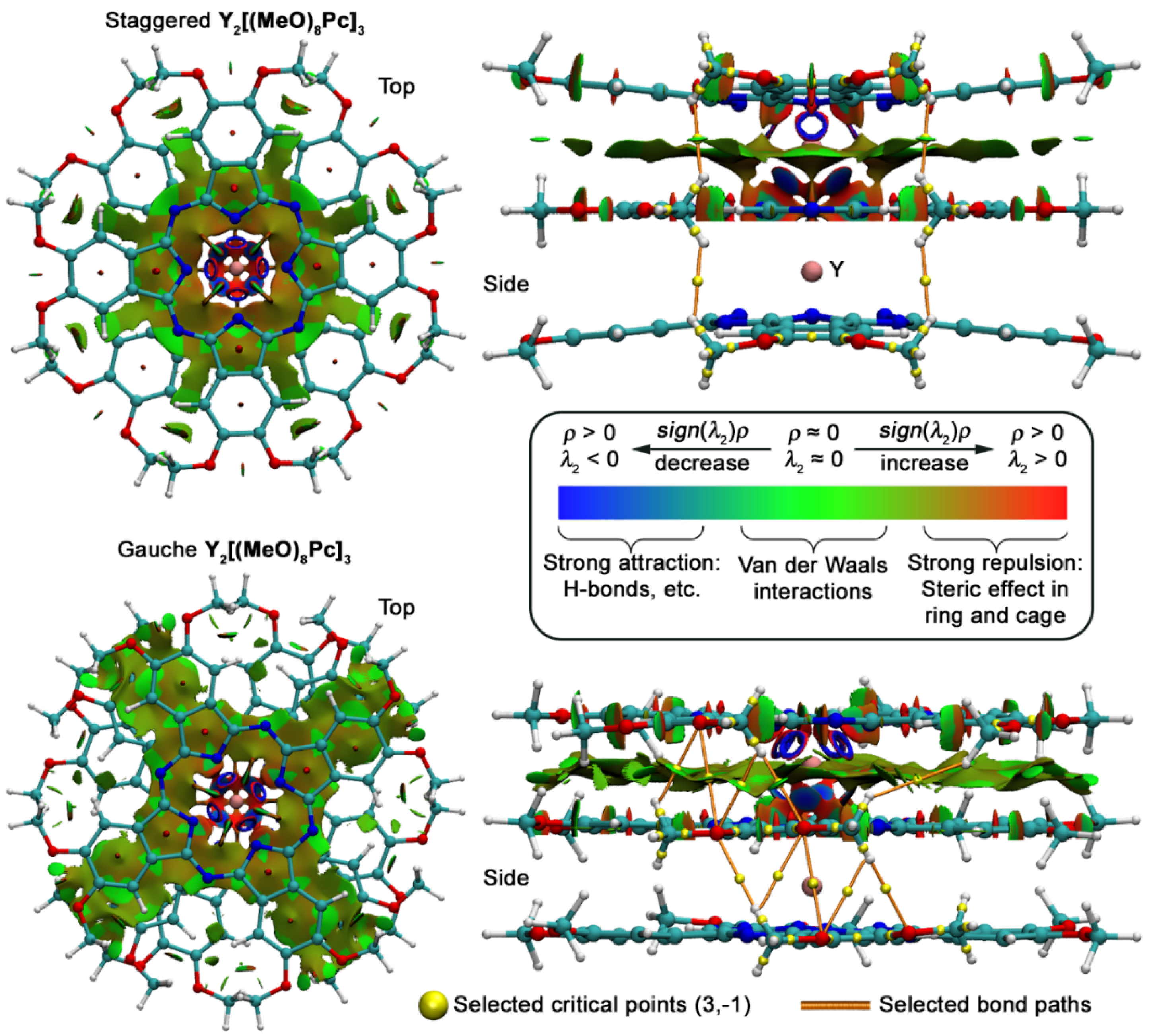
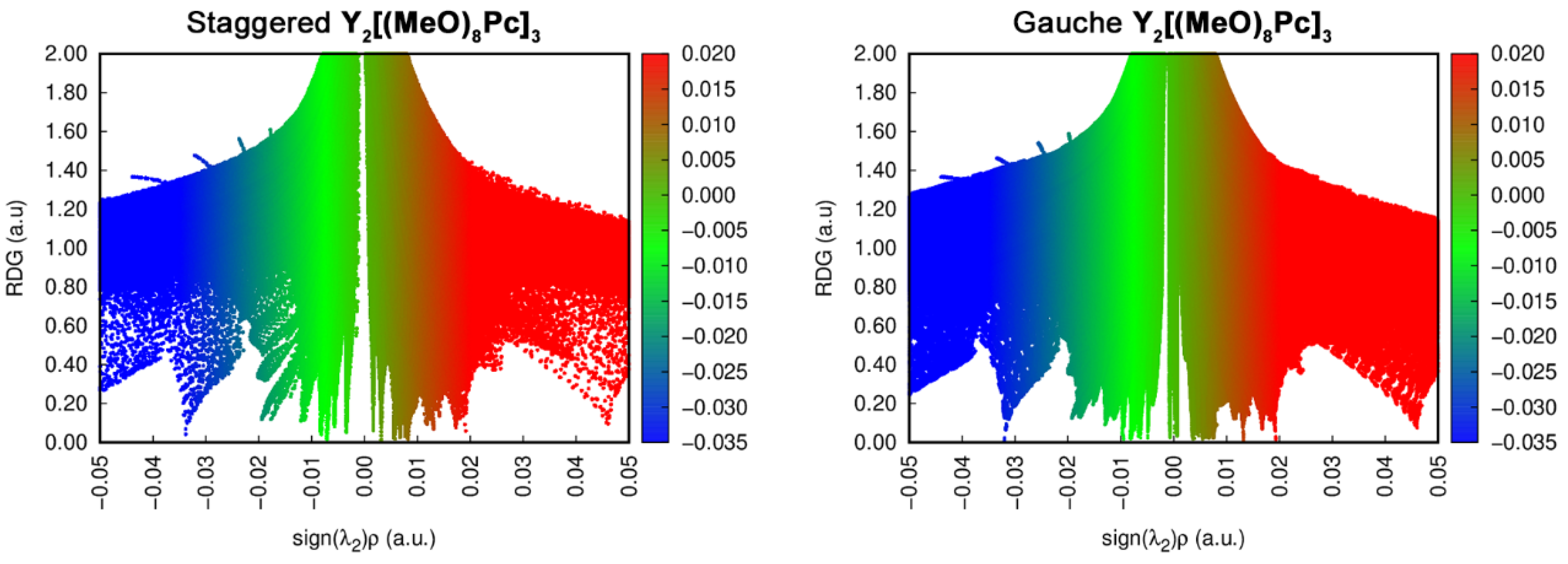
2.4. Analysis of Noncovalent Interactions Stabilizing Conformations of Trisphthalocyaninates
3. Materials and Methods
3.1. Physical-Chemical Measurements
3.2. Synthesis and Characterization of Phthalocyanines
3.3. Spectrophotometric Investigation of Trisphjthalocyaninates Oxidation
3.4. Quantum-Chemical Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koifman, O.I.; Ageeva, T.A.; Beletskaya, I.P.; Averin, A.D.; Yakushev, A.A.; Tomilova, L.G.; Dubinina, T.V.; Tsivadze, A.Y.; Gorbunova, Y.G.; Martynov, A.G.; et al. Macroheterocyclic Compounds—A Key Building Block in New Functional Materials and Molecular Devices. Macroheterocycles 2020, 13, 311–467. [Google Scholar] [CrossRef]
- Chan, W.L.; Xie, C.; Lo, W.S.; Bünzli, J.C.G.; Wong, W.K.; Wong, K.L. Lanthanide-tetrapyrrole complexes: Synthesis, redox chemistry, photophysical properties, and photonic applications. Chem. Soc. Rev. 2021, 50, 12189–12257. [Google Scholar] [CrossRef] [PubMed]
- Martynov, A.G.; Horii, Y.; Katoh, K.; Bian, Y.; Jiang, J.; Yamashita, M.; Gorbunova, Y.G. Rare-Earth Based Tetrapyrrolic Sandwiches: Chemistry, Materials and Applications. Chem. Soc. Rev. 2022; Accepted. [Google Scholar]
- Katoh, K.; Isshiki, H.; Komeda, T.; Yamashita, M. Multiple-decker phthalocyaninato Tb(III) single-molecule magnets and Y(III) complexes for next generation devices. Coord. Chem. Rev. 2011, 255, 2124–2148. [Google Scholar] [CrossRef]
- Wang, H.; Wang, B.W.; Bian, Y.; Gao, S.; Jiang, J. Single-molecule magnetism of tetrapyrrole lanthanide compounds with sandwich multiple-decker structures. Coord. Chem. Rev. 2016, 306, 195–216. [Google Scholar] [CrossRef]
- Urdampilleta, M.; Nguyen, N.V.; Cleuziou, J.P.; Klyatskaya, S.; Ruben, M.; Wernsdorfer, W. Molecular quantum spintronics: Supramolecular spin valves based on single-molecule magnets and carbon nanotubes. Int. J. Mol. Sci. 2011, 12, 6656–6667. [Google Scholar] [CrossRef]
- Martynov, A.G.; Safonova, E.A.; Tsivadze, A.Y.; Gorbunova, Y.G. Functional molecular switches involving tetrapyrrolic macrocycles. Coord. Chem. Rev. 2019, 387, 325–347. [Google Scholar] [CrossRef]
- Takamatsu, S.; Ishikawa, T.; Koshihara, S.; Ishikawa, N. Significant Increase of the Barrier Energy for Magnetization Reversal of a Single-4f-Ionic Single-Molecule Magnet by a Longitudinal Contraction of the Coordination Space. Inorg. Chem. 2007, 46, 7250–7252. [Google Scholar] [CrossRef]
