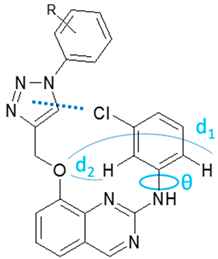
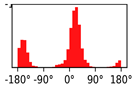
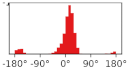
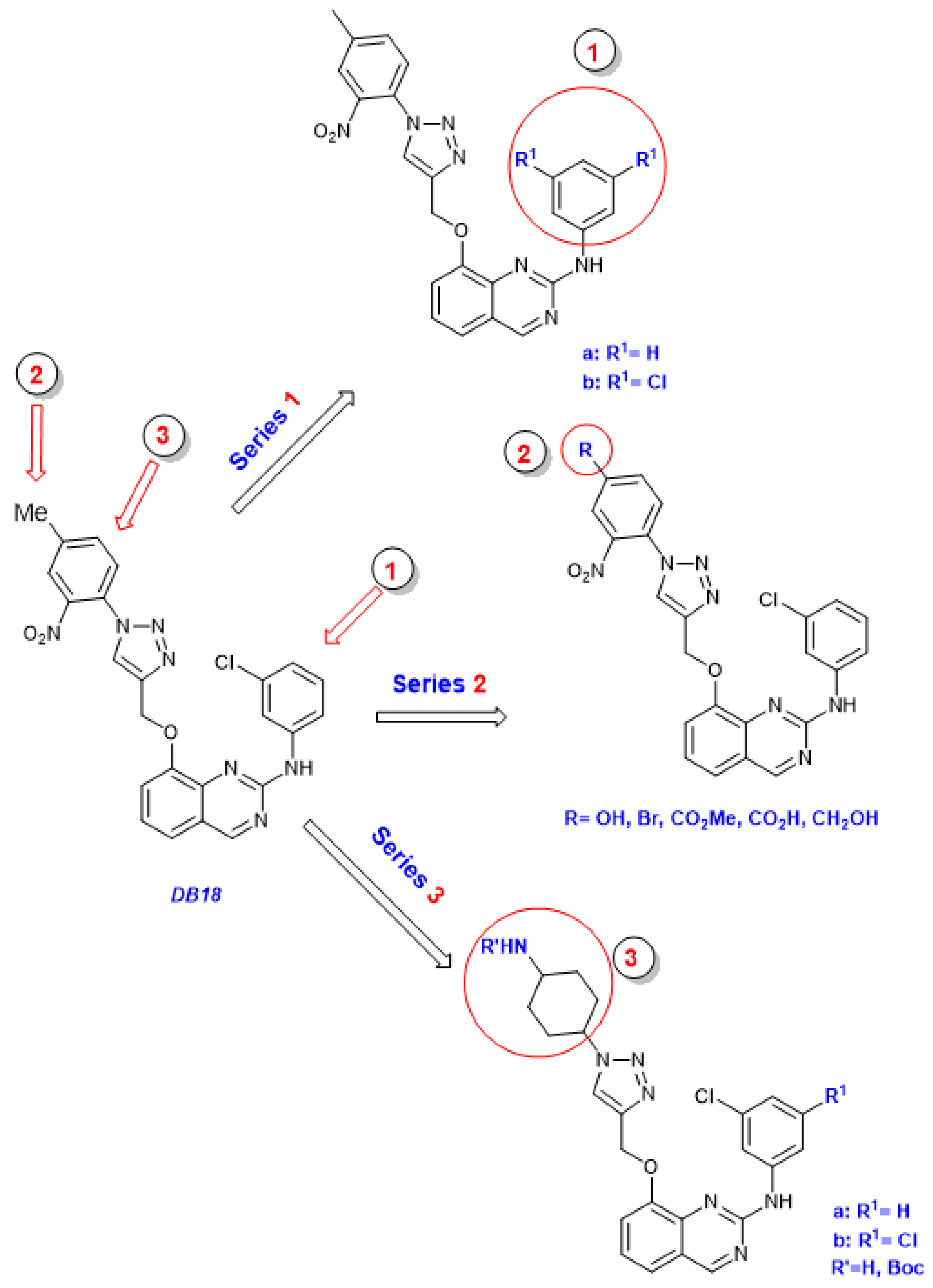
3.1. Chemical Syntheses
General information:
For this, see corresponding data in ref [
14]. All triazole compounds were purified by flash column chromatography on silica gel using 20% ethyl acetate in hexane, unless otherwise noted. For all final compounds (
7,
12,
16 and
19) a purity >95% was established by LC-MS.
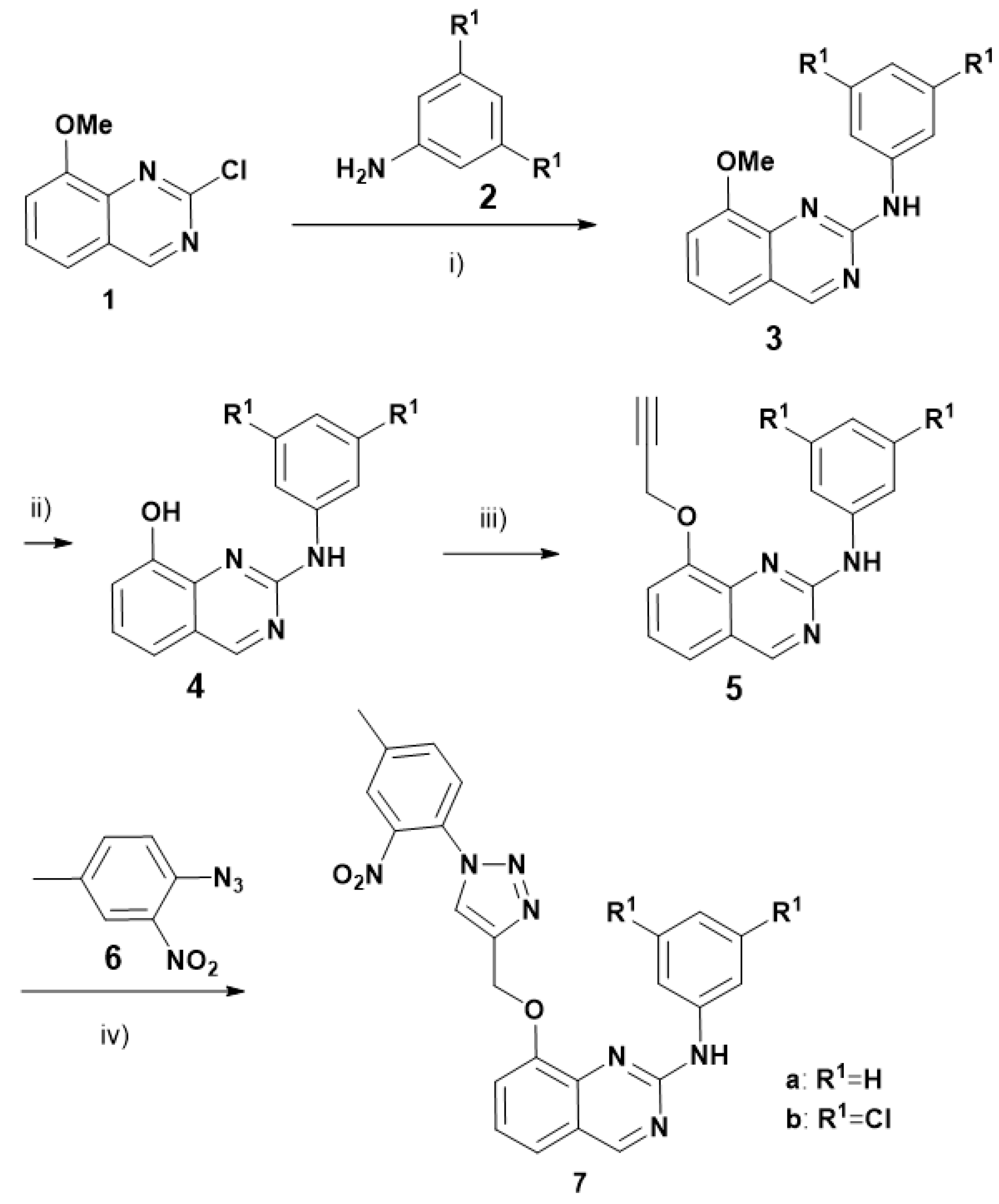
Preparation of compounds 3, 4, 7a, 7b, 13:
Compounds
3,
4,
5,
6,
7a,
7b,
13 were prepared as already described for
DB18 [
14].
