


Molybdenum, Vanadium, and Tungsten-Based Catalysts for Sustainable (ep)Oxidation



Abstract
:
1. Introduction
2. Tridentate Ligands and Related Complexes
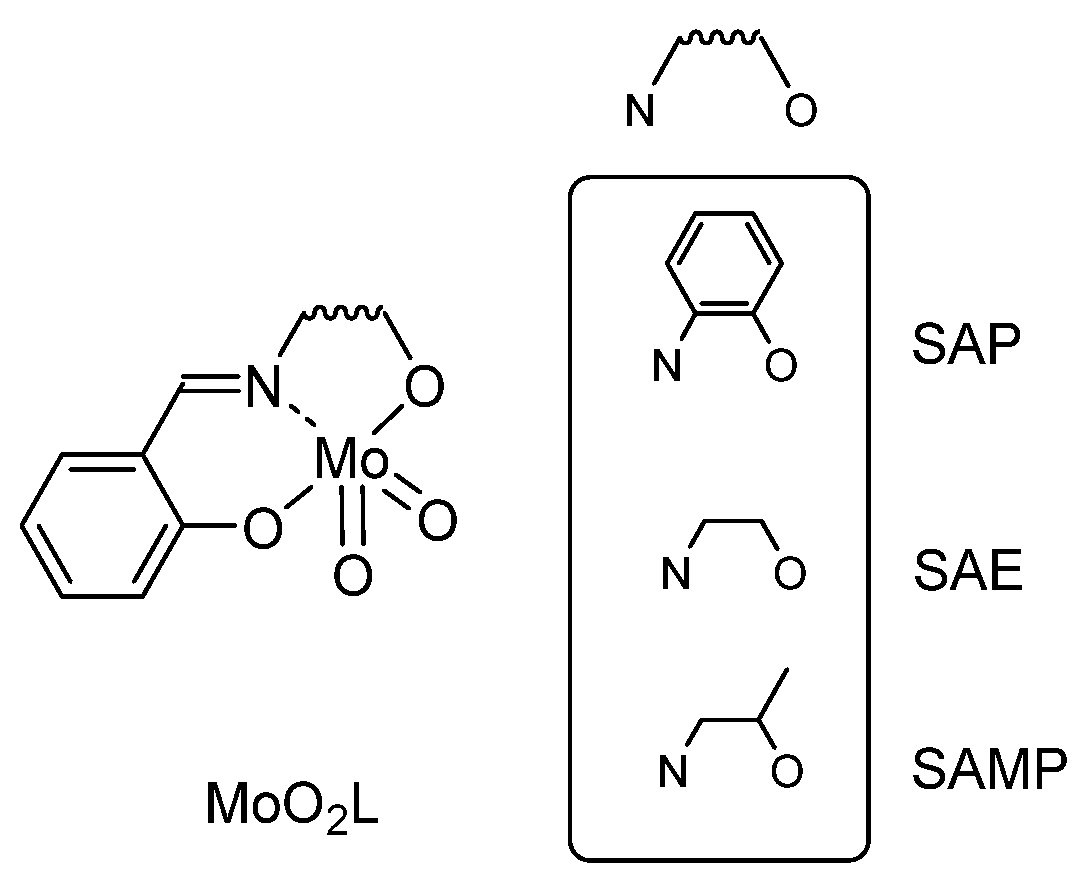
2.1. Molybdenum and Tungsten Complexes
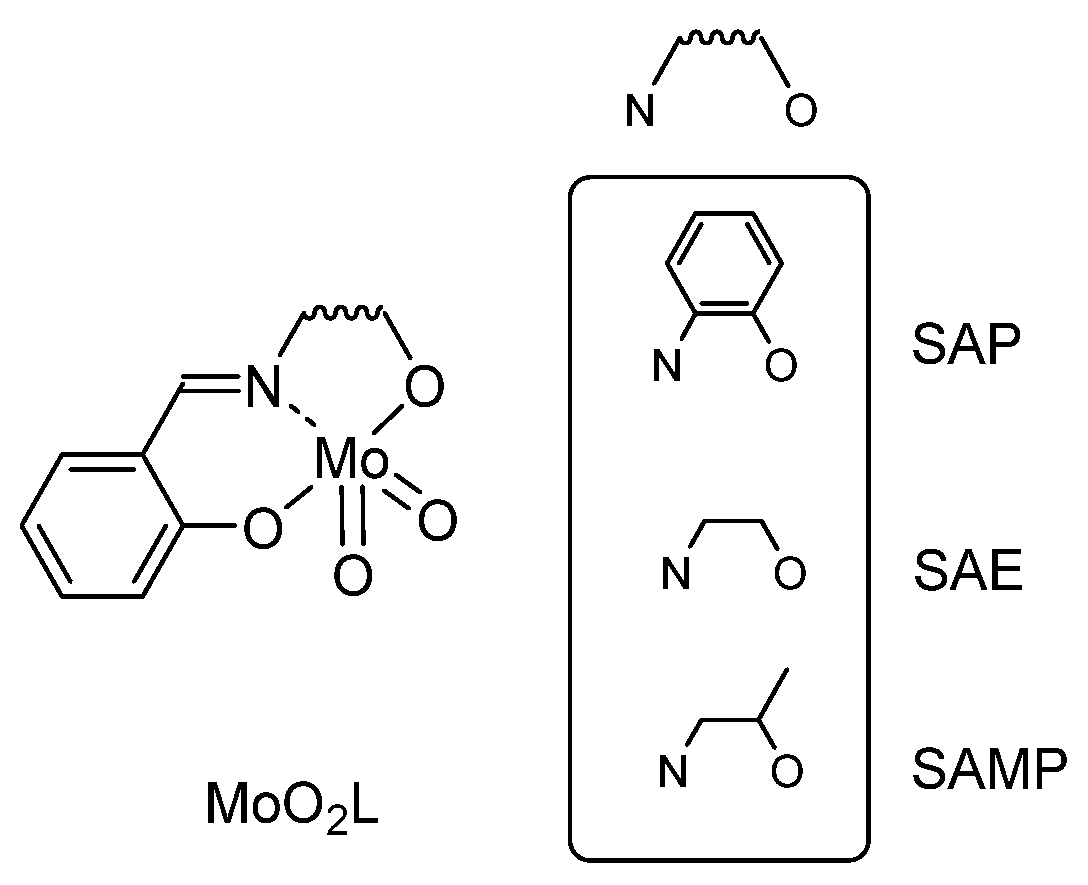