- Katoh, K.; Yasuda, N.; Damjanović, M.; Wernsdorfer, W.; Breedlove, B.K.; Yamashita, M. Manipulation of the Coordination Geometry along the C 4 Rotation Axis in a Dinuclear Tb 3+ Triple-Decker Complex via a Supramolecular Approach. Chem.–A Eur. J. 2020, 26, 4805–4815. [Google Scholar] [CrossRef]
- Sato, T.; Matsuzawa, S.; Katoh, K.; Breedlove, B.K.; Yamashita, M. Relationship between the Coordination Geometry and Spin Dynamics of Dysprosium(III) Heteroleptic Triple-Decker Complexes. Magnetochemistry 2019, 5, 65. [Google Scholar] [CrossRef]
- Bian, Y.; Wang, D.; Wang, R.; Weng, L.; Dou, J.; Zhao, D.; Ng, D.K.P.; Jiang, J. Structural studies of the whole series of lanthanide double-decker compounds with mixed 2,3-naphthalocyaninato and octaethylporphyrinato ligands. New J. Chem. 2003, 27, 844–849. [Google Scholar] [CrossRef]
- Takamatsu, S.; Ishikawa, N. A theoretical study of a drastic structural change of bis(phthalocyaninato)lanthanide by ligand oxidation: Towards control of ligand field strength and magnetism of single-lanthanide-ionic single molecule magnet. Polyhedron 2007, 26, 1859–1862. [Google Scholar] [CrossRef]
- Katoh, K.; Breedlove, B.K.; Yamashita, M. Symmetry of octa-coordination environment has a substantial influence on dinuclear Tb III triple-decker single-molecule magnets. Chem. Sci. 2016, 7, 4329–4340. [Google Scholar] [CrossRef] [PubMed]
- Horii, Y.; Damjanović, M.; Ajayakumar, M.R.; Katoh, K.; Kitagawa, Y.; Chibotaru, L.; Ungur, L.; Mas-Torrent, M.; Wernsdorfer, W.; Breedlove, B.K.; et al. Highly Oxidized States of Phthalocyaninato Terbium(III) Multiple-Decker Complexes Showing Structural Deformations, Biradical Properties and Decreases in Magnetic Anisotropy. Chem.–A Eur. J. 2020, 26, 8621–8630. [Google Scholar] [CrossRef] [PubMed]
- Martynov, A.G.; Polovkova, M.A.; Berezhnoy, G.S.; Sinelshchikova, A.A.; Khrustalev, V.N.; Birin, K.P.; Kirakosyan, G.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Heteroleptic Crown-Substituted Tris(phthalocyaninates) as Dynamic Supramolecular Scaffolds with Switchable Rotational States and Tunable Magnetic Properties. Inorg. Chem. 2021, 60, 9110–9121. [Google Scholar] [CrossRef]
- Martynov, A.G.; Gorbunova, Y.G.; Tsivadze, A.Y. Crown-substituted phthalocyanines—Components of molecular ionoelectronic materials and devices. Russ. J. Inorg. Chem. 2014, 59, 1635–1664. [Google Scholar] [CrossRef]