N-Phenyl-8-((1-(4-methyl-2-nitrophenyl)-1H-1,2,3-triazol-4-yl)methoxy)quinazolin-2-amine (7a) (58%):
1H NMR (300 MHz, DMSO-d6, δ ppm): 9.88 (br s, 1H, NH), 9.29 (s, 1H), 8.80 (s, 1H), 8.09 (br dd, J = 1.1, 0.5 Hz, 1H), 8.06–7.99 (m, 2H), 7.79 (ddd with a strong roof effect, J = 8.2, 1.7, 0.5 Hz, 1H), 7.75 (br d with a strong roof effect, J = 8.1 Hz, 1H), 7.58–7.49 (m, 2H), 7.33 (t, J = 7.9 Hz, 1H), 7.26–7.17 (m, 2H), 6.88 (ddt, J = 7.6, 7.0, 1.0 Hz, 1H), 5.48 (s, 2H), 2.53 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ ppm): 161.8 (CH), 156.2 (C), 151.3 (C), 143.8 (C), 143.6 (C), 142.9 (C), 142.0 (C), 140.5 (C), 134.6 (CH), 128.3 (2CH), 127.2 (CH), 126.6 (C), 125.8 (CH), 125.5 (CH), 123.2 (CH), 121.2 (CH), 121.0 (C), 120.1 (CH), 118.5 (2CH), 115.8 (CH), 62.1 (CH2), 20.3 (CH3); HRMS (CH3OH/CH2Cl2 = 9:1) m/z calcd for C24H20N7O3 [M + H]+: 454.16221, found: 454.1626, calcd for C24H19N7NaO3 [M+Na]+: 476.14416, found: 476.1445, calcd for C24H19N7KO3 [M+K]+: 492.11810, found: 492.1182.
N-(3,5-dichlorophenyl)-8-((1-(4-methyl-2-nitrophenyl)-1H-1,2,3-triazol-4-yl)methoxy)quinazolin-2-amine (7b) (54%):
1H NMR (400 MHz, DMSO-d6) δ ppm: 10.34 (s, 1H), 9.38 (s, 1H), 8.86 (s, 1H), 8.17 (d, J = 1.6 Hz, 2H), 8.07 (s, 1H), 7.80–7.78 (m, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.61–7.58 (m, 2H), 7.42 (t, J = 8.0 Hz, 1H), 7.02 (t, J = 2.0 Hz, 1H), 5.45 (s, 2H), 2.08 (s, 3H); Mass (m/z): 522.1 (M + H).
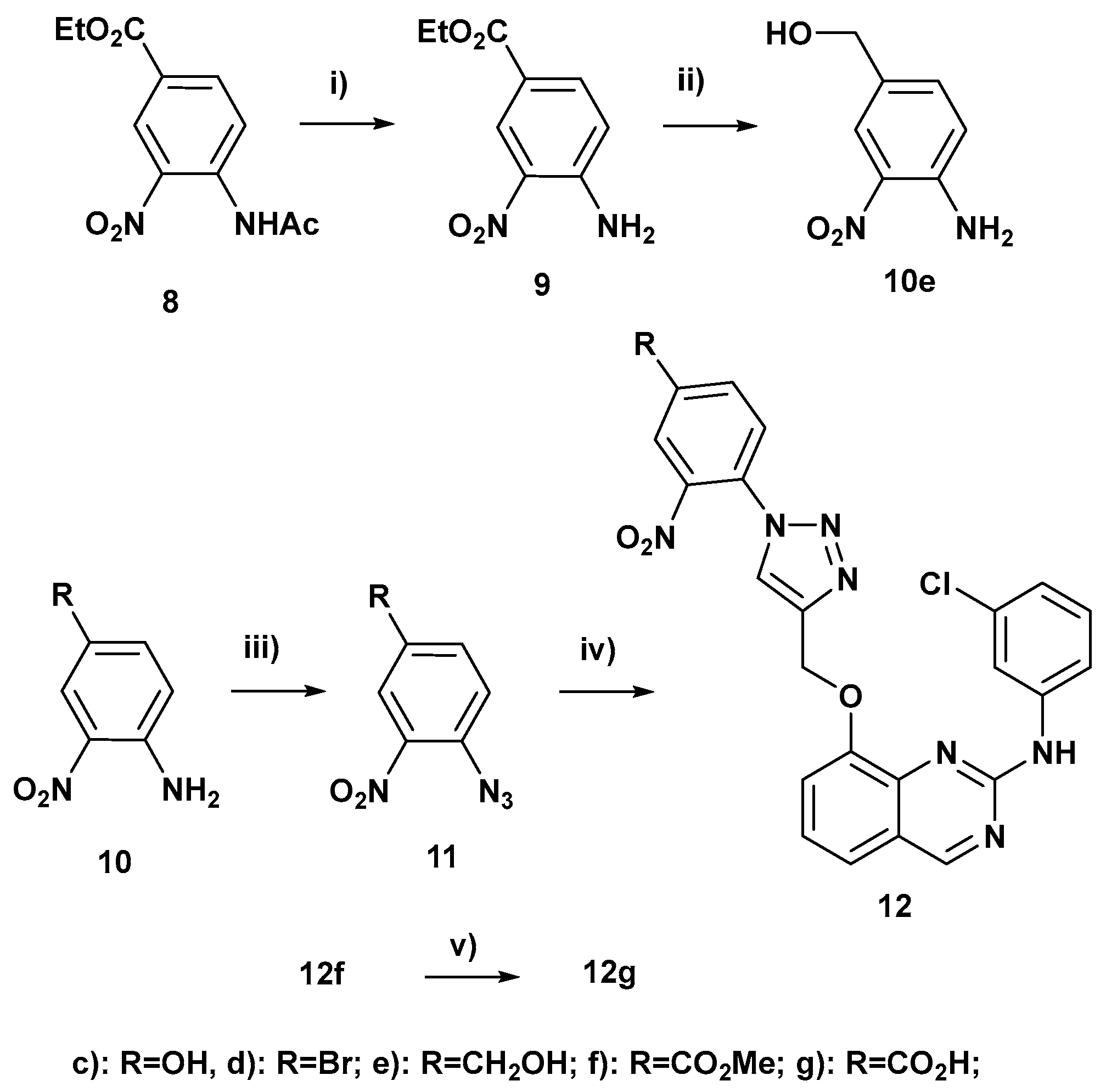
Ethyl 4-amino-3-nitrobenzoate (9):
In a 100 mL round-bottomed flask, ethyl 4-acetamido-3-nitrobenzoate (8) (1.04 g, 4 mmol), acetic acid (2 mL, 34.9 mmol, 8.72 equiv), 1,3-propanediol (6 mL), hydrazine monohydrate (1.785 g, 35 mmol, 8.75 equiv) and an olive-shaped magnetic stirring bar were successively introduced. This reaction flask was flushed under nitrogen, stoppered tightly and steeped in a preheated oil bath at 90 °C. It was left for 15 h at 90 °C under smooth stirring (160 rpm). After cooling down to rt, water (30 mL) was added and the resulting mixture was stirred at 200 rpm. It was filtered using a Büchner and the yellow precipitated solid was washed several times with water. It was transferred into a bigger flask and dried under vacuum with a pump. It afforded ethyl 4-amino-3-nitrobenzoate (9) (718 mg) whose NMR showed that it was pure. Rinsing by hot methanol the Büchner and the filter paper afforded additional 9 (60 mg) so the overall yield of 9 was 91%; 1H NMR (300 MHz, acetone-d6, δ ppm): 8.71 (d, J = 2.0 Hz, 1H), 7.94 (dd, J = 8.9, 2.0 Hz, 1H), 7.7–7.3 (envelope which topped at 7.52 ppm, 2H, NH2), 7.14 (d, J = 8.9 Hz, 1H), 4.33 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, acetone-d6, δ ppm): 165.4 (C), 149.6 (C), 136.0 (CH), 129.0 (CH), 120.0 (CH), 118.9 (C), 61.4 (CH2), 14.6 (CH3).