2.1.1. From SAP Ligands and Derivatives: Mechanistic Study
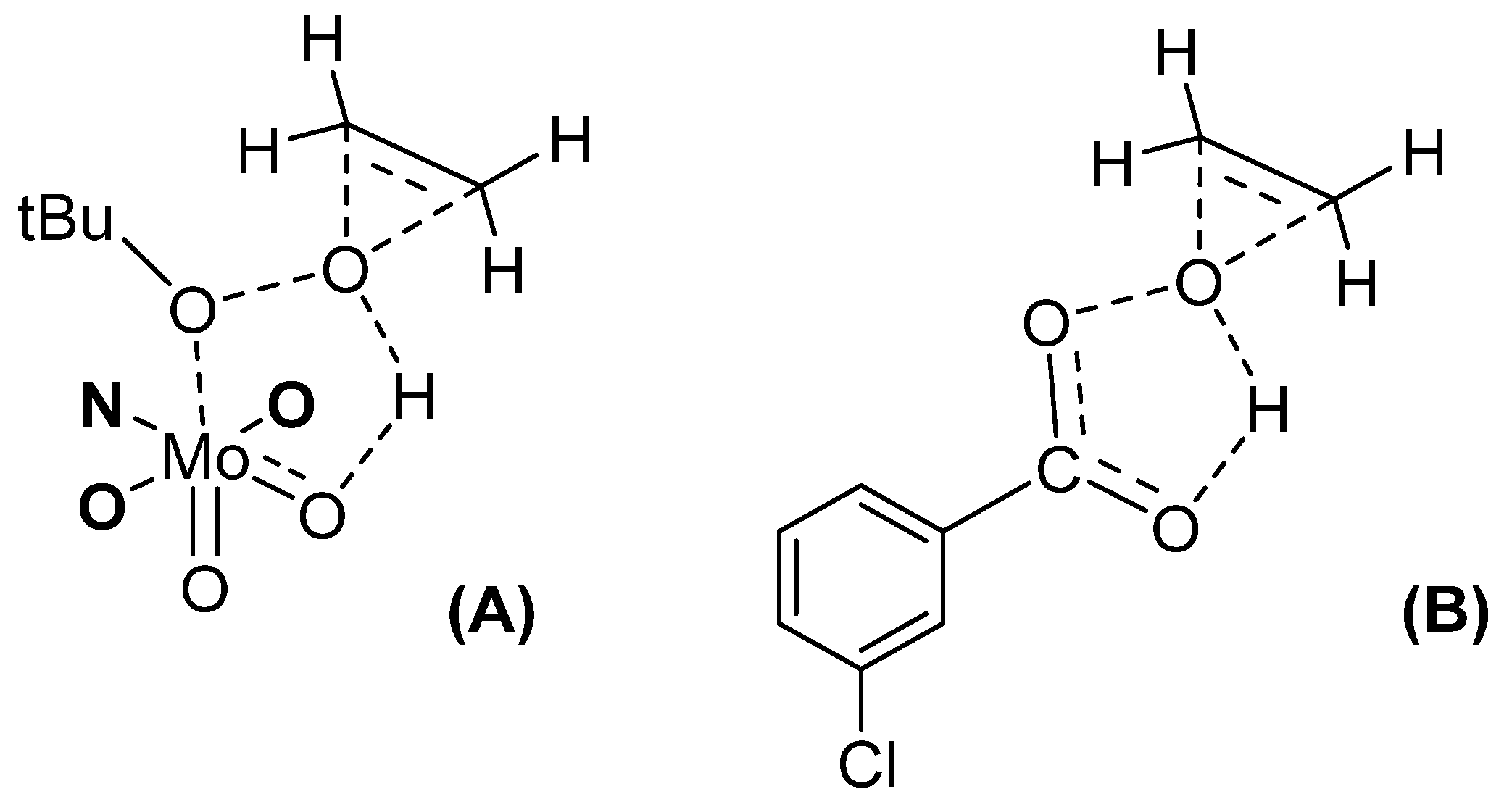
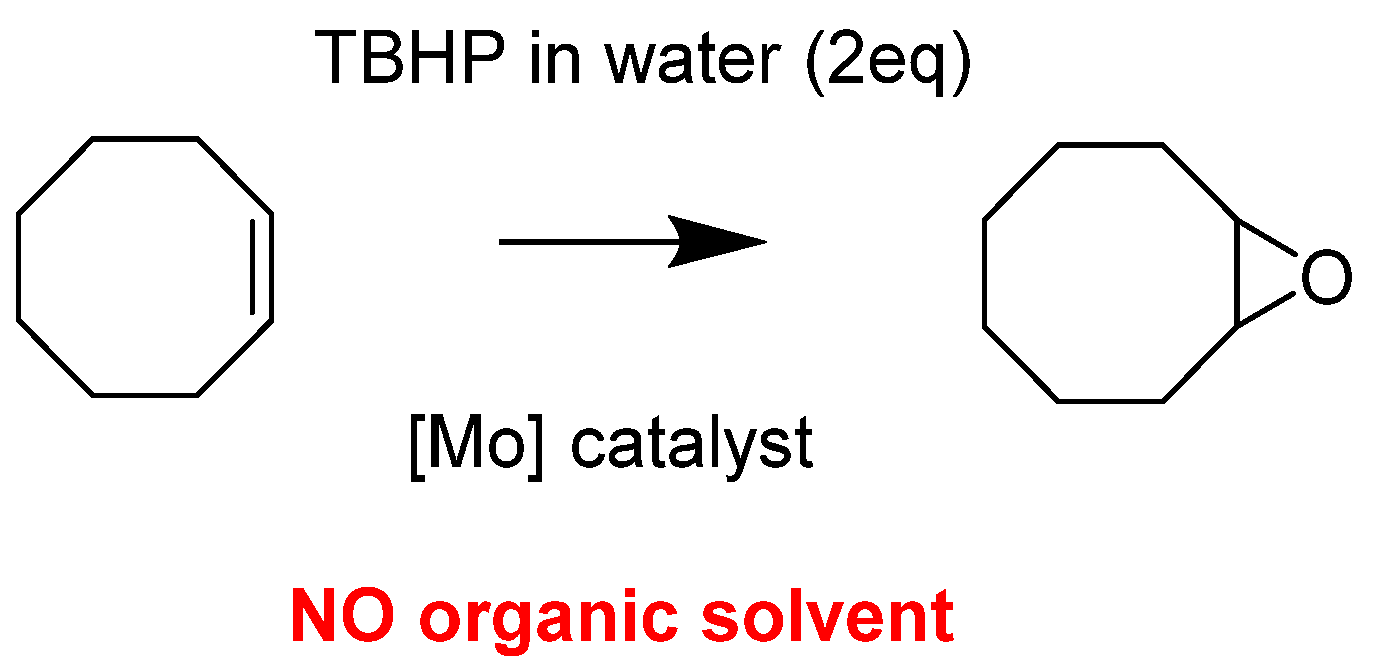
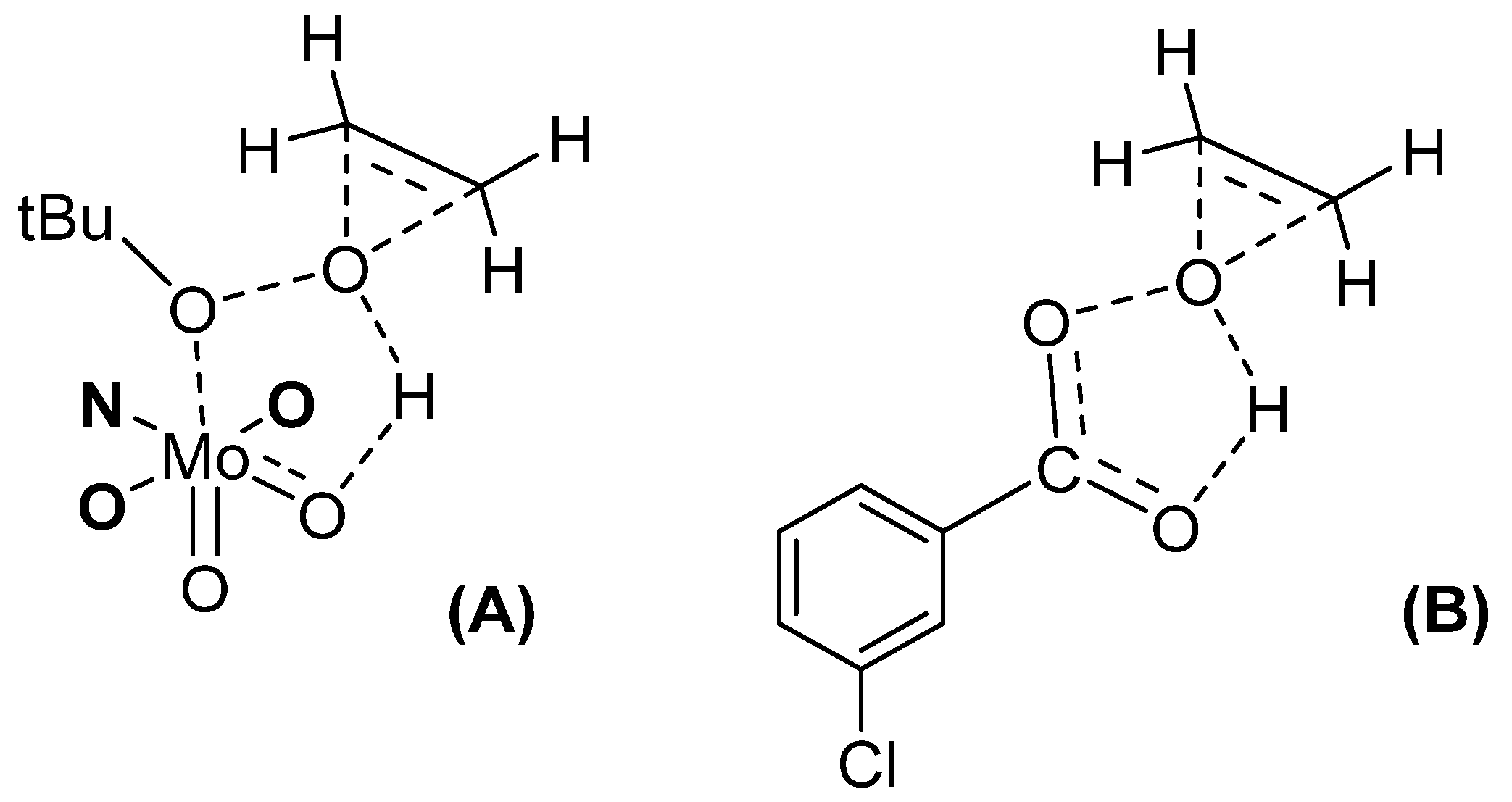
- Methodological Approach and Model Study with Cyclooctene
- Preliminary Studies toward the Best Backbone and Mechanistic Study