- Takahashi, K.; Itoh, M.; Tomita, Y.; Nojima, K.; Kasuga, K.; Isa, K. Preparation, Characterization, and Electrochemical Properties of Tris(2,3,9,10,16,17,23,24-octabutoxyphthalocyaninato)diytterbium(III) and-didysprosium(III). Chem. Lett. 1993, 22, 1915–1918. [Google Scholar] [CrossRef]
- Takahashi, K.; Shimoda, J.; Itoh, M.; Fuchita, Y.; Okawa, H. Synthesis and Characterization of Triple-decker Sandwich Dinuclear La(III) and Lu(III) Complexes of 2,3,9,10,16,17,23,24-Octabutoxyphthalocyanine. Chem. Lett. 1998, 2, 173–174. [Google Scholar] [CrossRef]
- Ferencz, A.; Neher, D.; Schulze, M.; Wegner, G.; Viaene, L.; De Schryver, F.C. Synthesis and spectroscopic properties of phthalocyanine dimers in solution. Chem. Phys. Lett. 1995, 245, 23–29. [Google Scholar] [CrossRef]
- Kleinwächter, J.; Hanack, M. Rotational Isomers in Stacked Macrocycles: Synthesis and Spectroscopic Properties of Peripherally Substituted (μ-Oxo)bis(phthalocyaninatosilicon) Compounds. J. Am. Chem. Soc. 1997, 119, 10684–10695. [Google Scholar] [CrossRef]
- Kudrik, E.V.; Afanasiev, P.; Bouchu, D.; Sorokin, A.B. Solvent-dependent rotational phenomena in μ-nitrido-[2,3,9,10,16,17,23,24-octa(n-pentoxy)phthalocyaninato]diiron complex. J. Porphyr. Phthalocyanines 2011, 15, 583–591. [Google Scholar] [CrossRef]
- Kroitor, A.P.; Martynov, A.G.; Gorbunova, Y.G.; Tsivadze, A.Y.; Sorokin, A.B. Exploring the Optimal Synthetic Pathways towards µ-Carbido Diruthenium(IV) Bisphthalocyaninates. Eur. J. Inorg. Chem. 2019, 2019, 1923–1931. [Google Scholar] [CrossRef]
- Dimer, H.; Oniwa, K.; Shimizu, S.; Shiina, Y.; Fukuda, T.; Kobayashi, N. A μ-oxo hetero dimer of silicon phthalocyanine and naphthalocyanine. Chem. Commun. 2013, 49, 8341–8343. [Google Scholar] [CrossRef]
- Shiina, Y.; Kage, Y.; Furukawa, K.; Wang, H.; Yoshikawa, H.; Furuta, H.; Kobayashi, N.; Shimizu, S. TTF-Annulated Silicon Phthalocyanine Oligomers and Their External-Stimuli-Responsive Orientational Ordering. Angew. Chem. Int. Ed. 2020, 59, 22721–22730. [Google Scholar] [CrossRef] [PubMed]
- Horii, Y.; Katoh, K.; Miyazaki, Y.; Damjanović, M.; Sato, T.; Ungur, L.; Chibotaru, L.F.; Breedlove, B.K.; Nakano, M.; Wernsdorfer, W.; et al. Coexistence of Spin–Lattice Relaxation and Phonon-Bottleneck Processes in GdIII–Phthlocyaninato Triple-Decker Complexes under Highly Diluted Conditions. Chem. Eur. J. 2020, 26, 8076–8082. [Google Scholar] [CrossRef]