4-Amino-3-nitrobenzyl alcohol (10e):
In a 250 mL round-bottomed flask, lithium chloride (initially 1.07 g, 0.813 g after drying, 19 mmol) was dried by flaming under vacuum. After cooling down rt, ethyl 4-amino-3-nitrobenzoate (201.9 mg, 0.96 mmol), 95% ethanol (14.4 mL), methanol (1.44 mL) and an olive-shaped magnetic stirring bar were successively introduced. This mixture was stirred at rt until LiCl was dissolved and sodium borohydride (735 mg, 19 mmol) was added. A red color appeared. The reaction flask was flushed under nitrogen, tightly stoppered and stirred at rt for 40 min. At this time, thickening was observed and LiBH(OMe)3 was formedin situas the reducing species. An excess of pressure (due to hydrogen formation) was evacuated by brief opening before tight stoppering again. This resulting mixture was stirred at 45 °C for 12 h upon which it changed from red to orange. TLC showed a complete reaction with a more polar spot of the expected alcohol (Rf = 0.22 with petroleum ether/acetone 70:30 vs. 0.48 for the starting ethyl ester). Ethyl acetate (60 mL) and water (15 mL) were added and stirring at 500 pm was continued for 20–30 min. The remaining mineral mass was washed more than 10 times with stirring at rt with ethyl acetate. Ethyl acetate was then removed in vacuo followed by ethanol and water under high vacuum. The remaining residue and intermediate impure fractions were subjected to 4 successive chromatographies on silica gel (8–9 g) columns with gradient elution with petroleum ether/acetone 85:15 to 75:25 (deposit on silica gel was made with toluene + ethanol). It afforded 4-amino-3-nitrobenzyl alcohol (10e) as a red crystallized solid (57.0 mg, 35%); 1H NMR (300 MHz, acetone-d6, δ ppm): 8.02 (ddd after improving the resolution with Traficante, J = 2.0, 1.2, 0.8 Hz, 1H), 7.41 (dd, J = 8.6, 2.0 Hz (ddt after improving the resolution with Traficante, J = 8.6, 2.0, 0.5 Hz), 1H), 7.04 (dd after improving the resolution with Traficante, J = 8.6, 0.4 Hz, 1H), 7.3–6.8 (envelope which topped at 6.94 ppm, 2H, NH2), 4.54 (d, J = 4.6 Hz, 2H), 4.33–4.15 (m which topped at 4.24 ppm, 1H, OH); 13C NMR (75 MHz, acetone-d6, δ ppm): 146.0 (C), 135.7 (CH), 132.0 (broad and small, C), 131.4 (C), 124.0 (CH), 120.0 (CH), 63.6 (CH2).
4-Azido-3-nitrophenol (11c):
37% Aqueous hydrochloric acid (3.2 equiv) was added to a stirred solution of 4-amino-3-nitrophenol (157.6 mg, 1 mmol) in water (2.2 mL). After cooling by an ice bath, an aqueous solution of sodium nitrite (91.7 mg, 1.31 mmol, 1.31 equiv) in water (0.41 mL) was added dropwise. Transfer of NaNO2 was completed by rinsing with water (0.12 mL). After 1 h upon which the bath temperature had risen from 0 to 2 °C, an aqueous solution of sodium azide (104.7 mg, 1.6 mmol, 1.6 equiv) in water (0.53 mL) was added dropwise under stirring. Transfer of NaN3 was completed by rinsing with water (0.15 mL). An abundant precipitation of yellow crystals immediately occurred. After 45 min further stirring at 0 °C, the ice bath was removed and the reaction mixture was left aside to stir at rt overnight. The aspect of the reaction mixture had not at all changed so the reaction of N3− was probably very fast. Extraction with ethyl acetate readily dissolved the precipitate and the organic extract was washed with water until neutral (3 times). Combined aqueous layers were reextracted with ethyl acetate and this second organic extract was washed with water until neutral (2 times). Combined organic extracts were dried (Na2SO4), concentrated and put under vacuum to afford a brown solid. TLC spot of the azide 11c (0.42 with hexane/acetone 7:3) was yellow at the start and became brownish after less than one hour contact with air. Chromatography of the brown crude product on silica gel (2.0 g) with elution with hexane/acetone 90:10 and then 85:15 afforded 11c as a yellow crystallized solid (174.8 mg, 97%); 1H NMR (300 MHz, acetone-d6, δ ppm): 12-8 (envelope which topped at 9.56 ppm, 1H, OH), 7.39 (d, J = 8.7 Hz, 1H), 7.39 (d, J = 2.9 Hz, 1H), 7.24 (dd, J = 8.9, 2.8 Hz, 1H); 13C NMR (100 MHz, acetone-d6, δ ppm): 155.8 (C), 142.7 (broad and small, C), 126.0 (C), 123.6 (CH), 122.4 (CH), 112.5 (CH).
1-Azido-4-bromo-2-nitrobenzene (11d):
To a stirred solution of 1-amino-4-bromo-2-nitrobenzene (11d) (500 mg, 2.30 mmol) in 6N HCl (10 mL) was added NaNO2 (317 mg, 4.60 mmol) in 2 mL water at 0 °C. Then stirred for 2 h at 0 °C, after then add NaN3 (299 mg, 4.60 mmol) lot wise at 0 °C, and stirred for overnight at rt. After completion of the reaction by TLC, the reaction mixture was poured into water and EtOAc. The phases were separated, and the organic layer was washed with water and concentrated to obtain the azide 11d as a yellow solid. Compound directly used for the next step without purification; 1H NMR (400 MHz, CDCl3, δ ppm): 8.07 (d, J = 2.3 Hz, 1H), 7.73 (dd, J = 8.6, 2.3 Hz, 1H), 7.22 (d, J = 8.6 Hz, 1H); 13C NMR (100 MHz, CDCl3, δ ppm): 141.1 (small, C), 136.9 (CH), 134.0 (C), 128.9 (CH), 122.2 (CH), 117.1 (C).