- Ligand Tuning and Optimization
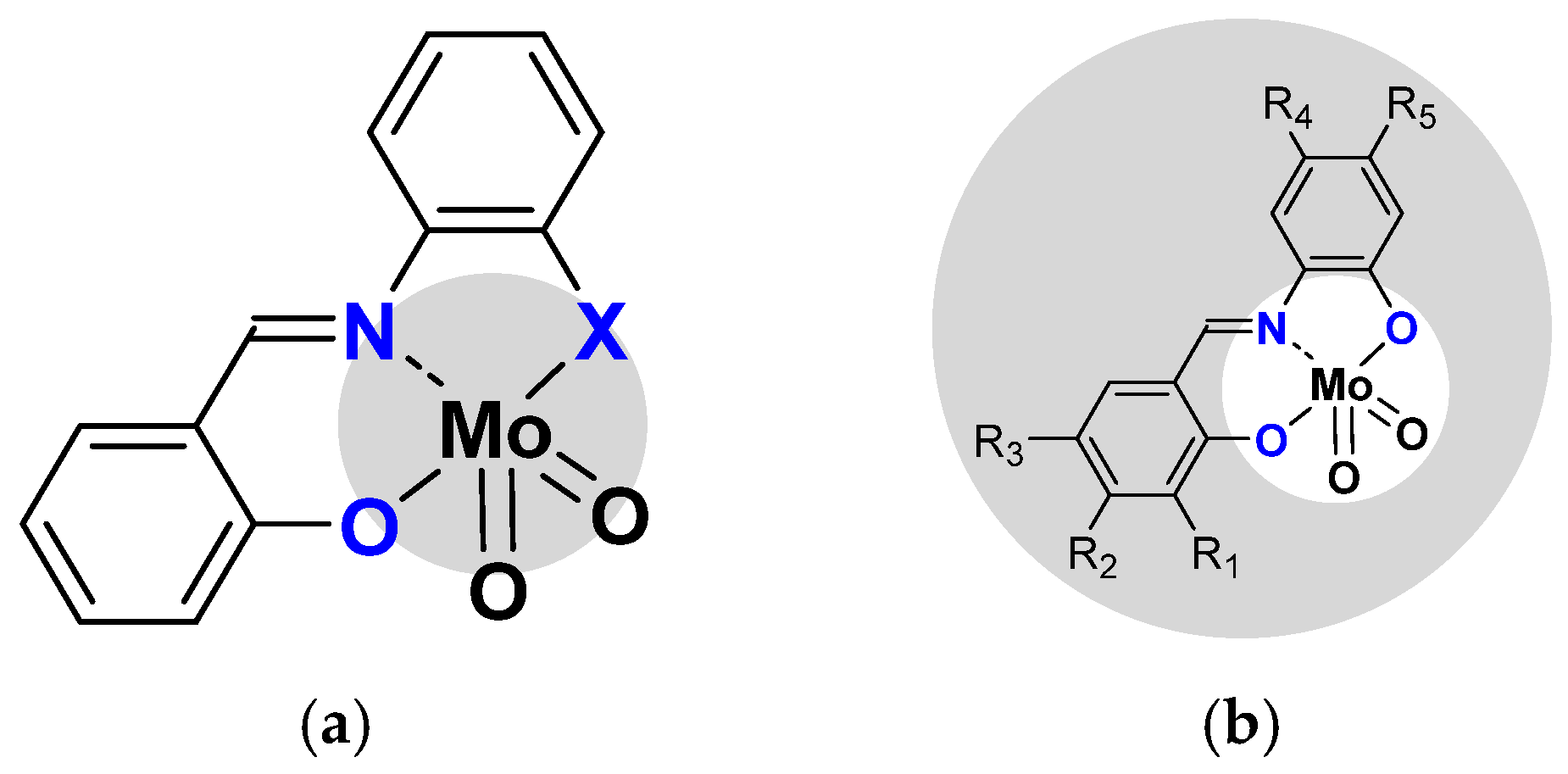
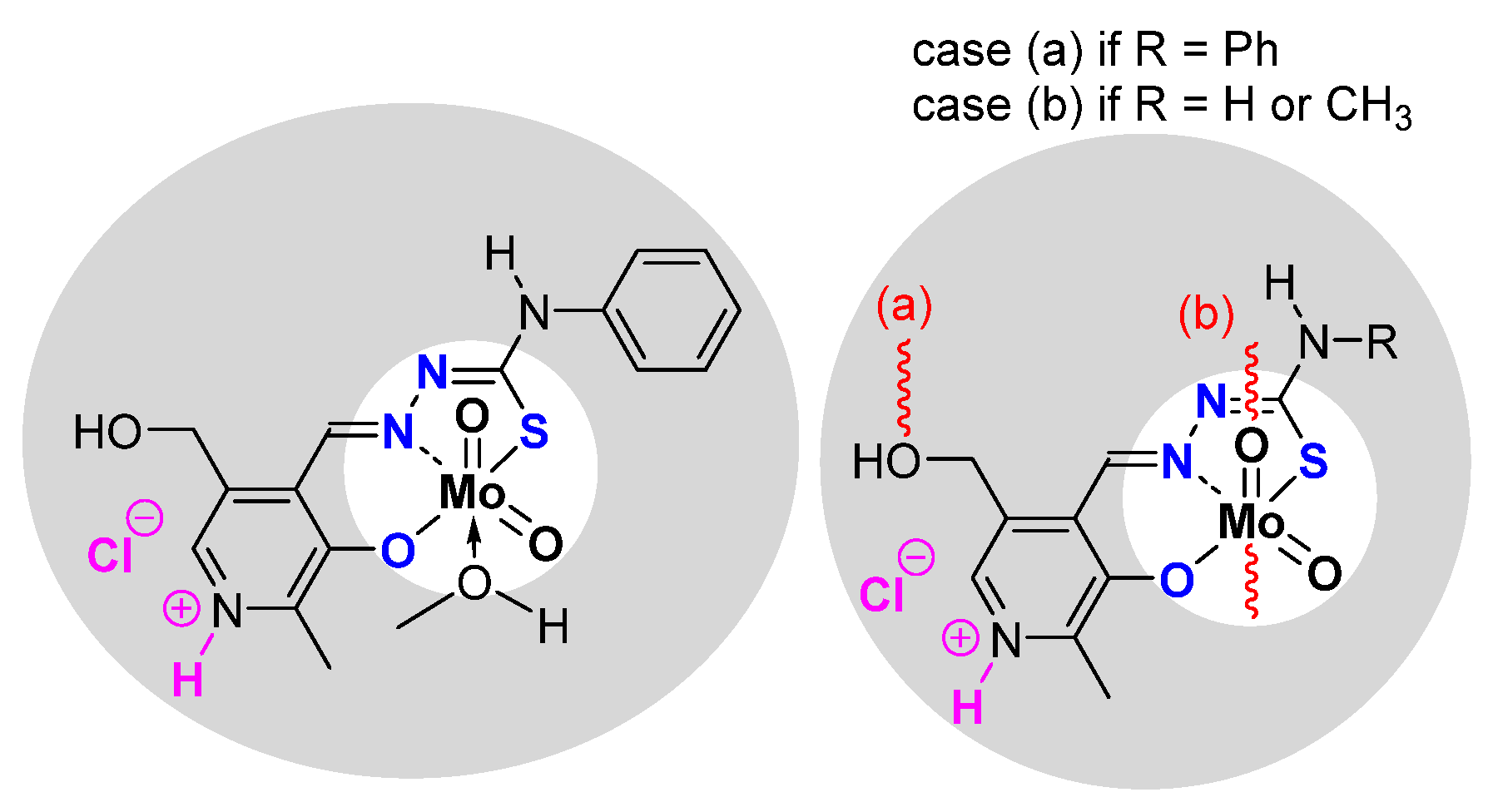
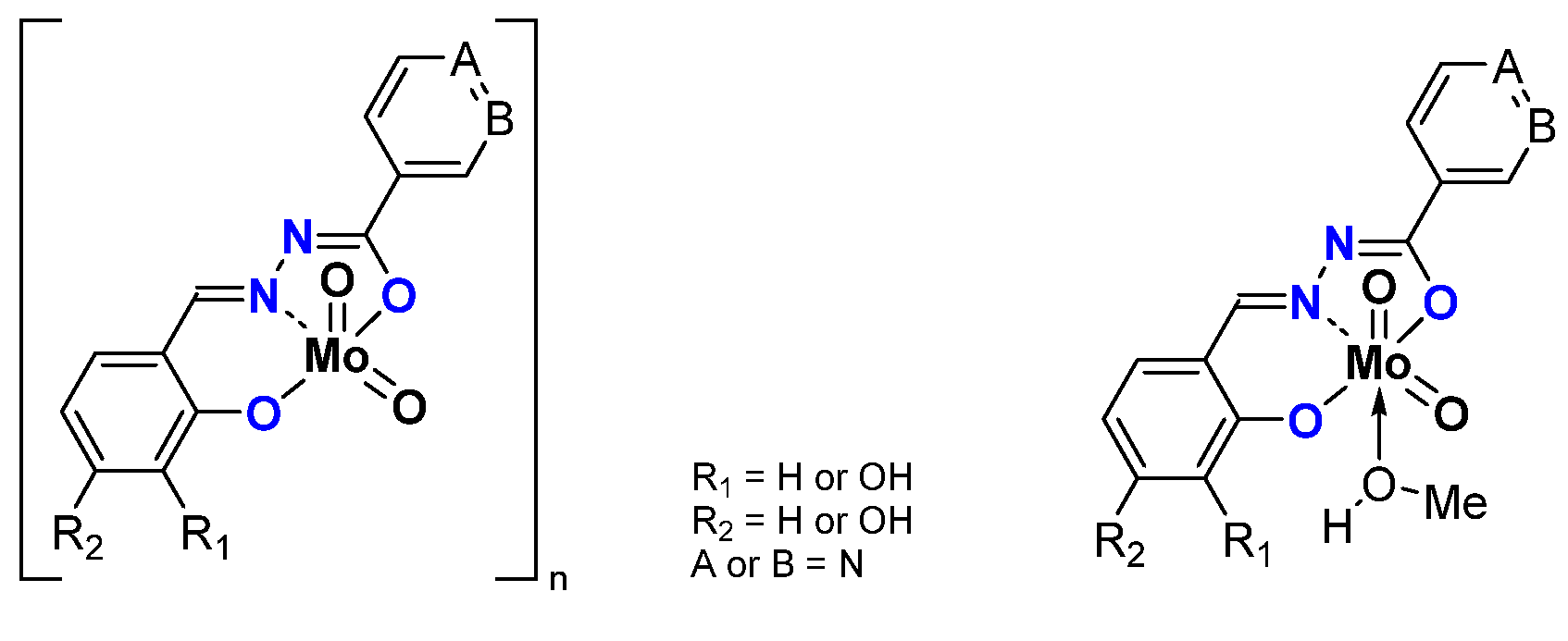
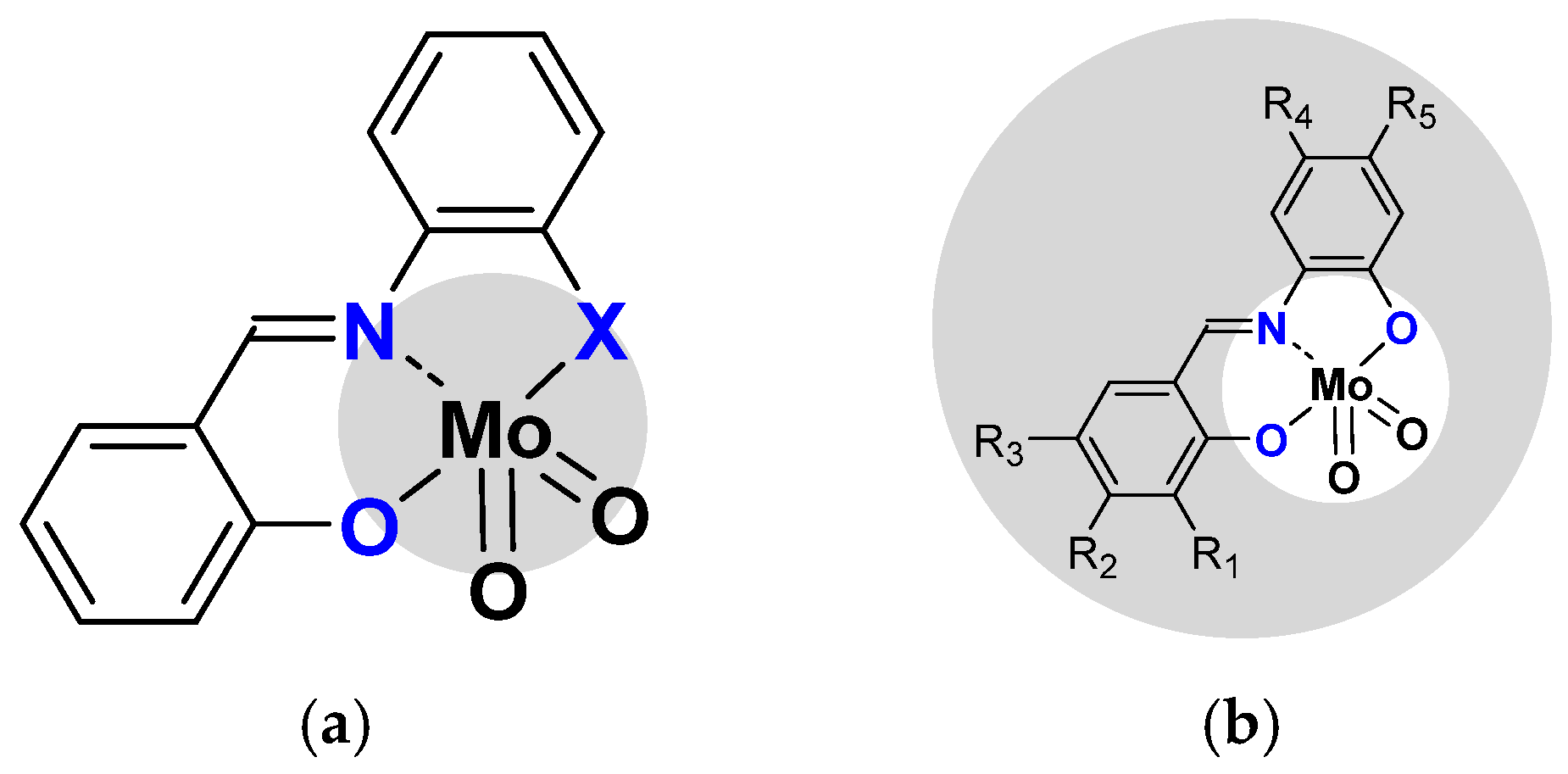
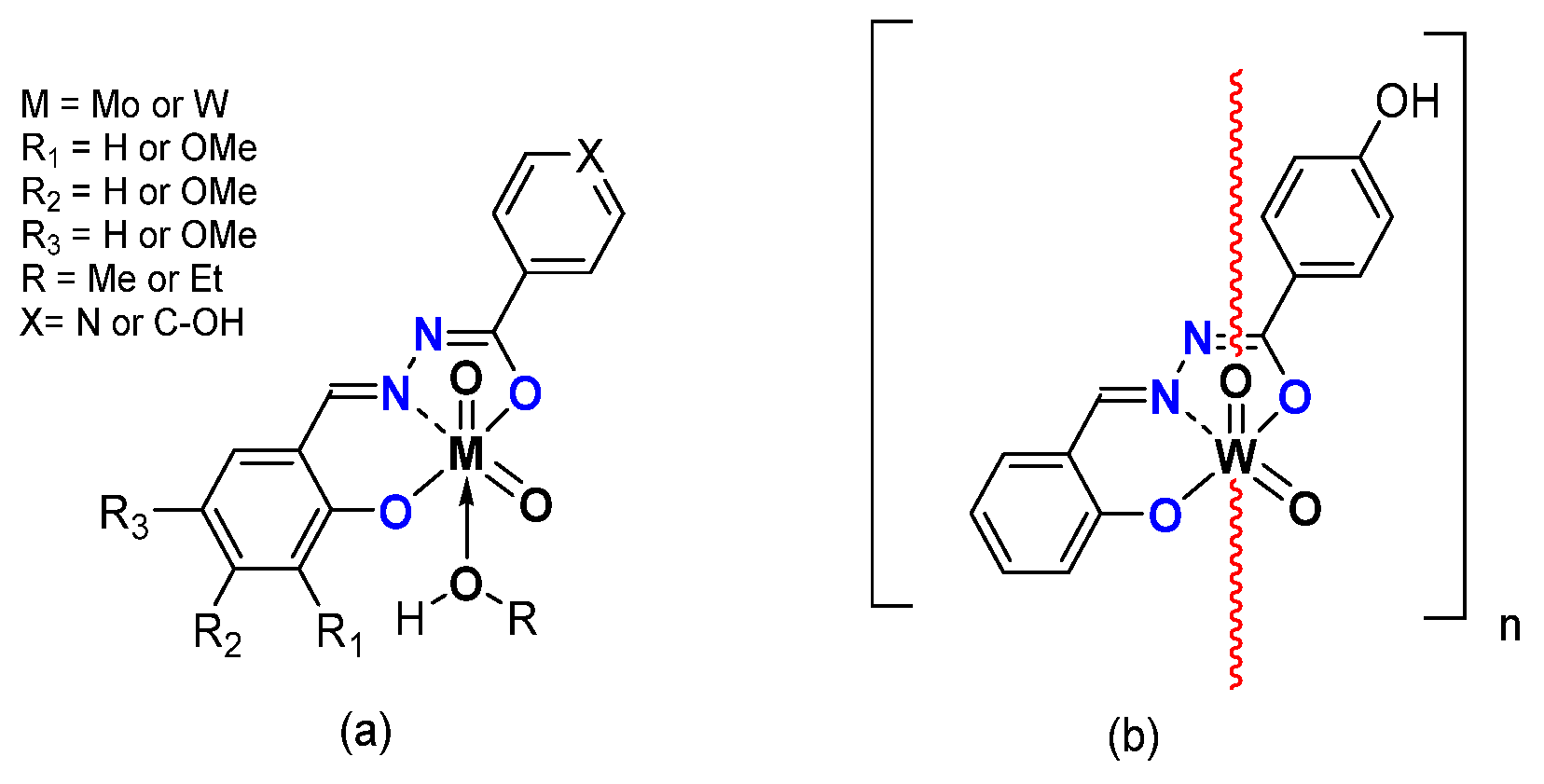
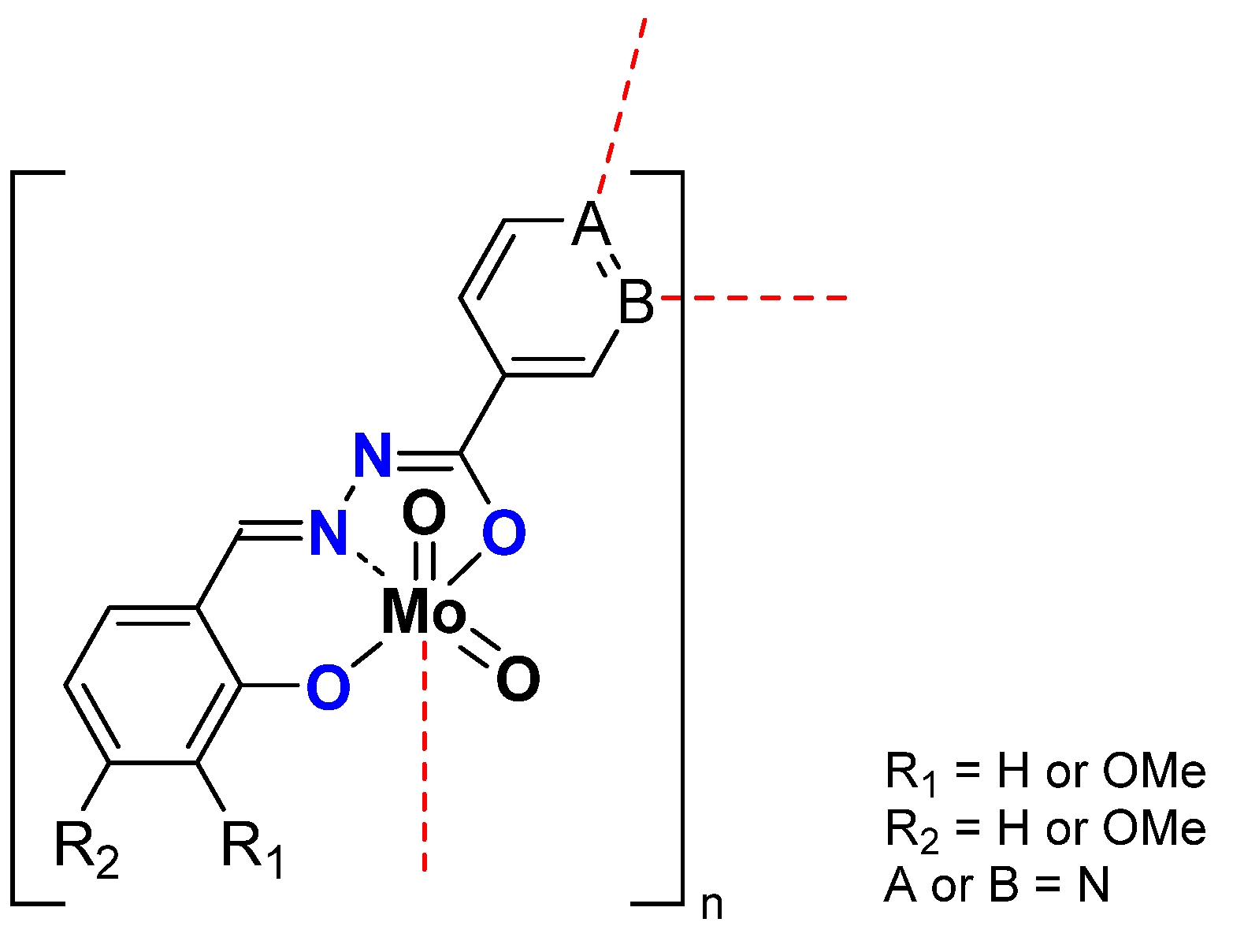