- Katoh, K.; Kajiwara, T.; Nakano, M.; Nakazawa, Y.; Wernsdorfer, W.; Ishikawa, N.; Breedlove, B.K.; Yamashita, M. Magnetic Relaxation of Single-Molecule Magnets in an External Magnetic Field: An Ising Dimer of a Terbium(III)-Phthalocyaninate Triple-Decker Complex. Chem. Eur. J. 2011, 17, 117–122. [Google Scholar] [CrossRef]
- Katoh, K.; Yamamoto, K.; Kajiwara, T.; Takeya, J.; Breedlove, B.K.; Yamashita, M. Magnetic properties of lanthanoid(III) phthalocyaninato triple-decker complexes in an external magnetic field and electronic transport properties for molecular spintronics. J. Phys. Conf. Ser. 2011, 303, 012035. [Google Scholar] [CrossRef]
- Chen, Y.; Bouvet, M.; Sizun, T.; Gao, Y.; Plassard, C.; Lesniewska, E.; Jiang, J. Facile approaches to build ordered amphiphilic tris(phthalocyaninato) europium triple-decker complex thin films and their comparative performances in ozone sensing. Phys. Chem. Chem. Phys. 2010, 12, 12851–12861. [Google Scholar] [CrossRef]
- Chen, Y.; Su, W.; Bai, M.; Jiang, J.; Li, X.; Liu, Y.; Wang, L.; Wang, S. High performance organic field-effect transistors based on amphiphilic tris(phthalocyaninato) rare earth triple-decker complexes. J. Am. Chem. Soc. 2005, 127, 15700–15701. [Google Scholar] [CrossRef]
- Martynov, A.G.; Zubareva, O.V.; Gorbunova, Y.G.; Sakharov, S.G.; Tsivadze, A.Y. Synthesis, spectral properties and supramolecular dimerisation of heteroleptic triple-decker phthalocyaninato complexes with one outer crown-substituted ligand. Inorg. Chim. Acta 2009, 362, 11–18. [Google Scholar] [CrossRef]
- Martynov, A.G.; Zubareva, O.V.; Gorbunova, Y.G.; Sakharov, S.G.; Nefedov, S.E.; Dolgushin, F.M.; Tsivadze, A.Y. Diphthalocyaninatolanthanum as a New Phthalocyaninato-Dianion Donor for the Synthesis of Heteroleptic Triple-Decker Rare Earth Element Crown-Phthalocyaninato Complexes. Eur. J. Inorg. Chem. 2007, 2007, 4800–4807. [Google Scholar] [CrossRef]
- Polovkova, M.A.; Martynov, A.G.; Birin, K.P.; Nefedov, S.E.; Gorbunova, Y.G.; Tsivadze, A.Y. Determination of the Structural Parameters of Heteronuclear (Phthalocyaninato)bis(crownphthalocyaninato)lanthanide(III) Triple-Deckers in Solution by Simultaneous Analysis of NMR and Single-Crystal X-ray Data. Inorg. Chem. 2016, 55, 9258–9269. [Google Scholar] [CrossRef] [PubMed]
- Troyanov, S.I.; Lapkina, L.A.; Larchenko, V.E.; Tsivadze, A.Y. Crystal structure of tristetra(15-crown-5)phthalocyaninatodilutetium(iii). Dokl. Chem. 1999, 367, 192–196. [Google Scholar]