4-Azido-3-nitrobenzyl alcohol (11e):
In a 25 mL round-bottomed flask, 4-amino-3-nitrobenzyl alcohol (19.7 mg, 0.117 mmol) was dissolved under stirring by adding water (1.25 mL), 37% aqueous hydrochloric acid (156 mg, 6 drops) and 95% ethanol (2.35 mL). The resulting orange solution was cooled at 0 °C. An aqueous solution of sodium nitrite (41.4 mg NaNO2, 0.594 mmol, 5.08 équiv + 462.4 mg water) was added dropwise under stirring. After 3 h further stirring at 0 °C, an aqueous solution of sodium azide (31.3 mg NaN3, 0.479 mmol, 4.1 équiv + 226.4 mg water) was added dropwise. Transfer of NaN3 was completed by rinsing with water (77.1 mg). Upon addition of NaN3, the color of the raction mixture changed from orange to yellow and a precipitation appeared. Stirring was then continued for 100 min at 0 °C and the resulting mixture was then left aside in a freezer overnight. After warming up to rt, TLC showed a less polar spot of azide (Rf = 0.35 with petroleum ether/acetone 70:30) and two minor more polar spots, the most polar seemed to be that of starting amine. Water (about 5 mL) and sodium bicarbonate (90 mg) were added. The resulting mixture was extracted 4 times with ethyl acetate. After drying over sodium sulfate and concentration, the remaining orange crude wax (22.4 mg) was subjected to a chromatography on a column of silica gel (5 g) with gradient elution with petroleum ether/acetone 95:5 to 80:20 (deposit on silica gel was made with toluene + a small amount of ethanol with heating). It afforded 4-azido-3-nitrobenzyl alcohol (11e) as a cream yellow crystallized solid (10.7 mg, 47%); 1H NMR (300 MHz, CDCl3, δ ppm): 7.95 (dt after improving the resolution with Traficante, J = 2.0, 0.9 Hz, 1H), 7.62 (ddt, J = 8.3, 2.0, 0.7 Hz), 1H), 7.32 (d, J = 8.3 Hz, 1H), 4.76 (s, 2H), 2.2-1.9 (envelope which topped at 2.06 ppm, 2H, OH); 13C NMR (100 MHz, CDCl3, δ ppm): 140.8 (broad and small, C), 138.5 (C), 133.8 (C), 132.1 (CH), 124.1 (CH), 120.9 (CH), 63.4 (CH2).
Methyl 4-azido-3-nitrobenzoate (11f):
To a stirred solution of methyl 4-amino-3-nitrobenzoate (10f) (500 mg, 2.55 mmol) in 6N HCl (10 mL) was added NaNO2 (352 mg, 5.1 mmol) in 2 mL water at 0 °C. Then stirred at 0 °C for 2 h, after then added NaN3 (331 mg, 5.1 mmol) lot wise at 0 °C, stirred for overnight at rt. After completion of the reaction by TLC, the reaction mixture was poured into water and EtOAc. The phases were separated, and the organic layer was washed with water and concentrated to obtain the azide 11f as a yellow solid. This compound was directly used for the next step without purification.
N-(3-Chlorophenyl)-8-((1-(4-hydroxy-2-nitrophenyl)-1H-1,2,3-triazol-4-yl)methoxy)quinazolin-2-amine (12c):
In a 10 mL round-bottomed flask containing 4-azido-3-nitrophenol (11c) (22.8 mg, 0.1266 mmol) was added alkyne 13 (37.7 mg, 0.1217 mmol), copper(II) sulfate pentahydrate (2.8 mg, 0.011 mmol, 0.09 equiv), sodium ascorbate (6.7 mg, 0.0335 mmol, 0.275 equiv), urea (2.3 mg, 0.0375 mmol, 0.31 equiv), water (89.0 mg, 3 drops), DMSO (149.0 mg, 8 drops), THF (0.655 mL) and a small olive-shaped magnetic stirring bar. That gave a mixture THF/DMSO/H2O about 15:3:2. The reaction flask was flushed under nitrogen, tightly stoppered and left under moderate stirring at rt (22–23 °C) for 5.5 h. In a separating funnel, water (0.42 mL), brine (saturated aq. NaCl: 1.57 mL) and 28–30% aqueous ammonium hydroxide (3 drops) were introduced to which was added the reaction mixture which was transferred by ethyl acetate and THF followed by diluted brine in order to transfer residual minerals which were remaining in the reaction flask. After partitioning, the aqueous phase was reextracted with ethyl acetate plus THF. Combined organic extracts were washed once with brine, dried (Na2SO4), concentrated and the remaining residue was put under vacuum. Chromatography on silica gel (2.1 g) column, which was loaded with dichloromethane (deposit of the crude product on the silica gel by dissolving in THF plus a little bit of methanol with smooth heating) eluting with 2% MeOH-chloroform, afforded triazole 12c as a light orange powder (65.2 mg, quant., Rf = 0.09 with 2% MeOH-chloroform). An analytically pure sample was obtained by crystallization in acetone/petroleum ether about 1:1 with final cooling in a freezer at about −35 °C to afford a yellow powder; 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.13 (br s, 1H, NH), 9.34 (s, 1H), 8.72 (s, 1H), 8.42 (t, J = 2.0 Hz, 1H), 7.80 (ddd, J = 8.3, 2.1, 0.9 Hz, 1H), 7.63 (d, J = 8.7 Hz, 1H), 7.57 (d, J = 7.9 Hz, 2H), 7.54 (d, J = 2.7 Hz, 1H), 7.38 (dd, J = 8.2, 7.5 Hz, 1H), 7.30 (dd, J = 8.7, 2.7 Hz, 1H), 7.24 (t, J = 8.1 Hz, 1H), 6.92 (ddd, J = 7.9, 2.1, 0.9 Hz, 1H), 5.46 (s, 2H); 13C NMR (100 MHz, DMSO-d6, δ ppm): 162.0 (CH), 159.2 (C), 155.9 (C), 151.5 (C), 144.9 (C), 143.1 (C), 142.5 (C), 142.1 (C), 133.0 (C), 129.8 (CH), 128.9 (CH), 126.0 (CH), 123.8 (CH), 121.2 (C), 120.6 (CH), 120.5 (C), 120.5 (CH), 119.9 (CH), 117.5 (CH), 116.8 (CH), 115.4 (CH), 111.6 (CH), 62.0 (CH2); HRMS (CH3OH/CH2Cl2 = 9:1) m/z calcd for C23H1635ClN7NaO4 [M + Na]+: 512.08445, found: 512.0848, calcd for C23H1635ClN5NaO4[M-N2 + Na]+: 484.07830, found: 484.0782, C23H1635ClN7KO4 [M + K]+: 528.05839, found: 528.0579.