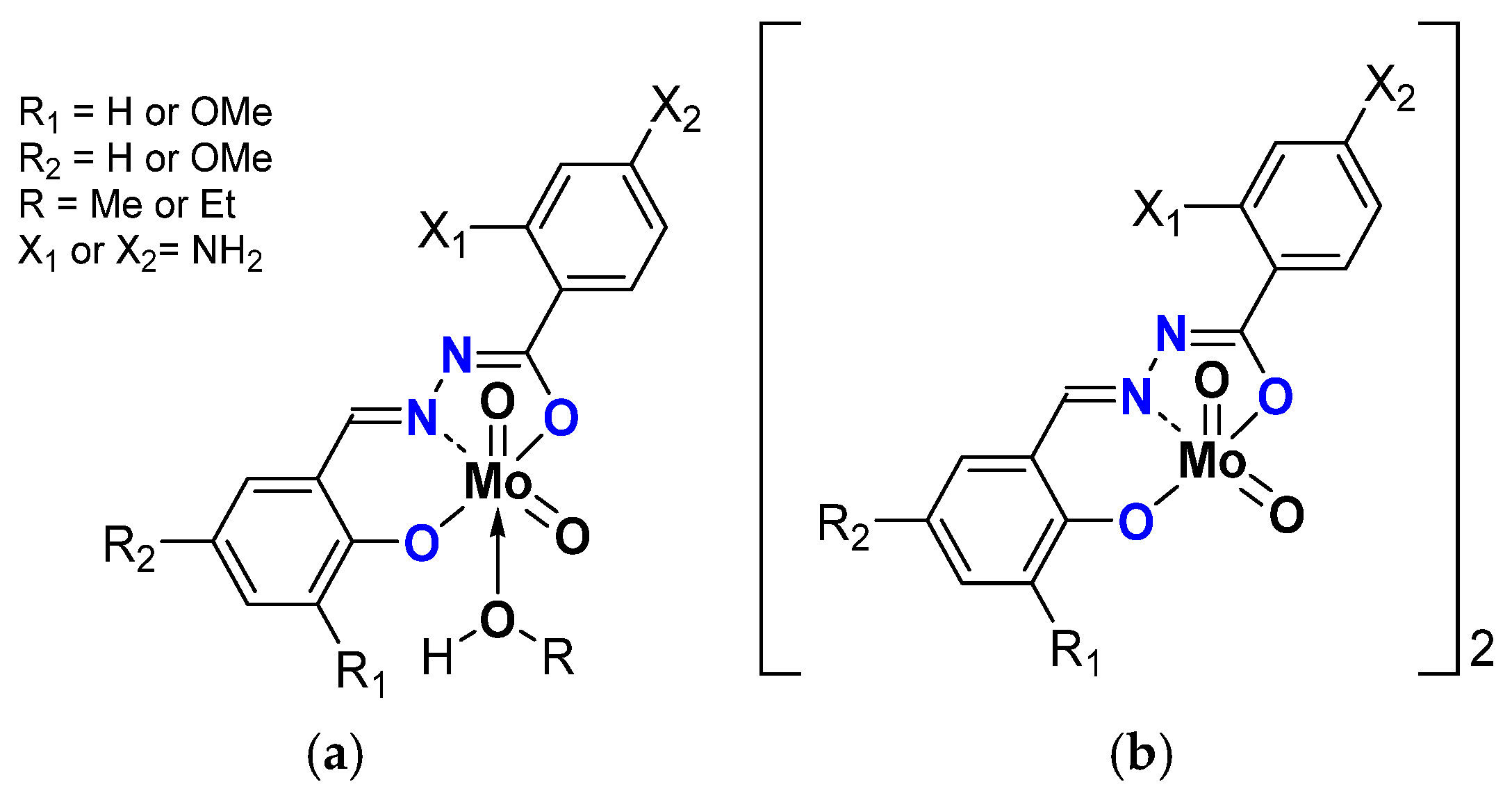
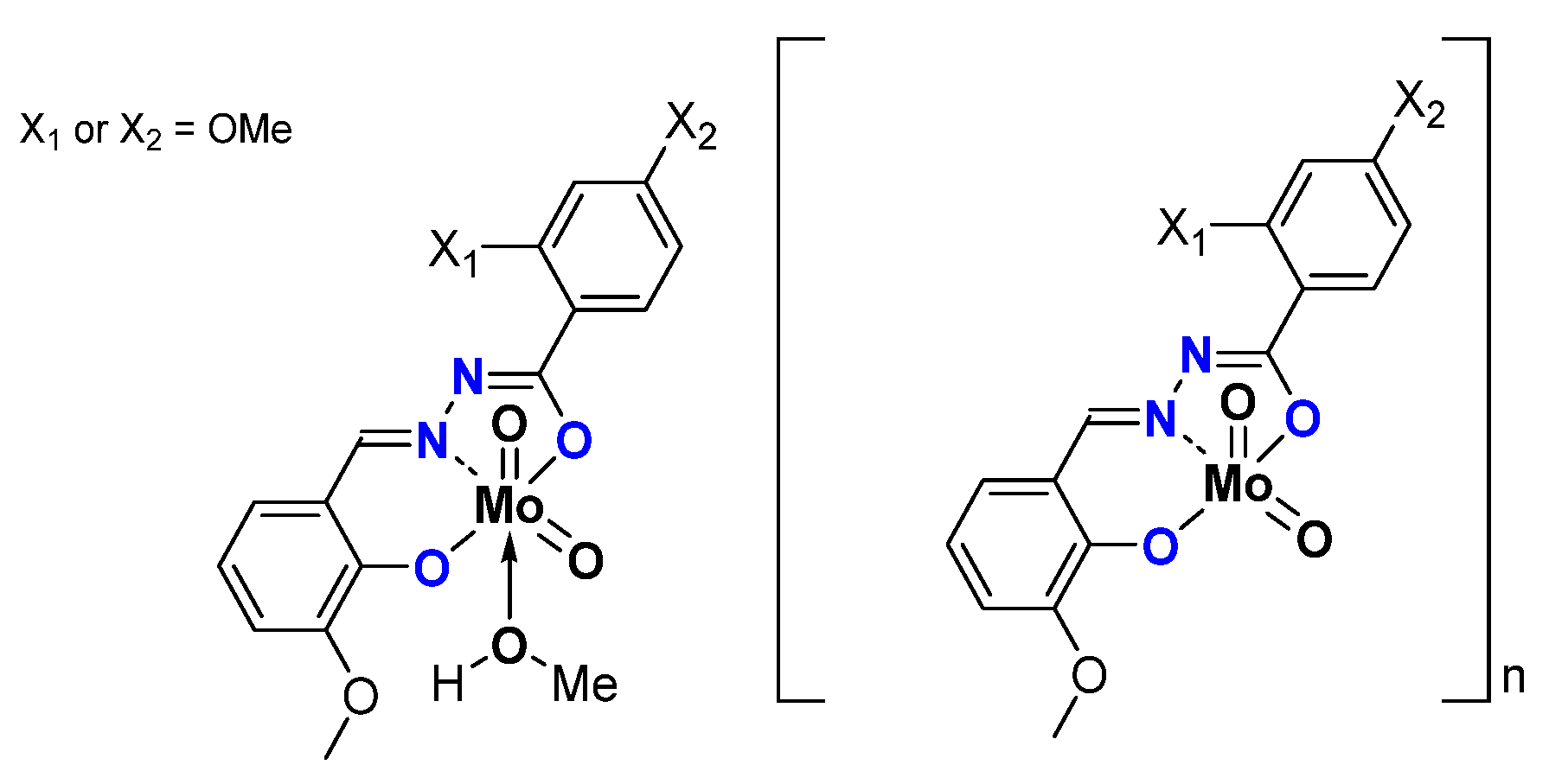
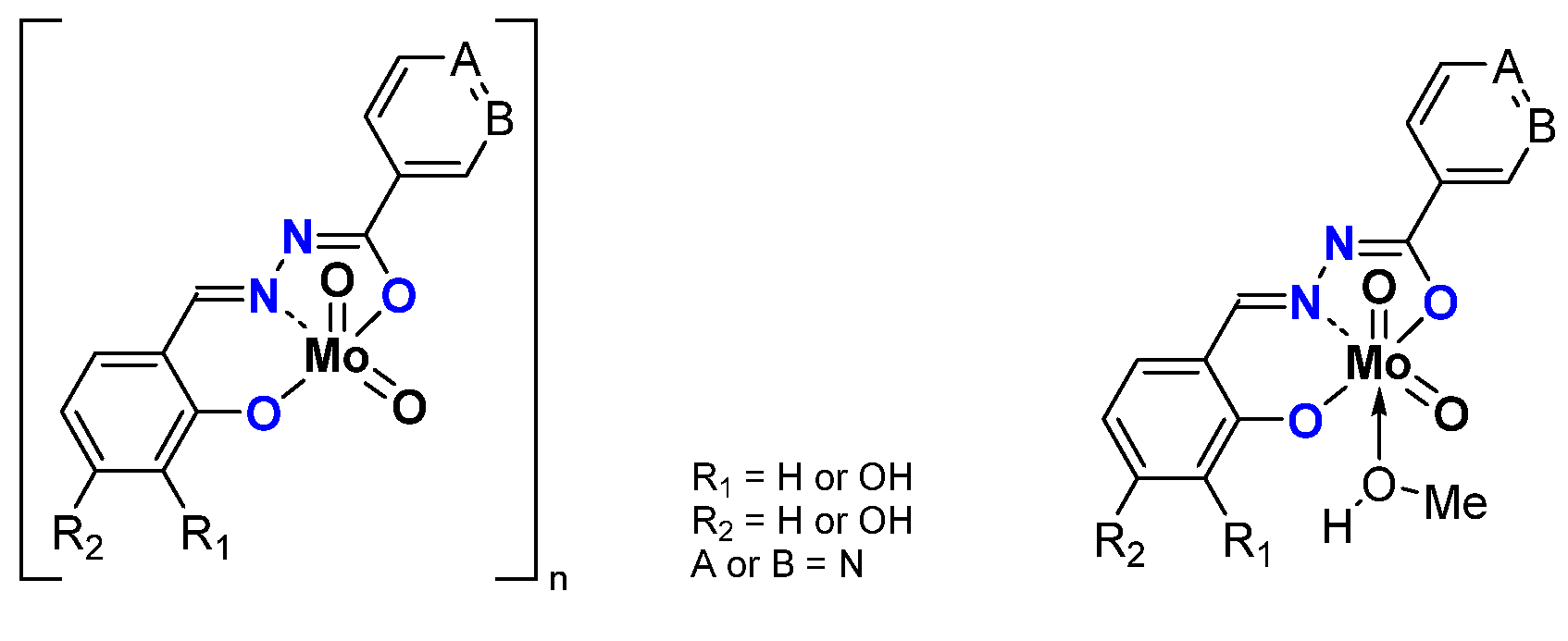
2.1.2. ONS, ONO Mo-Enlarging the Scope of Complexes through Collaboration
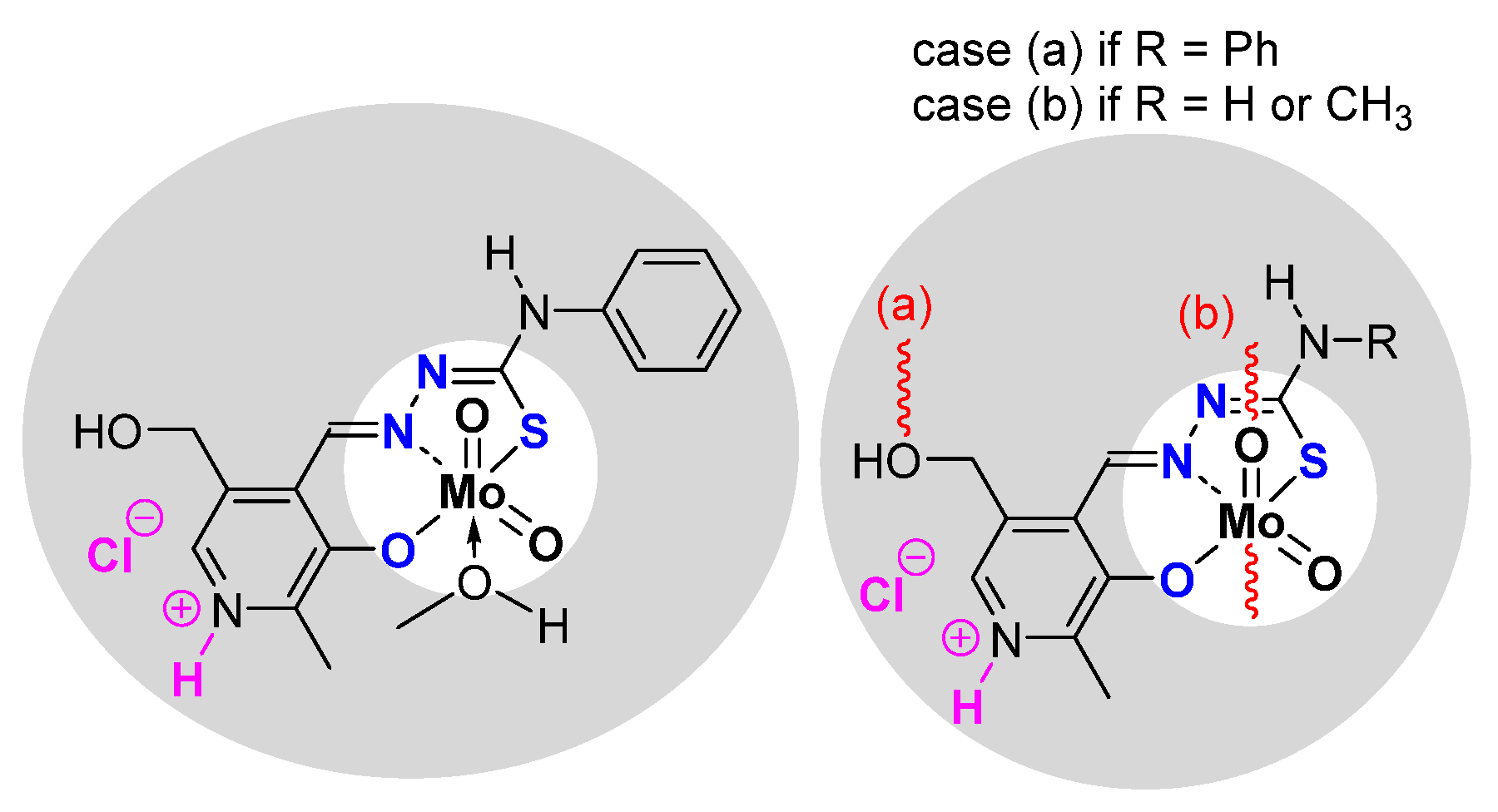
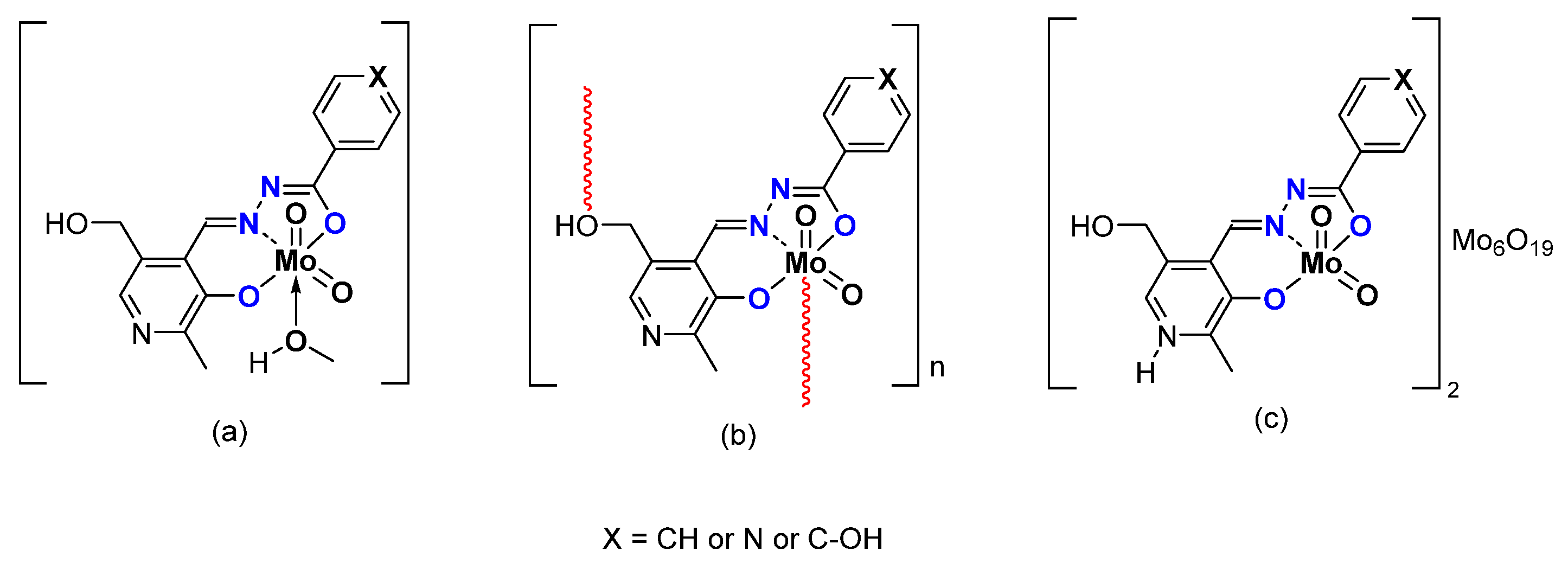