- Martynov, A.G.; Polovkova, M.A.; Berezhnoy, G.S.; Sinelshchikova, A.A.; Dolgushin, F.M.; Birin, K.P.; Kirakosyan, G.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Cation-Induced Dimerization of Heteroleptic Crown-Substituted Trisphthalocyaninates as Revealed by X-ray Diffraction and NMR Spectroscopy. Inorg. Chem. 2020, 59, 9424–9433. [Google Scholar] [CrossRef]
- Martynov, A.G.; Polovkova, M.A.; Kirakosyan, G.A.; Zapolotsky, E.N.; Babailov, S.P.; Gorbunova, Y.G. 1H NMR spectral analysis of structural features in a series of paramagnetic homoleptic binuclear triple-decker phthalocyaninato lanthanide complexes. Polyhedron 2022, 219, 115792. [Google Scholar] [CrossRef]
- Ishikawa, N.; Okubo, T.; Kaizu, Y. Spectroscopic and quantum chemical studies of excited states of one-and two-electron oxidation products of a lutetium triple-decker phthalocyanine complex. Inorg. Chem. 1999, 38, 3173–3181. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, X.; Zhou, Y.; Zhang, X.; Xu, H.; Liu, Z.; Li, X.; Jiang, J. Structures and spectroscopic properties of bis(phthalocyaninato) yttrium and lanthanum complexes: Theoretical study based on density functional theory calculations. J. Phys. Chem. A 2007, 111, 392–400. [Google Scholar] [CrossRef]
- Spyroulias, G.A.; Raptopoulou, C.P.; De Montauzon, D.; Mari, A.; Poilblanc, R.; Terzis, A.; Coutsolelos, A.G. Synthesis and physicochemical characterization of protonated and deprotonated forms in heteroleptic lanthanide(III) porphyrinate double-deckers. X-ray structure of GdIIIH(oep)(tpp) at 298 and 21 K. Inorg. Chem. 1999, 38, 1683–1696. [Google Scholar] [CrossRef]
- Fukuda, T.; Hata, K.; Ishikawa, N. Observation of Exceptionally Low-Lying π–π* Excited States in Oxidized Forms of Quadruple-Decker Phthalocyanine Complexes. J. Am. Chem. Soc. 2012, 134, 14698–14701. [Google Scholar] [CrossRef]
- Liu, C.; Yang, W.; Zhang, Y.; Jiang, J. Quintuple-Decker Heteroleptic Phthalocyanine Heterometallic Samarium–Cadmium Complexes. Synthesis, Crystal Structure, Electrochemical Behavior, and Spectroscopic Investigation. Inorg. Chem. 2020, 59, 17591–17599. [Google Scholar] [CrossRef]