N-(3-Chlorophenyl)-8-((1-(4-bromo-2-nitrophenyl)-1H-1,2,3-triazol-4-yl)methoxy)quinazolin-2-amine (12d):
To a stirred solution of alkyne 13 (100 mg, 0.32 mmol) in t-BuOH: water (1:1) (2 mL) was added azide 18d (157 mg, 0.64 mmol), sodium ascorbate (128 mg, 0.64 mmol), CuSO4.5H2O (161 mg, 0.64 mmol) in 4 mL water at RT. Then stirred for overnight at rt. After completion of the reaction by TLC, the reaction mixture was poured into water and EtOAc. The phases were separated, and the organic layer was washed with brine solution, dried over sodium sulphate and concentrated. Crude was purified by column chromatography (60–120 silica gel, plane ethyl acetate) to afford triazole 19d (100 mg, 56%) as a brown solid (LC-MS purity: 92.8%); 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.13 (br s, 1H, NH), 9.34 (s, 1H), 8.89 (s, 1H), 8.52 (d, J = 2.1 Hz, 1H), 8.43 (t, J = 2.0 Hz, 1H), 8.22 (dd, J = 8.5, 2.2 Hz, 1H), 7.85 (d, J = 8.5 Hz, 1H), 7.78 (ddd, J = 8.3, 2.1, 0.9 Hz, 1H), 7.60-7.53 (m, 2H), 7.38 (t, J = 7.9 Hz, 1H), 7.23 (t, J = 8.1 Hz, 1H), 6.92 (ddd, J = 7.9, 2.1, 0.9 Hz, 1H), 5.48 (s, 2H); 13C NMR (100 MHz, DMSO-d6, δ ppm): 162.0 (CH), 155.9 (C), 151.5 (C), 144.2 (C), 143.6 (C), 142.4 (C), 142.1 (C), 137.0 (CH), 132.9 (C), 129.8 (CH), 128.6 (CH), 128.2 (CH), 128.0 (C), 125.7 (CH), 123.7 (CH), 123.0 (C), 121.2 (C), 120.6 (CH), 120.0 (CH), 117.5 (CH), 116.7 (CH), 115.4 (CH), 61.9 (CH2); LC-MS: 554.1 [M + 2H] +; HRMS (CH3OH/CH2Cl2 = 9:1) m/z calcd for C23H1579Br35ClN7NaO3 [M + Na]+: 574.00005, found: 574.0005, calcd for C23H1579Br35ClN7KO3 [M + K]+: 589.97398, found: 589.9740, calcd for C23H1679Br35ClN7O3 [M + H]+: 552.01810, found: 552.0176.
4-(4-(((2-((3-Chlorophenyl)amino)quinazolin-8-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-3-nitrobenzyl alcohol (12e):
In a 25 mL round-bottomed flask containing 4-azido-3-nitrobenzyl alcohol (11e) (10.7 mg, 0,055 mmol) was added alkyne 5 (17.1 mg, 0.055 mmol), copper(II) sulfate pentahydrate (2.1 mg, 0.00824 mmol, 0.15 equiv), sodium ascorbate (5.2 mg, 0.026 mmol, 0.47 equiv), urea (1.8 mg, 0.0294 mmol, 0.53 equiv), water (53.1 mg, 2 drops), DMSO (105.0 mg, 6 drops), THF (0.553 mL) and a small olive-shaped magnetic stirring bar. The reaction flask was flushed under nitrogen, tightly stoppered and left under moderate stirring at rt for 4.5 h. TLC showed a complete reaction with the expected triazole as a very polar compound (Rf ~ 0.04 with petroleum ether/acetone 70:30 vs. 0.45 for the starting alkyne and 0.35 for the starting azide. In a separating funnel, water (1 mL) and 25% aqueous ammonium hydroxide (4 drops) were introduced to which was added the reaction mixture which was transferred by ethyl acetate (8.3 mL overall). A fine brick red precipitate appeared (presumably Cu2O). After partitioning, the aqueous phase was reextracted with ethyl acetate (3–4 mL). Combined yellow organic extracts were dried (Na2SO4), concentrated and the brown oily residue was put under vacuum. Chromatography on silica gel (6.6 g) eluting with ethyl acetate afforded triazole 12e (Rf = 0.45 with ethyl acetate) initially as a yellow oil which entirely crystallized in about 15 min at 50 °C to yield a yellow solid (22.4 mg, 81%).
1H NMR (300 MHz, DMSO-d6, δ ppm): 10.13 (br s, 1H, NH), 9.34 (s, 1H), 8.86 (s, 1H), 8.44 (t, J = 2.0 Hz, 1H), 8.15 (dt, J = 1.7, 0.9 Hz, 1H), 7.90 (ddt with a strong roof effect, J = 8.2, 1.8, 0.7 Hz, 1H), 7.82 (d with a strong roof effect, J = 8.2 Hz, 1H), 7.78 (ddd, J = 8.3, 2.1, 0.9 Hz, 1H), 7.57 (pseudo d, J = 8.0 Hz, 2H), 7.38 (dd, J = 8.4, 7.4 Hz, 1H), 7.23 (t, J = 8.1 Hz, 1H), 6.91 (ddd, J = 7.9, 2.1, 0.9 Hz, 1H), 5.68 (t, J = 5.7 Hz, 1H), 5.48 (s, 2H), 4.72 (d, J = 5.7 Hz, 2H); 13C NMR (100 MHz, DMSO-d6, δ ppm): 162.0 (CH), 155.9 (C), 151.5 (C), 146.6 (C), 143.7 (C), 143.4 (C), 142.4 (C), 142.1 (C), 132.9 (C), 131.4 (CH), 129.8 (CH), 127.3 (C), 127.1 (CH), 125.8 (CH), 123.7 (CH), 122.6 (CH), 121.2 (C), 120.6 (CH), 119.9 (CH), 117.5 (CH), 116.7 (CH), 115.3 (CH), 61.9 (CH2), 61.3 (CH2)); HRMS (CH3OH/CH2Cl2 = 9:1) m/z calcd for C24H1835ClN7NaO4 [M + Na]+: 526.10010, found: 526.1008, C24H1835ClN7KO4 [M + K]+: 542.07404, found: 542.0743.
Methyl 4-(4-(((2-((3-chlorophenyl)amino)quinazolin-8-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-3-nitrobenzoate (12f):
To a stirred solution of alkyne 13 (100 mg, 0.32 mmol) in t-BuOH: water (1:1) (2 mL) was added azide 11f (134 mg, 0.64 mmol), sodium ascorbate (128 mg, 0.64 mmol), CuSO4.5H2O (161 mg, 0.64 mmol) in 4 mL water at rt. Then stirred at rt for overnight. After completion of the reaction by TLC, the reaction mixture was poured into water and EtOAc. The phases were separated, and the organic layer was washed with brine solution, dried over sodium sulphate and concentrated. Crude purified by column chromatography (60–120 silica gel, plane ethyl acetate) to afford triazole 12f (40 mg, 23%) as a brown solid. 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.13 (br s, 1H, NH), 9.34 (s, 1H), 8.98 (s, 1H), 8.64 (d, J = 1.8 Hz, 1H), 8.49–8.42 (m, 2H), 8.06 (d, J = 8.3 Hz, 1H), 7.77 (ddd, J = 8.3, 2.0, 0.8 Hz, 1H), 7.58 (pseudo dm, J = 7.8 Hz, 2H), 7.38 (t, J = 7.9 Hz, 1H), 7.23 (t, J = 8.1 Hz, 1H), 6.90 (ddd, J = 7.9, 2.1, 0.8 Hz, 1H), 5.50 (s, 2H), 3.97 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ ppm): 163.7 (C), 162.0 (CH), 155.9 (C), 151.5 (C), 143.8 (C), 143.6 (C), 142.5 (C), 142.0 (C), 134.3 (CH), 132.9 (C), 131.9 (C), 131.6 (C), 129.8 (CH), 127.5 (CH), 126.1 (CH), 125.7 (CH), 123.7 (CH), 121.2 (C), 120.6 (CH), 120.0 (CH), 117.6 (CH), 116.7 (CH), 115.4 (CH), 61.8 (CH2), 53.0 (CH3); HRMS (CH3OH/CH2Cl2=9:1) m/z calcd for C25H1835ClN7NaO5 [M + Na]+: 554.09501, found: 554.0953, calcd for C25H1835ClN7KO5 [M + K]+: 570.06895, found: 570.0689, calcd for C25H1935ClN7O5 [M + H]+: 532.11307, found: 532.1129.