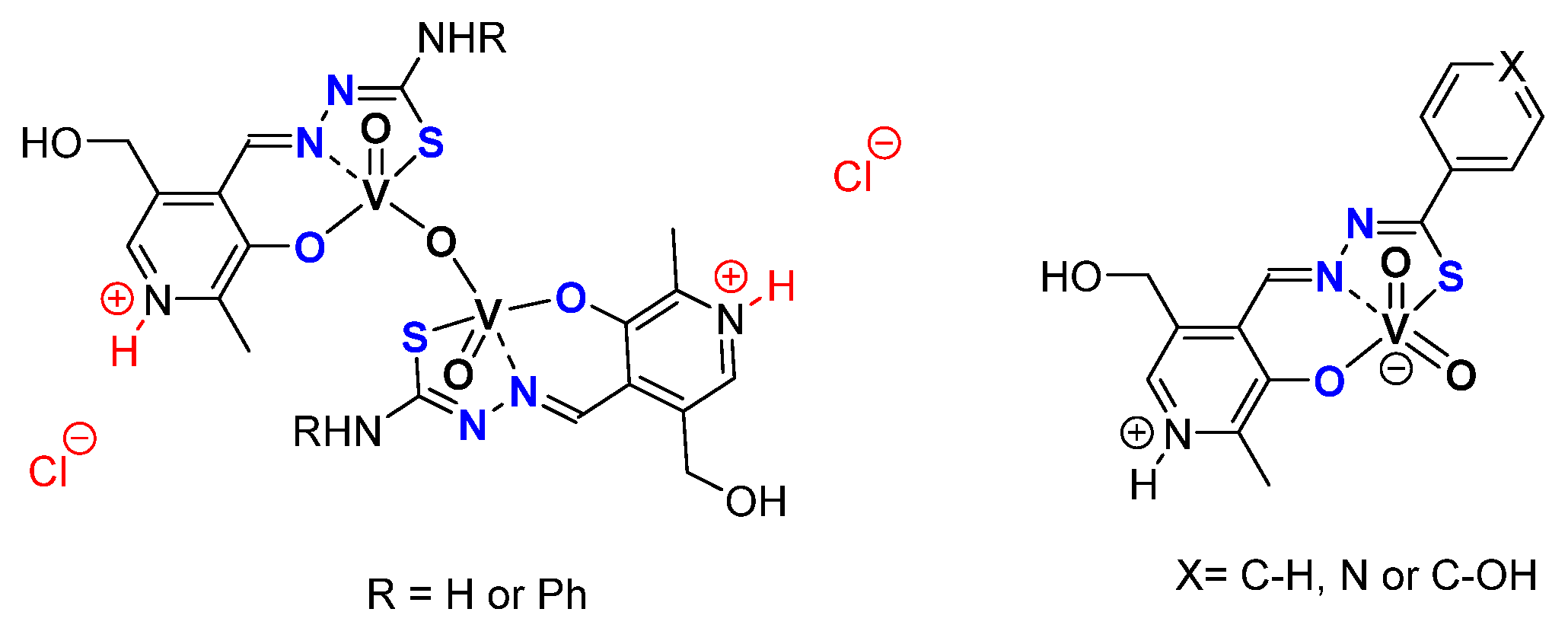
- Pyridoxal Fragment within the Ligand
- From Mo to W Complexes with Salicylaldehyde Part: Some Experimental Features
2.2. Extension to High-Valued Species
2.2.1. Other Olefins
2.2.2. Application to Biomass Substrates
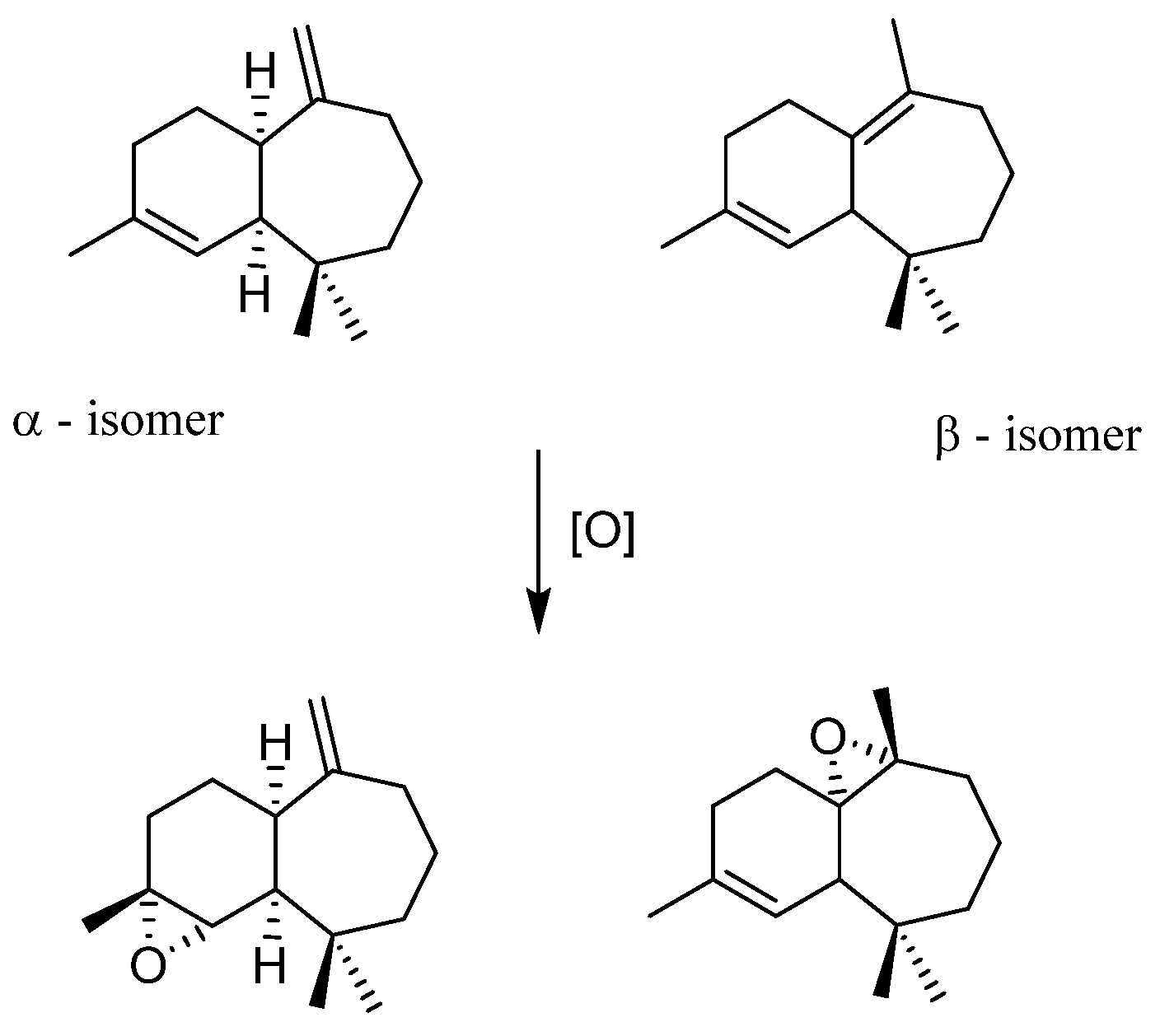