- Markovitsi, D.; Tran-Thi, T.-H.; Even, R.; Simon, J. Near infrared absorption spectra of lanthanide bis-phthalocyanines. Chem. Phys. Lett. 1987, 137, 107–112. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, F.; Huang, H.; Wang, J.; Yu, H.; Jiang, J.; Yan, D.; Wang, Z. Near infrared electrochromism of lutetium phthalocyanine. Synth. Met. 2005, 148, 123–126. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Nemykin, V.N.; Hadt, R.G.; Belosludov, R.V.; Mizuseki, H.; Kawazoe, Y. Influence of Molecular Geometry, Exchange-Correlation Functional, and Solvent Effects in the Modeling of Vertical Excitation Energies in Phthalocyanines Using Time-Dependent Density Functional Theory (TDDFT) and Polarized Continuum Model TDDFT Methods: Ca. J. Phys. Chem. A 2007, 111, 12901–12913. [Google Scholar] [CrossRef] [PubMed]
- Martynov, A.G.; Mack, J.; May, A.K.; Nyokong, T.; Gorbunova, Y.G.; Tsivadze, A.Y. Methodological Survey of Simplified TD-DFT Methods for Fast and Accurate Interpretation of UV-Vis-NIR Spectra of Phthalocyanines. ACS Omega 2019, 4, 7265–7284. [Google Scholar] [CrossRef]
- Damjanovic, M.; Morita, T.; Katoh, K.; Yamashita, M.; Enders, M. Ligand π-Radical Interaction with f-Shell Unpaired Electrons in Phthalocyaninato-Lanthanoid Single-Molecule Magnets: A Solution NMR Spectroscopic and DFT Study. Chem. Eur. J. 2015, 21, 14421–14432. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Kawakami, T.; Yamanaka, S.; Okumura, M. DFT and DFT-D studies on molecular structure of double-decker phthalocyaninato-terbium(III) complex. Mol. Phys. 2014, 112, 995–1001. [Google Scholar] [CrossRef]
- May, A.; Majumdar, P.; Martynov, A.G.; Lapkina, L.A.; Troyanov, S.I.; Gorbunova, Y.G.; Tsivadze, A.Y.; Mack, J.; Nyokong, T. Optical limiting properties, structure and simplified TD-DFT calculations of scandium tetra-15-crown-5 phthalocyaninates. J. Porphyr. Phthalocyanines 2020, 24, 589–601. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Ehlert, S.; Mewes, J.M. r2SCAN-3c: A “swiss army knife” composite electronic-structure method. J. Chem. Phys. 2021, 154, 064103. [Google Scholar] [CrossRef]
- Risthaus, T.; Hansen, A.; Grimme, S. Excited states using the simplified Tamm–Dancoff-Approach for range-separated hybrid density functionals: Development and application. Phys. Chem. Chem. Phys. 2014, 16, 14408–14419. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. A simplified Tamm-Dancoff density functional approach for the electronic excitation spectra of very large molecules. J. Chem. Phys. 2013, 138, 244104. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Grimme, S. A simplified time-dependent density functional theory approach for electronic ultraviolet and circular dichroism spectra of very large molecules. Comput. Theor. Chem. 2014, 1040–1041, 45–53. [Google Scholar] [CrossRef]