4-(4-(((2-((3-Chlorophenyl)amino)quinazolin-8-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-3-nitrobenzoic acid (12g):
To a stirred solution of methyl ester 12f (30 mg, 0.0564 mol) in MeOH/THF/water (3:2:1) (3 mL) was added LiOH.H2O (7.0 mg, 0.169 mol) and stirring was continued at rt for 16 h. After completion of the reaction by TLC, the reaction mixture was concentrated, diluted with water and acidified with 2N aqueous HCl. Then the solid which was formed was filtered and dried to obtain carboxylic acid 12g as a yellow solid (15 mg, 52%). 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.13 (br s, 1H, NH), 9.34 (s, 1H), 8.96 (s, 1H), 8.61 (d, J = 1.8 Hz, 1H), 8.47–8.41 (m, 2H), 8.02 (d, J = 8.3 Hz, 1H), 7.78 (ddd, J = 8.3, 2.1, 0.9 Hz, 1H), 7.58 (pseudo dm, J = 7.8 Hz, 2H), 7.38 (t, J = 7.9 Hz, 1H), 7.23 (t, J = 8.1 Hz, 1H), 6.91 (ddd, J = 7.9, 2.1, 0.9 Hz, 1H), 5.50 (s, 2H) [Remark: H of CO2H was not detected]; 13C NMR (75 MHz, DMSO-d6, δ ppm): 164.6 (C), 162.0 (CH), 155.9 (C), 151.5 (C), 143.8 (C), 143.6 (C), 142.5 (C), 142.0 (C), 134.4 (CH), 133.0 (C), 132.9 (C), 131.6 (C), 129.8 (CH), 127.5 (CH), 126.1 (CH), 125.7 (CH), 123.7 (CH), 121.2 (C), 120.6 (CH), 120.0 (CH), 117.6 (CH), 116.8 (CH), 115.5 (CH), 61.9 (CH2); LC-MS purity: 95%, LC-MS: 518.1 [M + H]+; HRMS (CH3OH/CH2Cl2 = 9:1) m/z calcd for C24H1535ClN7O5 [M-H]−: 516.08287, found: 516.0831, calcd for C24H1635Cl2N7O5 [M + Cl]−: 552.05955, found: 552.0598, calcd for C24H1535Cl2N7NaO5 [M-H + Na + Cl]−: 574.04149, found: 574.0417.
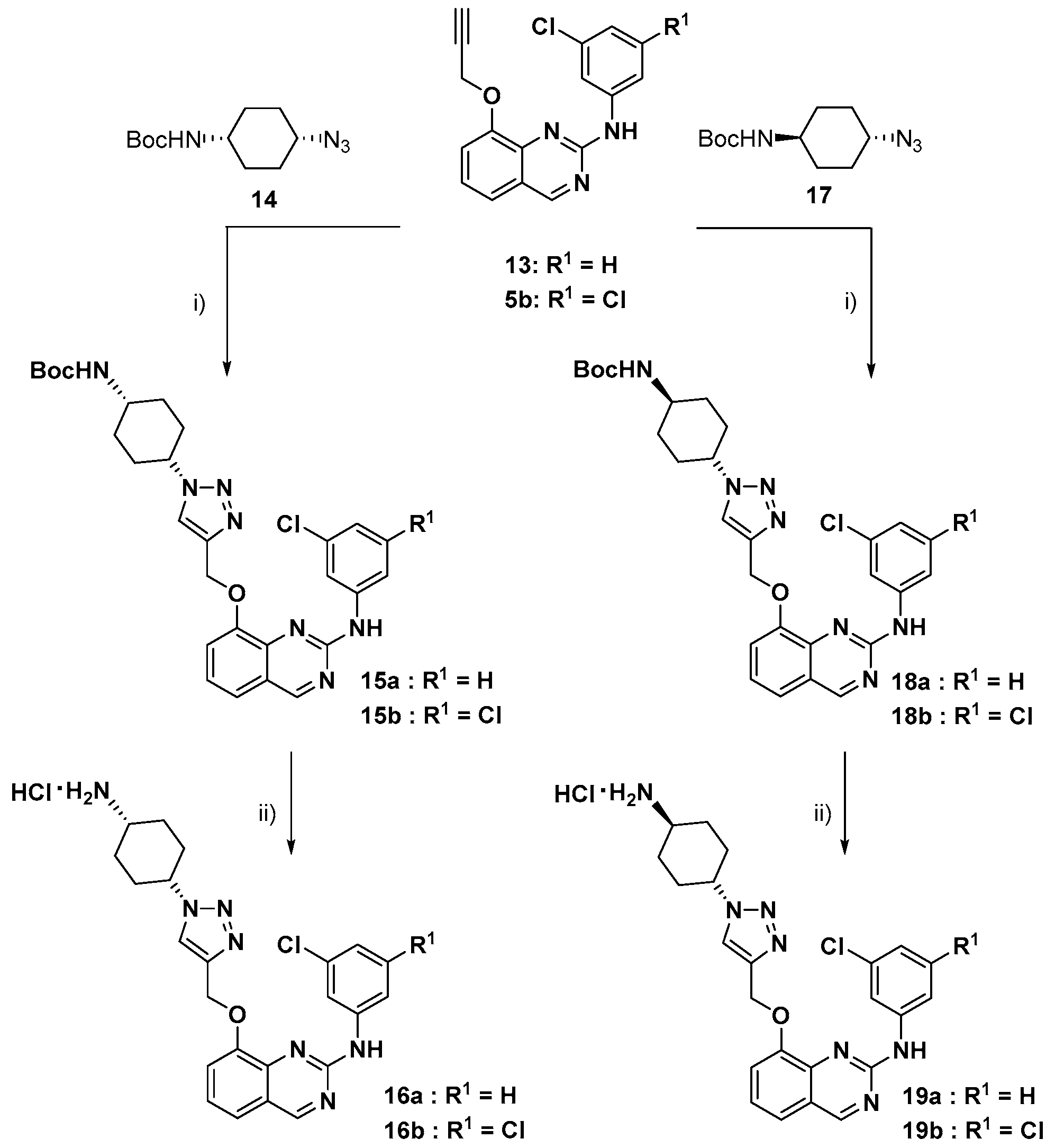
tert-Butyl (cis-4-azidocyclohexyl)carbamate (14):
Step 1. Synthesis of trans-4-(tert-butoxycarbonylamino)cyclohexyl methanesulfonate:
To a stirred solution of trans-4-Boc-aminocyclohexanol (500 mg, 2.32 mmol) in DCM (5 mL) was added TEA (1 mL, 6.97 mmol) at rt and stirred for 15 min. Then added Ms-Cl (0.3 mL, 3.48 mmol) drop wisely, stirred at RT for 6 h. After completion of the reaction by TLC, the reaction mixture was poured into water (5 mL). The phases were separated, and the organic layer was washed with a sat aq. NaHCO3 solution (5 mL), dried over sodium sulfate and concentrated and dried to obtain the trans-4-(tert-butoxycarbonylamino)cyclohexyl methanesulfonate as a colorless liquid.