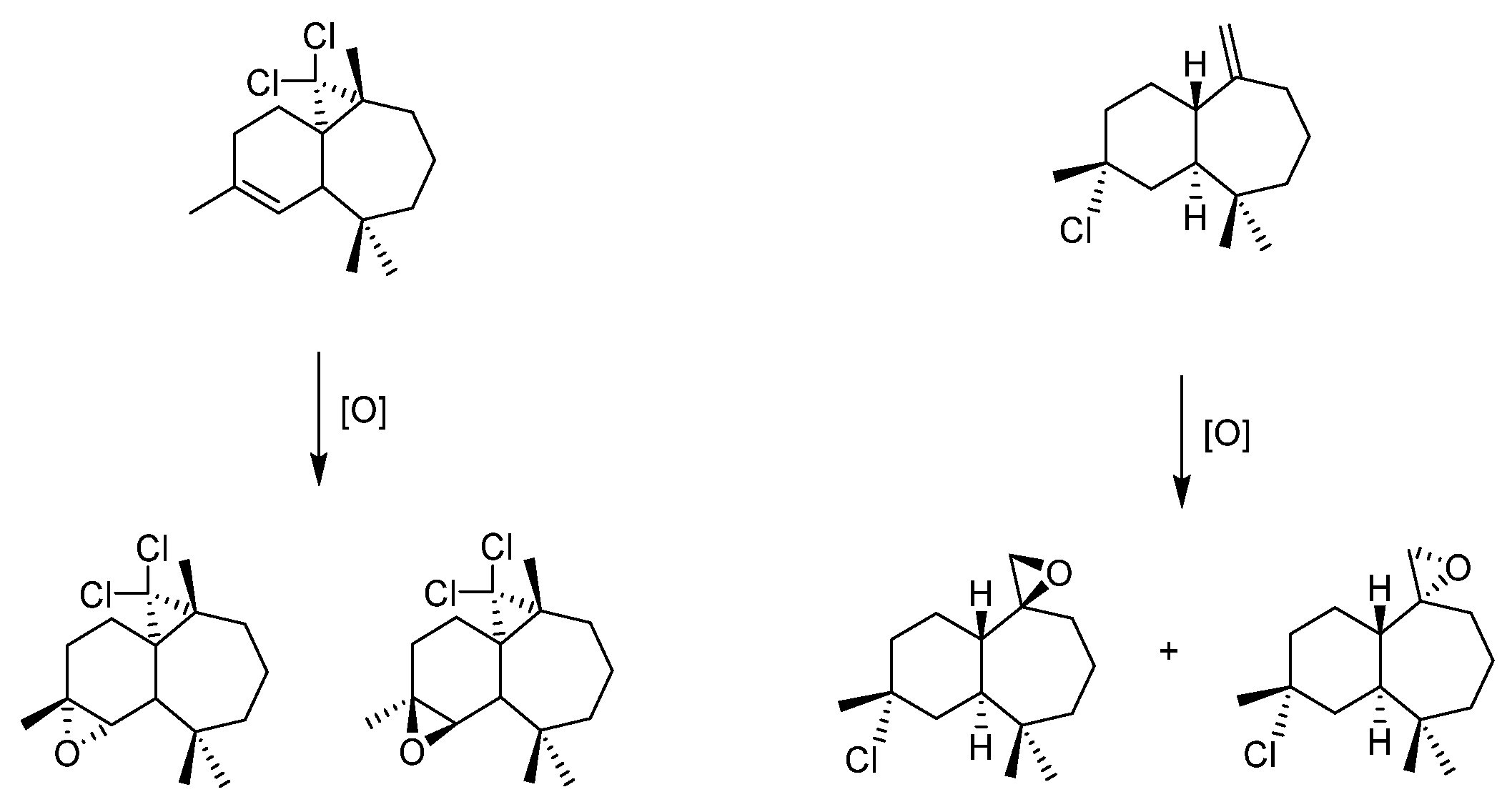
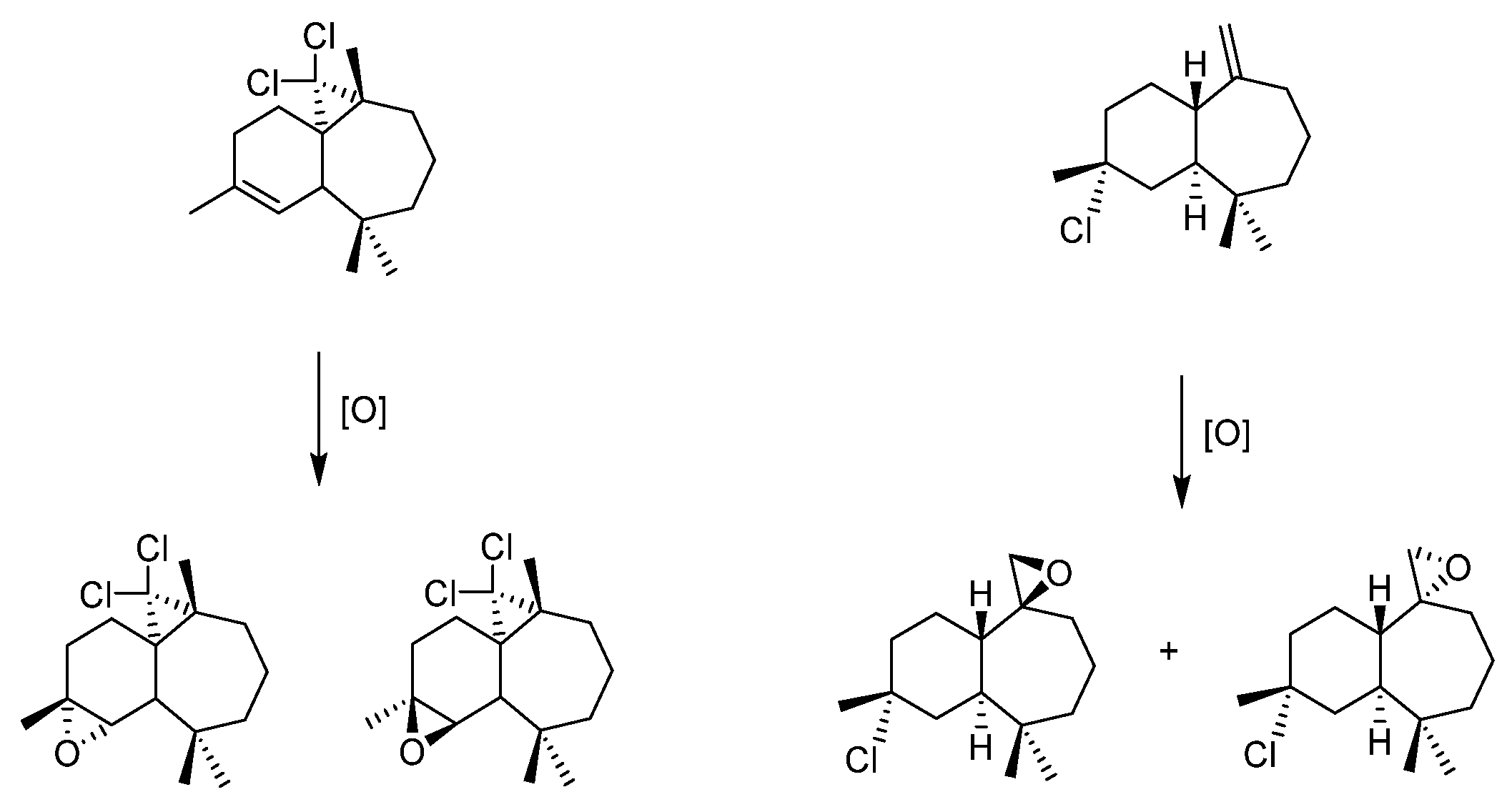
- Himachalenes
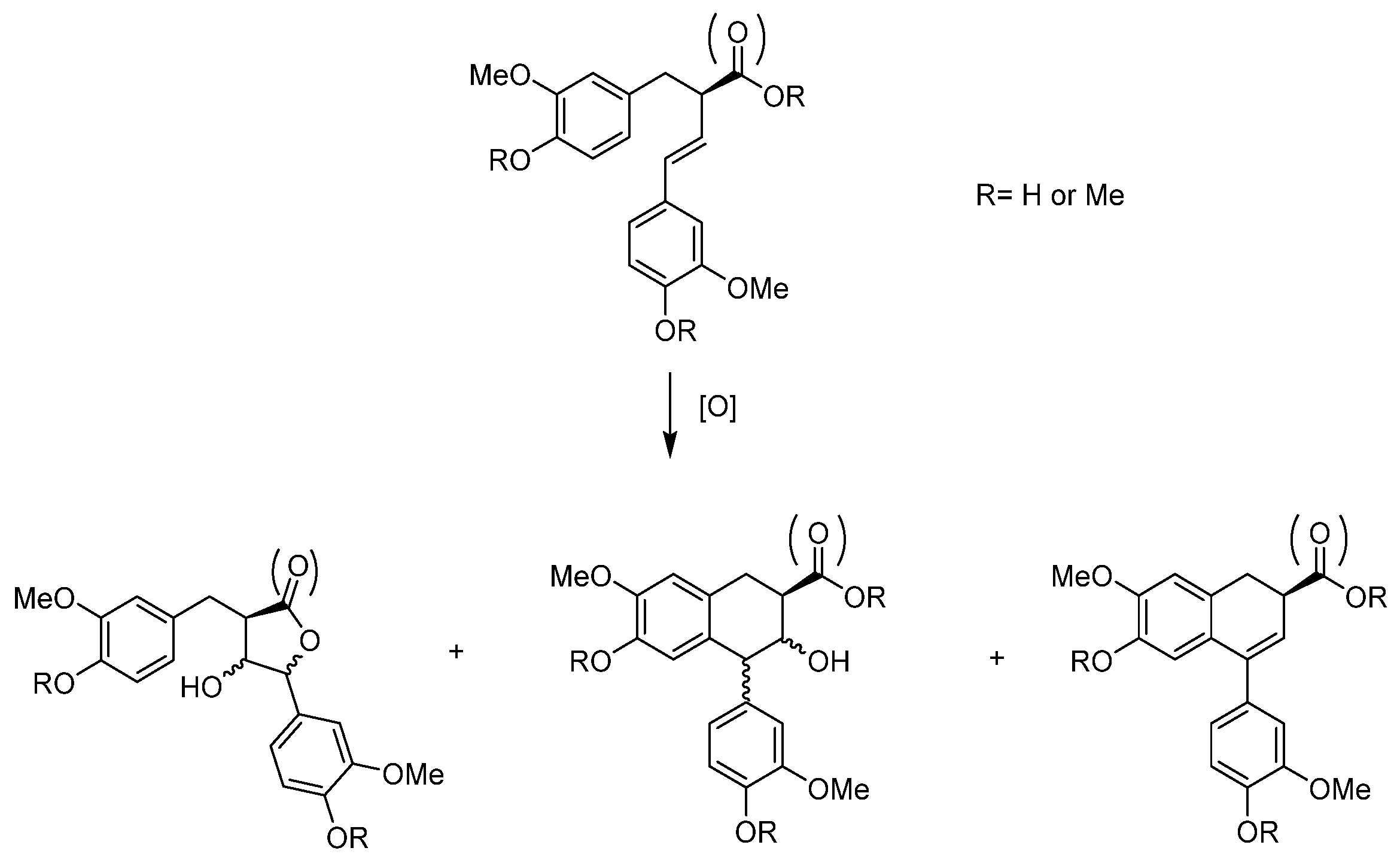
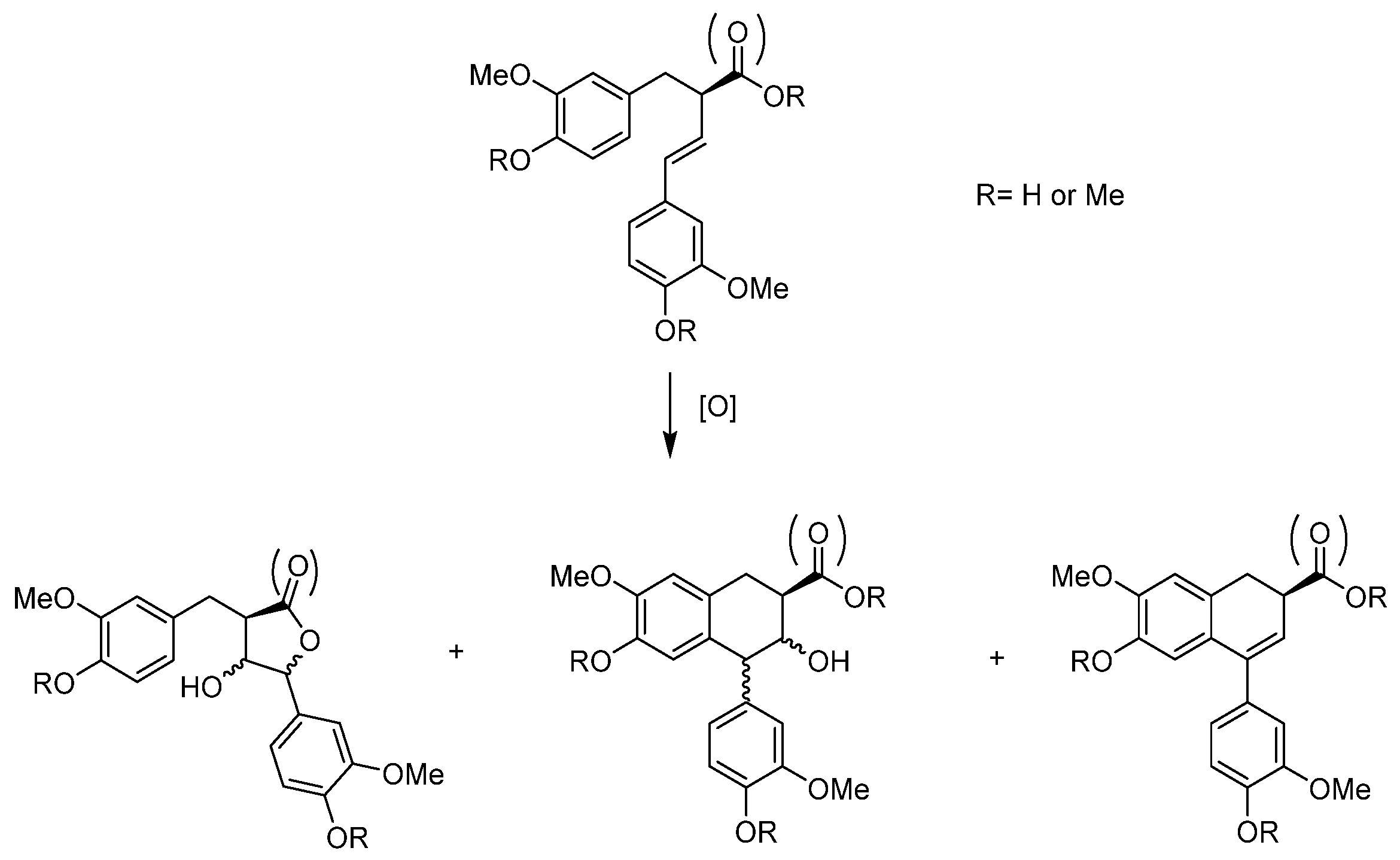
- Lignans
- Limonene
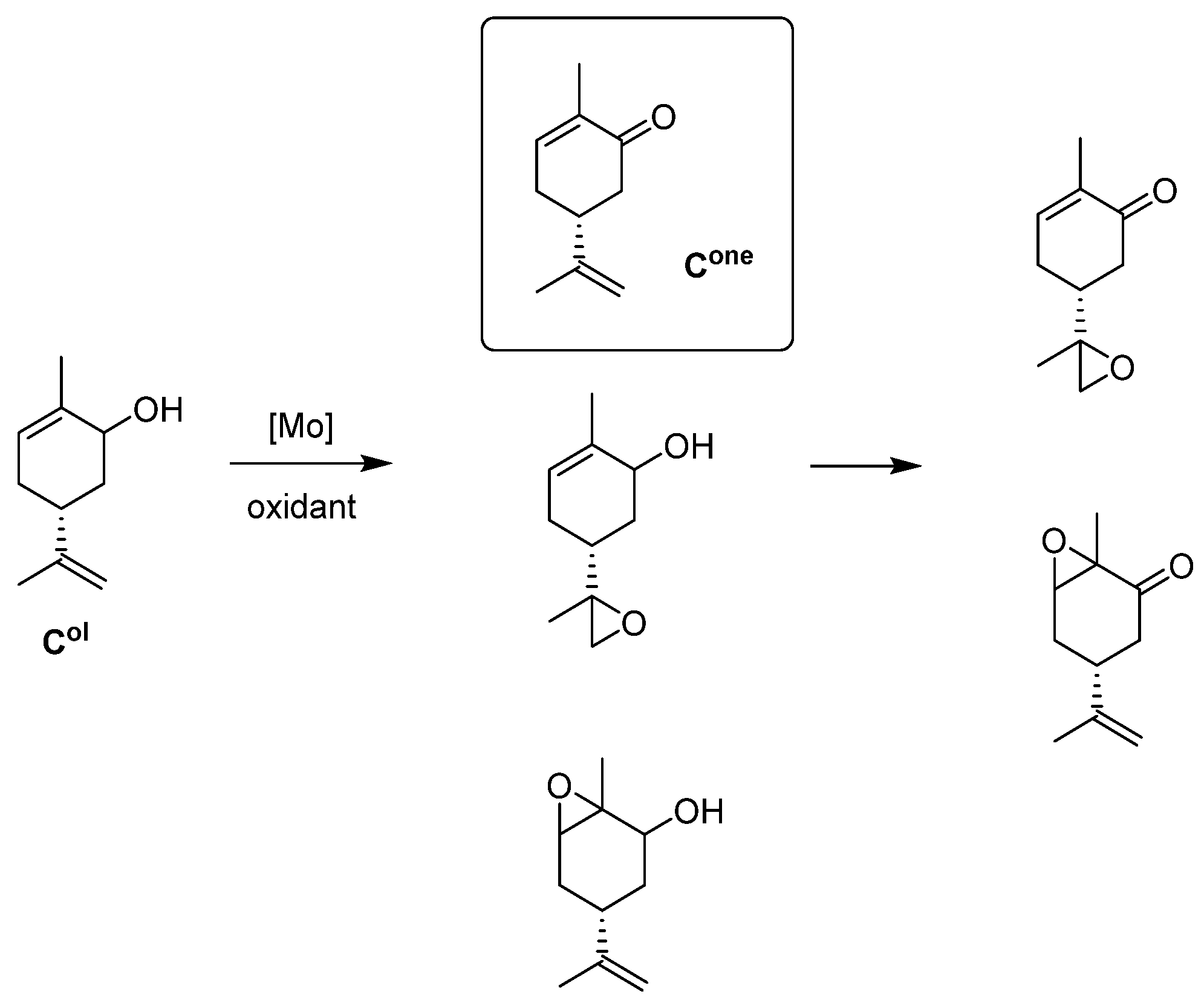
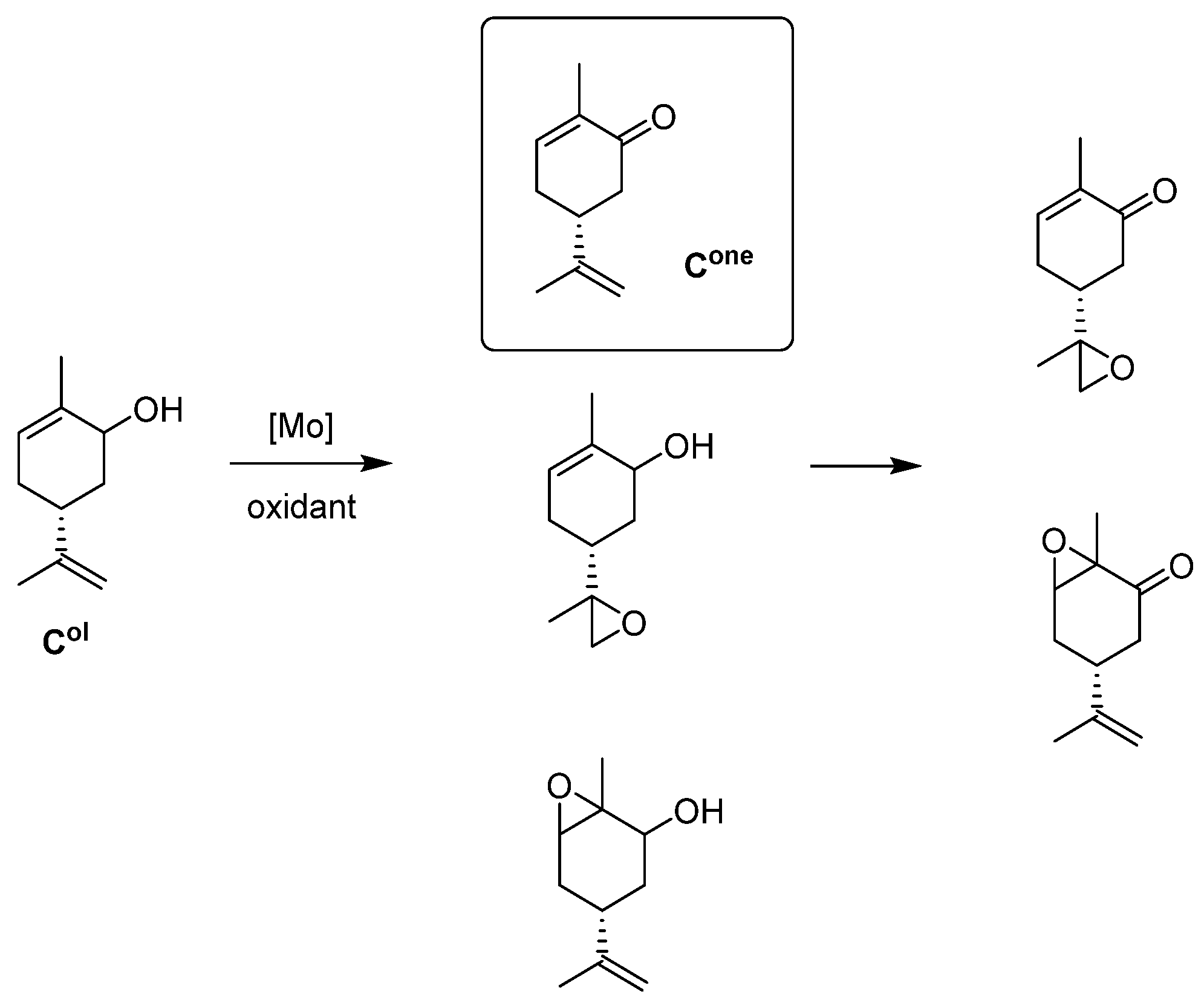