- Belosludov, R.V.; Nevonen, D.; Rhoda, H.M.; Sabin, J.R.; Nemykin, V.N. Simultaneous Prediction of the Energies of Q x and Q y Bands and Intramolecular Charge-Transfer Transitions in Benzoannulated and Non-Peripherally Substituted Metal-Free Phthalocyanines and Their Analogues: No Standard TDDFT Silver Bullet Yet. J. Phys. Chem. A 2019, 123, 132–152. [Google Scholar] [CrossRef] [PubMed]
- Usoltsev, S.; Shagurin, A.; Marfin, Y. Semi-Empirical Calculation of Bodipy Aggregate Spectroscopic Properties through Direct Sampling of Configurational Ensembles. Int. J. Mol. Sci. 2022, 23, 10955. [Google Scholar] [CrossRef] [PubMed]
- Schies, C.; Alemayehu, A.B.; Vazquez-Lima, H.; Thomas, K.E.; Bruhn, T.; Bringmann, G.; Ghosh, A. Metallocorroles as inherently chiral chromophores: Resolution and electronic circular dichroism spectroscopy of a tungsten biscorrole. Chem. Commun. 2017, 53, 6121–6124. [Google Scholar] [CrossRef] [PubMed]
- Yagodin, A.V.; Martynov, A.G.; Gorbunova, Y.G.; Tsivadze, A.Y. Synthesis, electronic structure and NH-tautomerism of novel mono- and dibenzoannelated phthalocyanines. Dyes Pigments 2020, 181, 108564. [Google Scholar] [CrossRef]
- Qi, D.; Zhang, L.; Wan, L.; Zhang, Y.; Bian, Y.; Jiang, J. Conformational effects, molecular orbitals, and reaction activities of bis(phthalocyaninato) lanthanum double-deckers: Density functional theory calculations. Phys. Chem. Chem. Phys. 2011, 13, 13277–13286. [Google Scholar] [CrossRef]
- Ishikawa, N.; Kaizu, Y. Excited States of the Lutetium Phthalocyanine Trimer: Semiempirical Molecular Orbital and Localized Orbital Study. J. Phys. Chem. 1996, 100, 8722–8730. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Martynov, A.G.; Berezhnoy, G.S.; Safonova, E.A.; Polovkova, M.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Aromatic Nucleophilic Substitution as a Side Process in the Synthesis of Alkoxy- and Crown-Substituted (Na)phthalocyanines. Macroheterocycles 2019, 12, 75–81. [Google Scholar] [CrossRef]
- Damjanovic, M.; Katoh, K.; Yamashita, M.; Enders, M. Combined NMR Analysis of Huge Residual Dipolar Couplings and Pseudocontact Shifts in Terbium(III)-Phthalocyaninato Single Molecule Magnets. J. Am. Chem. Soc. 2013, 135, 14349–14358. [Google Scholar] [CrossRef] [PubMed]