Step 2. Synthesis of tert-butyl (cis-4-azidocyclohexyl)carbamate (14):
To a stirred solution of trans-4-(tert-butoxycarbonylamino)cyclohexyl methanesulfonate (500 mg, 1.70 mmol) in DMF (3 mL) was added NaN3 (1.1 g, 17.06 mmol) at rt. Then heated to 80 °C for 24 h. After completion of the reaction by TLC, the reaction mixture was poured into ice water, solid was formed, filtered and dried to obtain the azide 14 as an off-white solid. This compound was directly used for the next step without purification.
tert-Butyl ((cis)-4-(4-(((2-((3-chlorophenyl)amino)quinazolin-8-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)cyclohexyl)carbamate (15a):
To a stirred solution of alkyne 13 (100 mg, 0.32 mmol) in t-BuOH: water (1:1) (2 mL) was added azide 14 (77.6 mg, 0.32 mmol), followed by sodium ascorbate (64 mg, 0.32 mmol) and CuSO4.5H2O (26 mg, 0.104 mmol) in 4 mL water at RT. Then stirred for overnight at RT. After completion of the reaction by TLC, the reaction mixture was poured into water and EtOAc. The phases were separated, and the organic layer was washed with brine solution, dried over sodium sulphate and concentrated. Crude was purified by column chromatography (60–120 silica gel, plane ethyl acetate) to afford triazole 15a as an off-white solid (120 mg, 67%); 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.10 (br s, 1H, NH), 9.32 (s, 1H), 8.40 (t, J = 2.0 Hz, 1H), 8.35 (s, 1H), 7.78 (ddd, J = 8.3, 2.1, 0.9 Hz, 1H), 7.57–7.49 (m, 2H: 1 dd with a roof effect at 7.55 ppm, J = 8.0, 1.2 Hz and 1 dd with a roof effect at 7.53 ppm, J = 7.8, 1.2 Hz), 7.36 (t, J = 7.9 Hz, 1H), 7.24 (t, J = 8.1 Hz, 1H), 6.96 (ddd, J = 7.9, 2.1, 0.9 Hz, 1H), 6.99–6.87 (broad envelope which topped at 6.92 ppm, 1H), 5.35 (s, 2H), 4.56 (tt, J = 9.9, 3.9 Hz, 1H), 3.72–3.59 (m which topped at 3.66 ppm, 1H), 2.26–2.08 (m, 2H), 1.94–1.81 (m, 2H), 1.77–1.59 (m, 4H), 1.39 (s, 9H); 13C NMR (75 MHz, DMSO-d6, δ ppm): 161.9 (CH), 155.8 (C), 155.0 (C), 151.7 (C), 142.5 (C), 142.2 (C), 142.1 (C), 133.0 (C), 129.8 (CH), 123.7 (CH), 122.8 (CH), 121.2 (C), 120.6 (CH), 119.7 (CH), 117.6 (CH), 116.7 (CH), 115.2 (CH), 77.6 (C), 62.2 (CH2), 57.6 (CH), 44.7 (broad and small, CH), 28.4 (2CH2), 28.2 (3CH3), 27.4 (2CH2); HRMS (CH3OH/CH2Cl2 = 9:1) m/z calcd for C28H3235ClN7NaO3 [M + Na]+: 572.21474, found: 572.2150, calcd for C24H1735Cl3N6KO [M + K]+: 588.18867, found: 588.1888, calcd for C28H3335ClN7O3 [M + H]+: 550.23279, found: 550.2330.
tert-Butyl ((cis)-4-(4-(((2-((3,5-dichlorophenyl)amino)quinazolin-8-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)cyclohexyl)carbamate (15b):
Reaction of alkyne 5b (100 mg, 0.29 mmol) with azide 14 (70 mg, 0.29 mmol) gave triazole 15b (100 mg, 59%); 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.34 (br s, 1H, NH), 9.37 (s, 1H), 8.37 (s, 1H), 8.17 (d, J = 1.9 Hz, 2H), 7.57 (nearly d with a roof effect, J = 7.9 Hz, 2H), 7.41 (nearly dd with a roof effect (protons at 7.41 and 7.57 ppm are coupled with each other), J = 8.2, 7.6 Hz, 1H), 7.06 (t, J = 1.9 Hz, 1H), 6.98 (br d, J = 7.0 Hz, 1H, NH), 5.33 (s, 2H), 4.54 (tt, J = 10.1, 3.9 Hz, 1H), 3.72–3.59 (m which topped at 3.66 ppm, 1H), 2.25–2.08 (m, 2H), 1.93–1.79 (m, 2H), 1.77–1.59 (m, 4H), 1.39 (s, 9H); 13C NMR (100 MHz, DMSO-d6, δ ppm): 162.1 (CH), 155.5 (C), 155.0 (broad and small, C), 151.8 (C), 143.0 (C), 142.1 (C), 141.9 (C), 133.7 (2C), 124.2 (CH), 123.0 (CH), 121.3 (C), 119.8 (CH), 119.5 (CH), 116.1 (2CH), 114.8 (CH), 77.5 (C), 62.0 (CH2), 57.6 (CH), 44.6 (broad and small, CH), 28.4 (2CH2), 28.2 (3CH3), 27.4 (2CH2); HRMS (CH3OH/CH2Cl2 = 9:1) m/z calcd for C28H3135Cl2N7NaO3 [M + Na]+: 606.17576, found: 606.1761, calcd for C28H3135Cl2N7KO3 [M + K]+: 622.14970, found: 622.1490, calcd for C28H3035Cl2N7Na2O3 [M-H + 2Na]+: 628.15771, found: 628.1571.
8-((1-((cis)-4-Aminocyclohexyl)-1H-1,2,3-triazol-4-yl)methoxy)-N-(3-chlorophenyl)quinazolin-2-amine hydrochloride (16a):
To a stirred solution of carbamate 15a (50 mg) in methanol (2.0 mL) was added 4 M HCl in dioxane (1 mL) dropwise and stirred at room temperature for 1 h, After completion of the reaction by TLC, the reaction mixture was concentrated to obtain the hydrochloride 16a as an off white solid in 76% yield. 1H NMR (400 MHz, DMSO-d6) δ ppm: 10.28 (s, 1H), 9.36 (s, 1H), 8.58 (s, 1H), 8.39 (s, 1H), 8.31 (s, 3H), 7.75 (dd, J = 1.6, 8.4 Hz, 1H), 7.58–7.55 (m, 2H), 7.38 (t, J = 8.0 Hz, 1H), 7.26 (t, J = 8.0 Hz, 1H), 6.98 (dd, J = 1.6, 8.0 Hz, 1H), 5.36 (s, 2H), 4.69–4.66 (m, 1H), 3.34 (bs, 1H), 2.39–2.34 (m, 2H), 1.97–1.86 (m, 5H); 13C NMR (400 MHz, DMSO-d6): δ 163.0, 156.2, 151.8, 142.9, 142.4, 133.6, 130.4, 124.5, 123.9, 121.6, 121.4, 120.3, 118.4, 117.5, 115.9, 66.8, 62.7, 56.6, 29.5, 27.2, 26.5: Mass (m/z): 450.3 (M + H).