- Carveol Oxidation
3. Vanadium Species
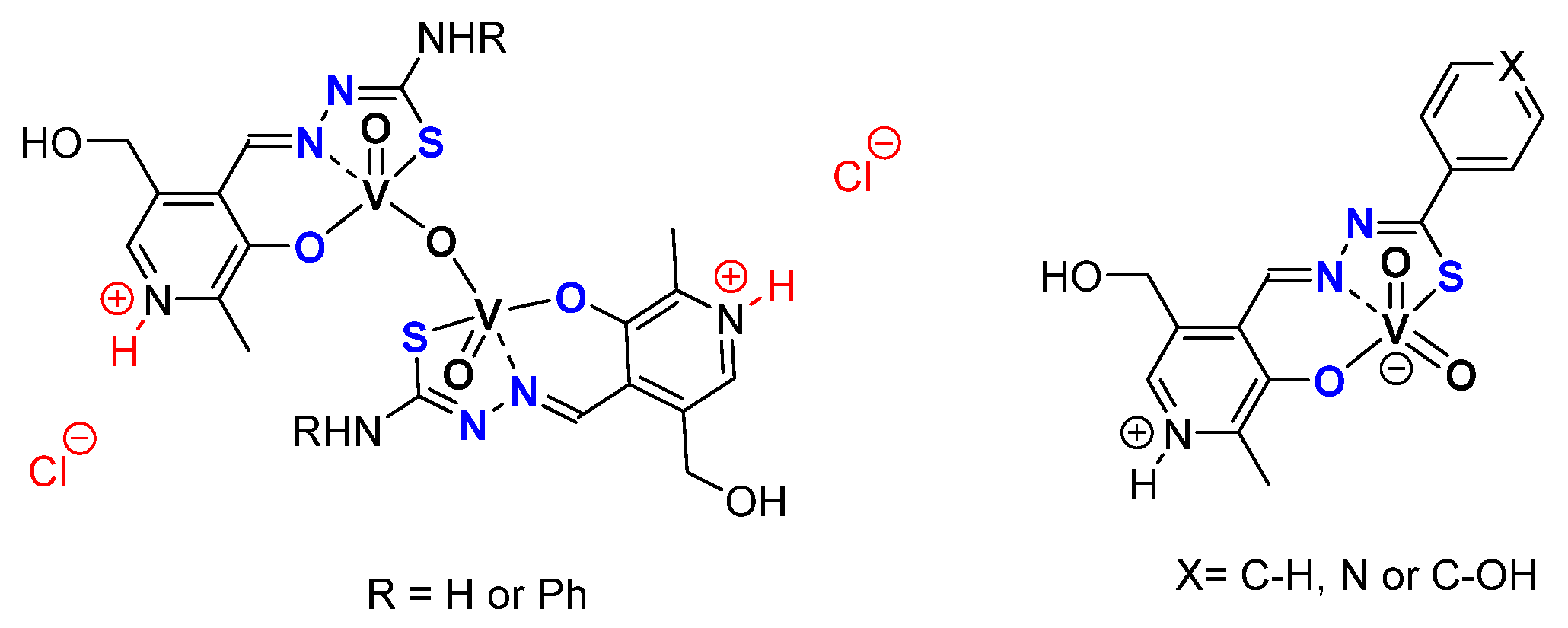
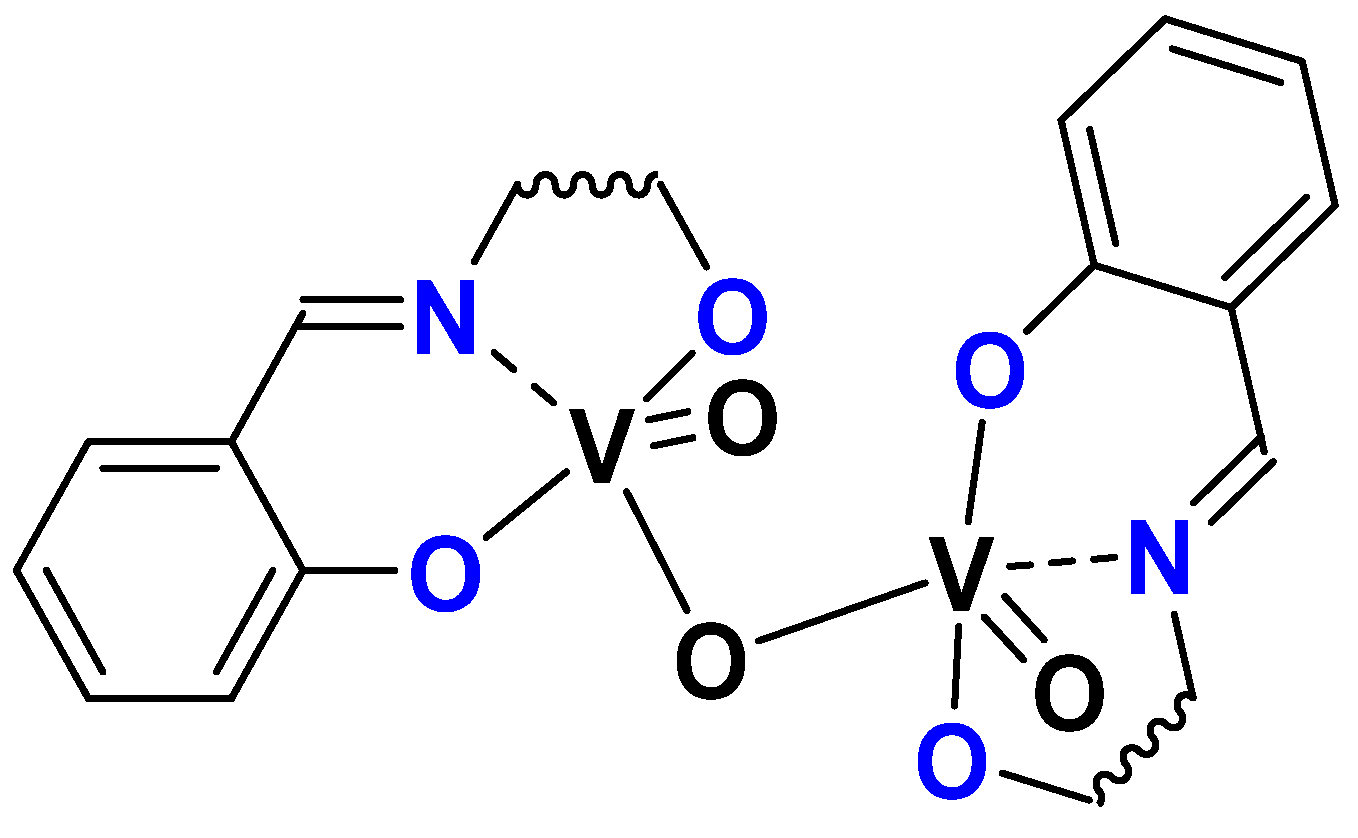
3.1. SAP
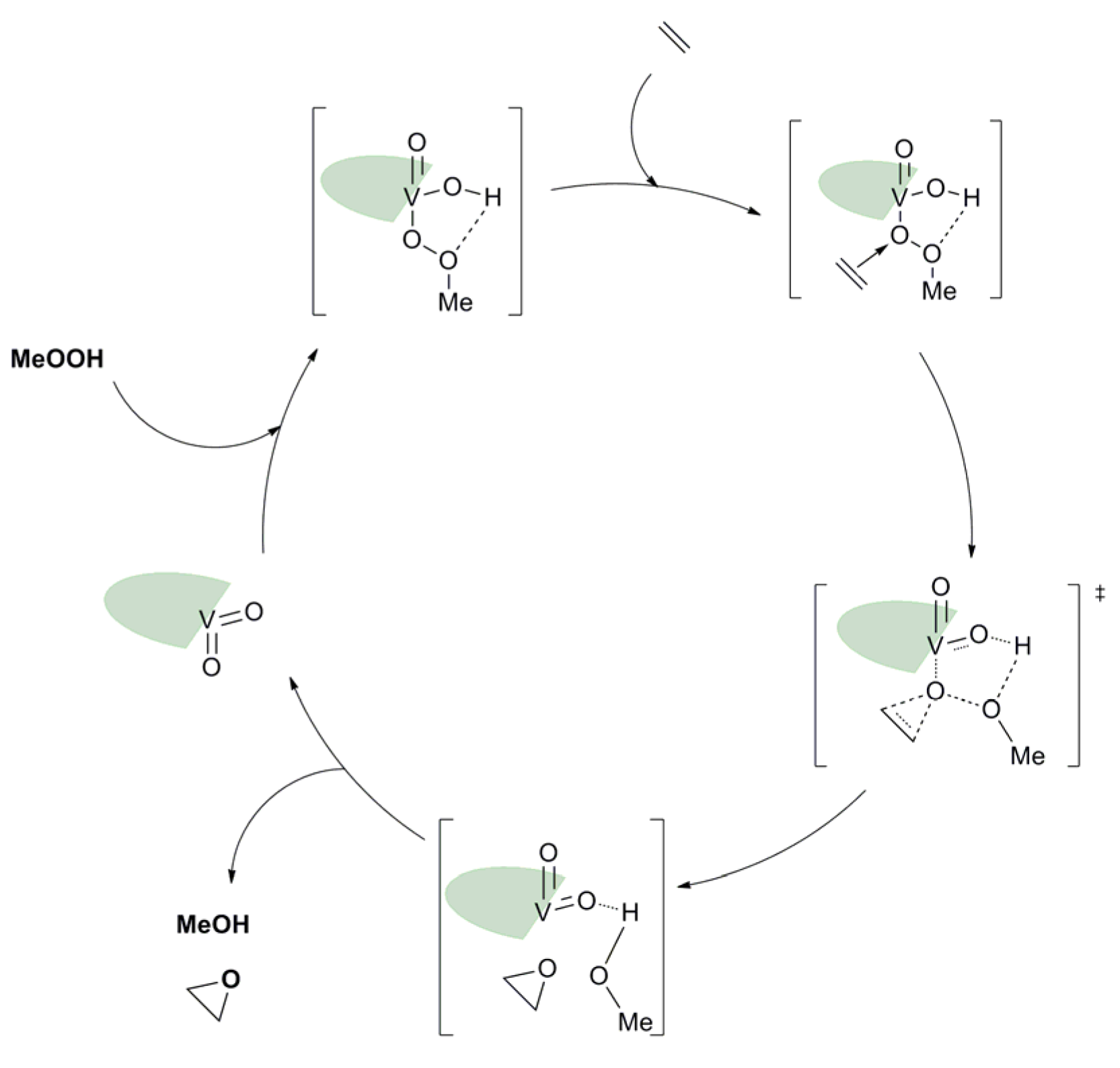
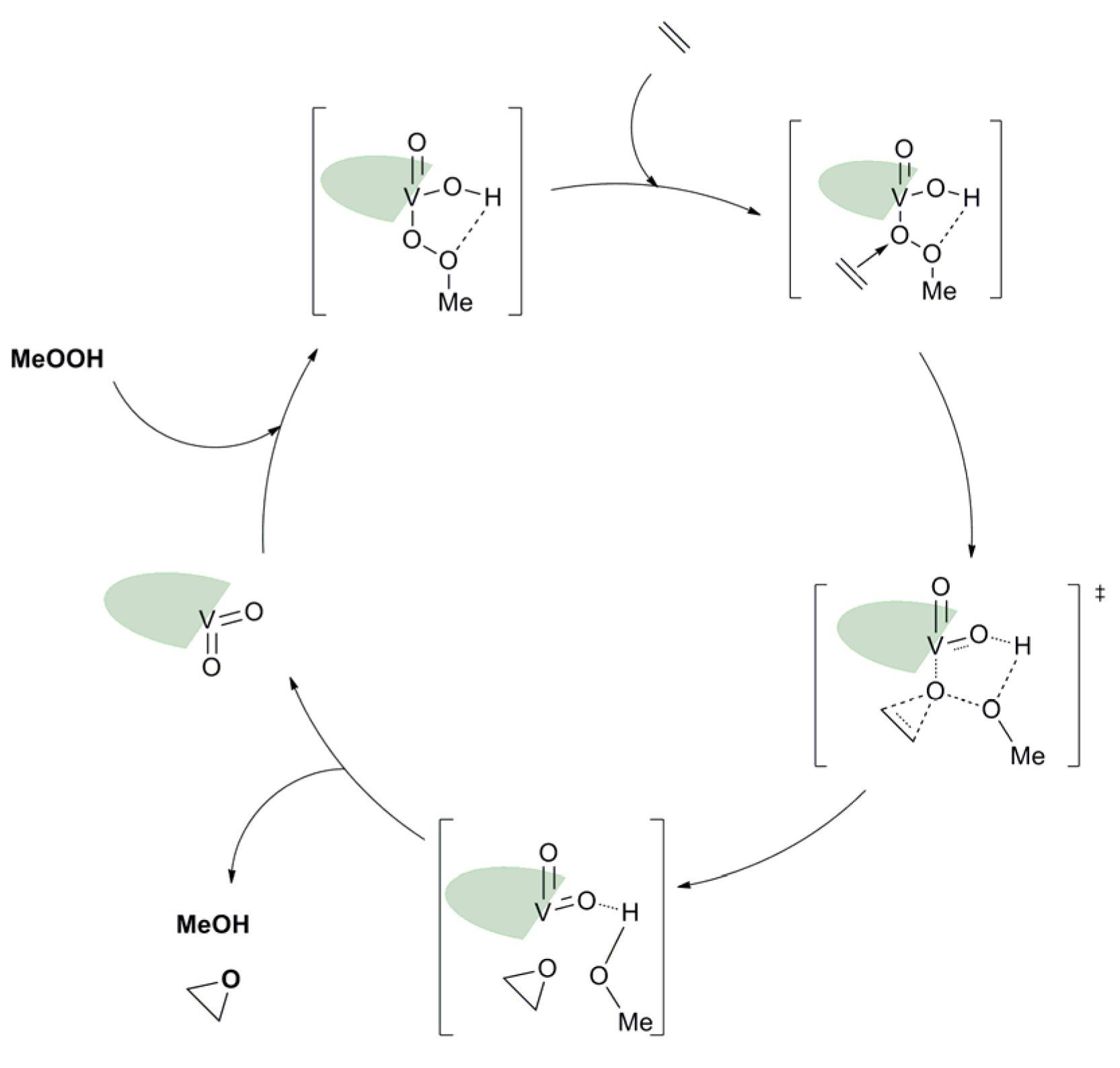
3.2. ONO, ONS, and Mechanism
4. Keggin-Type Polyoxometalates as (ep)Oxidation Catalyst