- Babailov, S.P.; Polovkova, M.A.; Kirakosyan, G.A.; Martynov, A.G.; Zapolotsky, E.N.; Gorbunova, Y.G. NMR thermosensing properties on binuclear triple-decker complexes of terbium(III) and dysprosium(III) with 15-crown-5-phthalocyanine. Sens. Actuators A Phys. 2021, 331, 112933. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Allouche, A.-R. Gabedit-A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Qi, D.; Jiang, J. Nature of the Intense Second-Order Nonlinear Optical Activity: DFT Studies on the Octupolarization of Sandwich-Type Bis(phthalocyaninato) Yttrium Skeletons. ChemPhysChem 2015, 16, 1889–1897. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ∆E(gas), kcal/mol | ∆E(C6H6), kcal/mol | ∆E(CH2Cl2), kcal/mol | |
|---|---|---|---|
| Y2[(MeO)8Pc]3 | −13.3 | −3.8 | −2.5 |
| Y2[(MeO)8Pc]3+ | −5.6 | 3.2 | 3.8 |
| Y2[(MeO)8Pc]32+ | 2.4 | 10.0 | 10.0 |
| d(Y…Y), Å | d(N4…N4), Å | θ, ° | d(Y…Y), Å | d(N4…N4), Å | θ, ° | |
|---|---|---|---|---|---|---|
| s-Conformer | g-Conformer | |||||
| Y2[(MeO)8Pc]3 | 3.403 (3.429) 2 | 2.923 (2.952) 2 | 45 1 (43.9) 2 | 3.486 (3.517) 4 | 2.983 (3.028) 4 | 22.6 (33.0) 4 |
| Y2[(MeO)8Pc]3+ | 3.385 | 2.898 | 451 | 3.459 | 2.97 | 23.0 |
| Y2[(MeO)8Pc]32+ | 3.366 (3.435) 3 | 2.873 (2.980)3 | 45 1 (44.2) 3 | 3.442 | 2.956 | 22.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martynov, A.G.; Polovkova, M.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Redox-Triggered Switching of Conformational State in Triple-Decker Lanthanide Phthalocyaninates. Molecules 2022, 27, 6498. https://doi.org/10.3390/molecules27196498
Martynov AG, Polovkova MA, Gorbunova YG, Tsivadze AY. Redox-Triggered Switching of Conformational State in Triple-Decker Lanthanide Phthalocyaninates. Molecules. 2022; 27(19):6498. https://doi.org/10.3390/molecules27196498
Chicago/Turabian StyleMartynov, Alexander G., Marina A. Polovkova, Yulia G. Gorbunova, and Aslan Yu. Tsivadze. 2022. "Redox-Triggered Switching of Conformational State in Triple-Decker Lanthanide Phthalocyaninates" Molecules 27, no. 19: 6498. https://doi.org/10.3390/molecules27196498
APA StyleMartynov, A. G., Polovkova, M. A., Gorbunova, Y. G., & Tsivadze, A. Y. (2022). Redox-Triggered Switching of Conformational State in Triple-Decker Lanthanide Phthalocyaninates. Molecules, 27(19), 6498. https://doi.org/10.3390/molecules27196498