8-((1-((cis)-4-Aminocyclohexyl)-1H-1,2,3-triazol-4-yl)methoxy)-N-(3,5-dichlorophenyl)quinazolin-2-amine hydrochloride (16b):
Reaction of carbamate 15b (50 mg, 0.085 mmol) with HCl in dioxane gave hydrochloride 16b (31 mg, 74%). 1H NMR (400 MHz, DMSO-d6) δ ppm: 10.36 (s, 1H), 9.37 (s, 1H), 8.55 (s, 1H), 8.29 (s, 2H), 8.17 (d, J = 1.6 Hz, 2H), 7.59–7.56 (m, 2H), 7.40 (t, J = 7.6 Hz, 1H), 7.06 (t, J = 1.6Hz, 1H), 5.33 (s, 2H), 4.69–4.63 (m, 1H), 3.66 (bs, 1H), 2.38–2.33 (m, 2H), 1.96–1.85 (m, 6H); 13C NMR (400 MHz, DMSO-d6): δ 162.7, 156.1, 152.2, 143.5, 142.6, 134.3, 124.8, 124.1, 121.8, 120.4, 120.1, 116.8, 115.3, 62.4, 56.65, 46.6, 31.8, 27.2, 26.5: Mass (m/z): 484.0 (M + H).
tert-Butyl ((trans)-4-(4-(((2-((3-chlorophenyl)amino) quinazolin-8-yl) oxy) methyl)-1H-1,2,3-triazol-1-yl)cyclohexyl)carbamate (18a):
Reaction of alkyne 13 (100 mg, 0.32 mmol) with azide 17 (77.6 mg, 0.32 mmol) gave triazole 18a (100 mg, 56%); 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.11 (br s, 1H, NH), 9.32 (s, 1H), 8.44 (t, J = 2.0 Hz, 1H), 8.34 (s, 1H), 7.74 (ddd, J = 8.3, 2.1, 0.9 Hz, 1H), 7.57–7.50 (m, 2H), 7.36 (t, J = 7.9 Hz, 1H), 7.24 (t, J = 8.1 Hz, 1H), 6.97 (ddd, J = 7.9, 2.1, 0.9 Hz, 1H), 6.82 (br d, J = 7.3 Hz, 1H, exchanged by D2O), 5.32 (s, 2H), 4.47 (tt, J = 11.6, 3.8 Hz, 1H), 3.42–3.26 (m, 1H, superimposed to the signal of water which was disclosed by addition of D2O), 2.16–2.04 (m, 2H), 1.99–1.78 (m, 4H), 1.40 (s, 9H) and superimposed 2H m at 1.50–1.30 ppm; 13C NMR (100 MHz, DMSO-d6, δ ppm): 161.9 (CH), 155.8 (C), 154.8 (C), 151.7 (C), 142.5 (C), 142.1 (2C), 133.0 (C), 129.7 (CH), 123.7 (CH), 122.9 (CH), 121.1 (C), 120.5 (CH), 119.6 (CH), 117.6 (CH), 116.7 (CH), 115.0 (CH), 77.4 (C), 62.1 (CH2), 58.3 (CH), 48.0 (small, CH), 31.4 (2CH2), 30.9 (2CH2), 28.2 (3CH3); HRMS (CH3OH/CH2Cl2 = 9:1) m/z calcd for C28H3235ClN7NaO3 [M + Na]+: 572.21474, found: 572.2148, calcd for C24H1735Cl3N6KO [M + K]+: 588.18867, found: 588.1889.
tert-Butyl ((trans)-4-(4-(((2-((3,5-dichlorophenyl)amino)quinazolin-8-yl)oxy) methyl)-1H-1,2,3-triazol-1-yl)cyclohexyl)carbamate (18b):
Reaction of alkyne 5b (100 mg, 0.29 mmol) with azide 17 (70 mg, 0.29 mmol) gave triazole 18b (100 mg, 59%); 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.30 (br s, 1H, NH), 9.36 (s, 1H), 8.35 (s, 1H), 8.16 (d, J = 1.9 Hz, 2H), 7.59–7.53 (m: approximately br d at 7.56 ppm with a roof effect, J ~ 8 Hz, 2H), 7.40 (dd plus 2 small inner bands with a roof effect, J = 8.1, 7.7 Hz, 1H), 7.07 (t, J = 1.9 Hz, 1H), 6.82 (br d, J = 7.3 Hz, 1H, exchanged by D2O), 5.31 (s, 2H), 4.46 (tt, J = 11.7, 3.8 Hz, 1H), 3.42–3.22 (m, 1H, superimposed to the signal of water which was disclosed by addition of D2O), 2.16–2.04 (m, 2H), 1.99–1.78 (m, 4H), 1.40 (s, 9H) and superimposed 2H m at 1.45–1.31 ppm; 13C NMR (100 MHz, DMSO-d6, δ ppm): 162.1 (CH), 155.5 (C), 154.8 (C), 151.8 (C), 143.0 (C), 142.1 (C), 141.8 (C), 133.7 (2C), 124.2 (CH), 123.1 (CH), 121.3 (C), 119.8 (CH), 119.5 (CH), 116.1 (2CH), 114.6 (CH), 77.4 (C), 61.9 (CH2), 58.3 (CH), 48.0 (small, CH), 31.4 (2CH2), 30.9 (2CH2), 28.2 (3CH3); HRMS (CH3OH/CH2Cl2 = 9:1) m/z calcd for C28H3135Cl2N7NaO3 [M + Na]+: 606.17576, found: 606.1752, calcd for C24H2335Cl2N7NaO3 [M-C4H8 + Na]+: 550.11316, found: 550.1120.
8-((1-((trans)-4-Aminocyclohexyl)-1H-1,2,3-triazol-4-yl)methoxy)-N-(3-chlorophenyl)quinazolin-2-amine hydrochloride (19a):
Reaction of carbamate 18a (50 mg, 0.091 mmol) with HCl in dioxane gave hydrochloride 19a (30 mg, 72%). 1H NMR (400 MHz, DMSO-d6): δ 10.23 (s,1H), 9.34 (s, 1H), 8.43 (s, 1H), 8.37 (s, 1H), 8.28 (d, J = 4.0 Hz, 3H), 7.73 (dd, J = 1.2 Hz, 8.0 Hz, 1H), 7.56–7.53 (m, 2H), 7.37 (t, J = 8.0 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 6.98 (dd, J = 1.6 Hz, 8.0 Hz, 1H), 5.34 (s, 2H), 4.56–4.50 (m, 1H), 3.17–3.12 (m, 1H), 2.15 (t, J = 9.2 Hz, 4H), 1.98–1.88 (m, 2H), 1.67–1.60 (m, 2H). 13C NMR (400 MHz, DMSO-d6): δ 162.8, 156.26, 152.0, 142.6, 133.6, 130.3, 124.4, 123.6, 121.7, 121.3, 120.28, 118.3, 117.4, 115.7, 66.8, 62.7, 58.2, 48.5, 30.9, 29.2; Mass (m/z): 450.0 (M + H).
8-((1-((trans)-4-Aminocyclohexyl)-1H-1,2,3-triazol-4-yl)methoxy)-N-(3,5-dichlorophenyl)quinazolin-2-amine hydrochloride (19b):
Reaction of carbamate 18b (50 mg, 0.085 mmol) with HCl in dioxane gave hydrochloride 19b (31 mg, 74%). 1H NMR (400 MHz, DMSO-d6): δ 10.37 (s, 1H), 9.37 (s, 1H), 8.43 (s, 1H), 8.37 (s, 1H), 8.28 (d, J = 3.6 Hz, 2H), 8.17 (d, J = 1.6 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 7.40 (t, J = 8.0 Hz, 1H), 7.07 (t, J = 2.0 Hz, 1H), 5.31 (s, 2H), 4.55–4.50 (m, 1H), 3.17–3.12 (m, 1H), 2.51 (t, J = 1.6 Hz, 4H), 2.17–2.12 (m, 2H), 1.97–1.89 (m, 2H). 13C NMR (400 MHz, DMSO-d6): δ 162.7, 156.0, 152.3, 143.5, 142.4, 134.3, 124.8, 123.9, 121.8, 120.4, 120.0, 116.7, 115.2, 66.8, 62.4, 58.2, 48.5, 30.9, 29.2; Mass (m/z): 484.2 (M + H).


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}