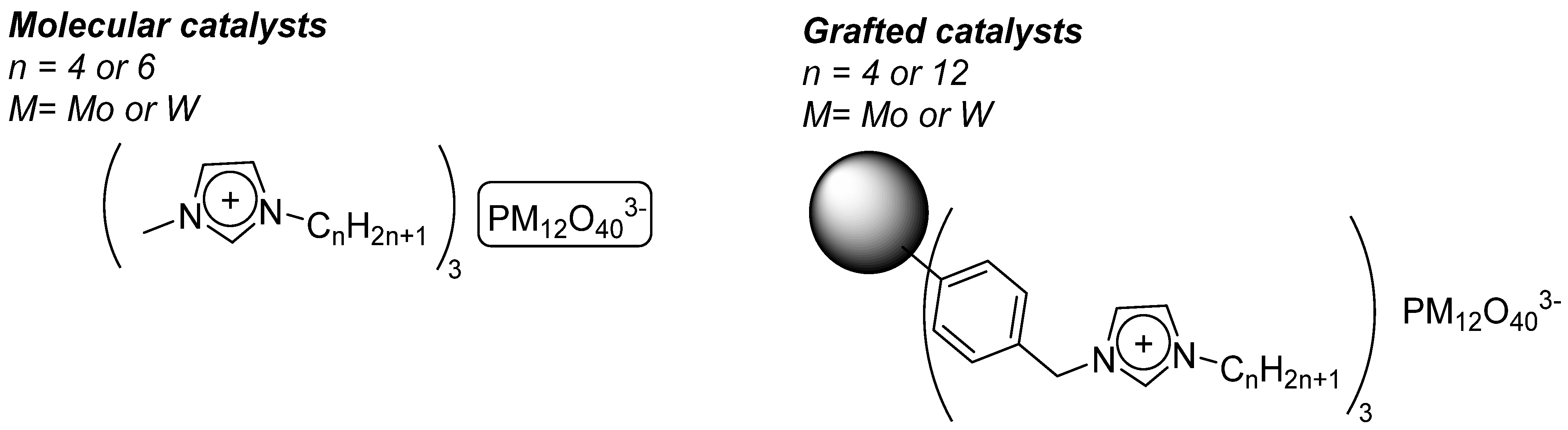
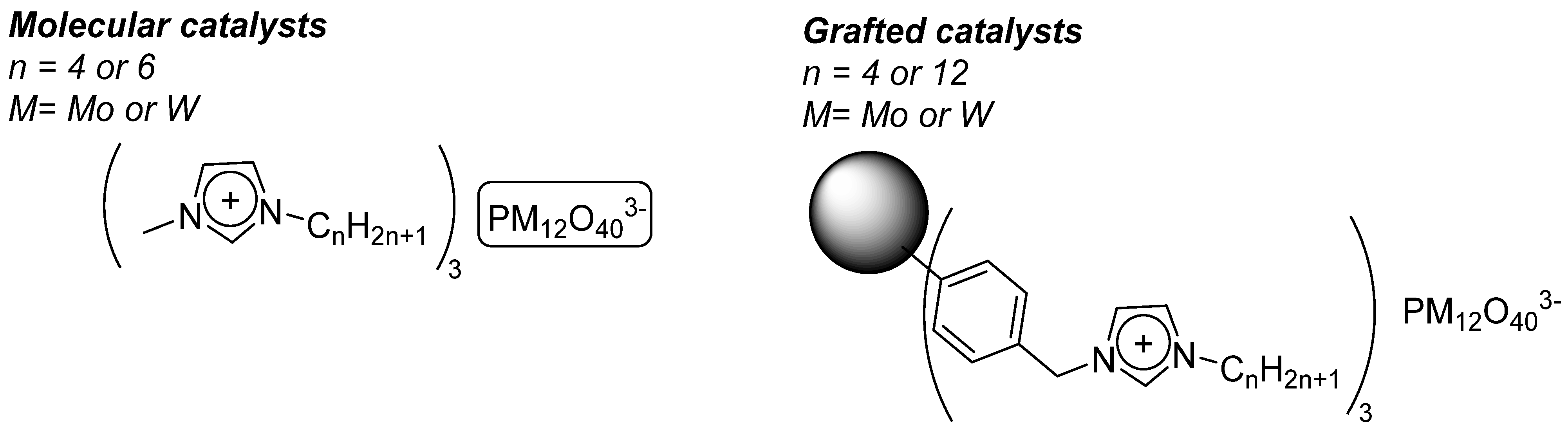
4.1. Pyridinium Salts
4.2. Supported Catalysts
4.2.1. Grafted POMs on Merrifield Resins [65]
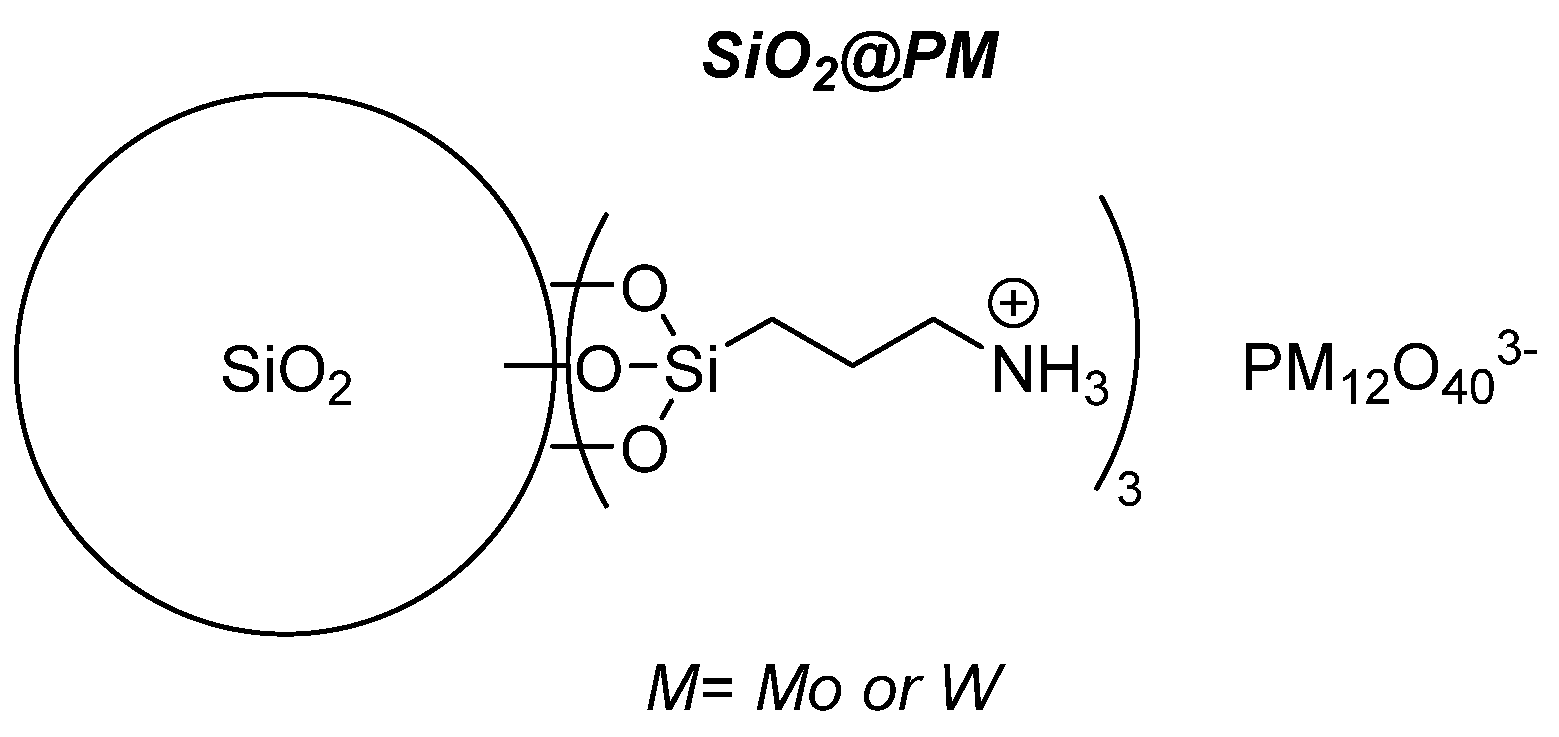
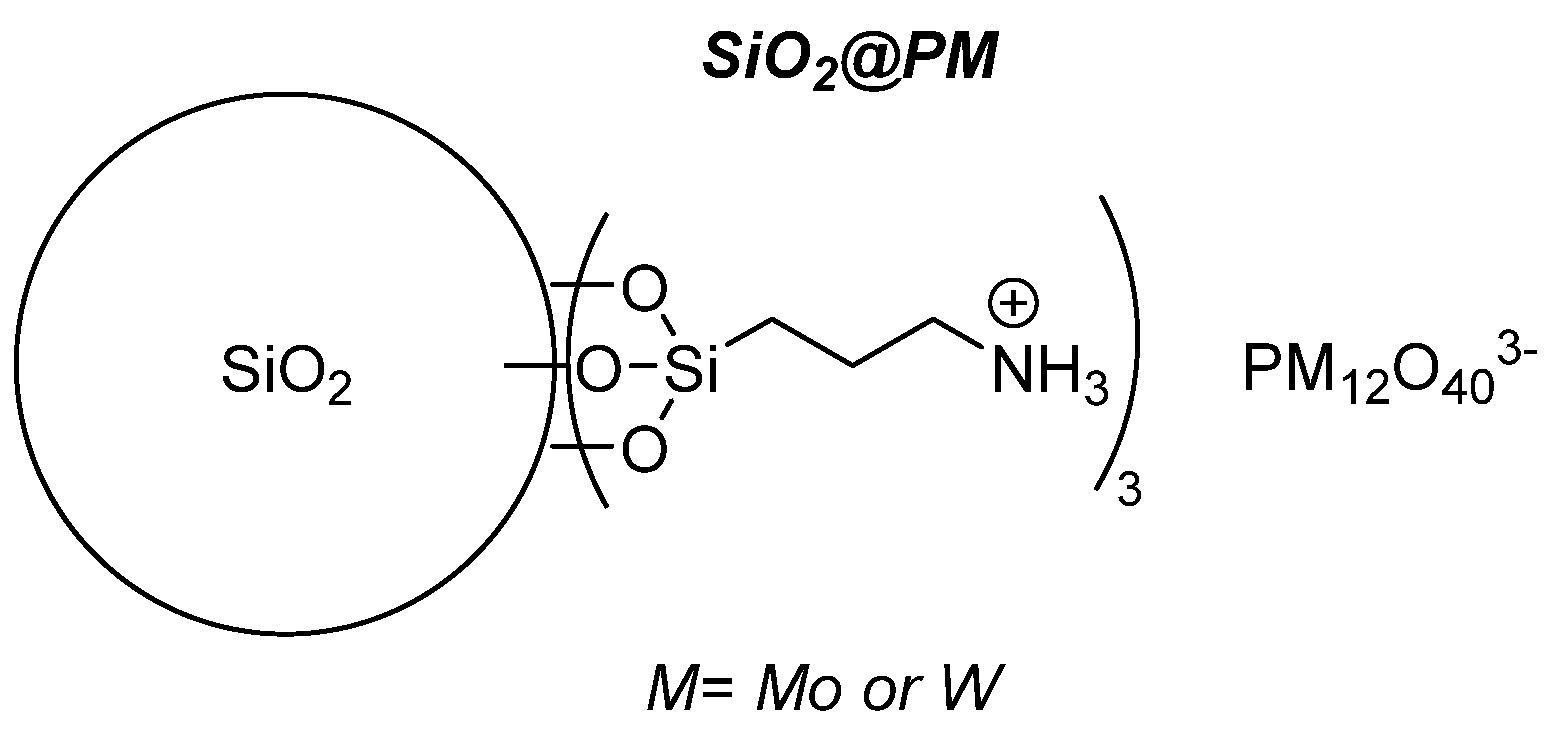
4.2.2. Grafted POMs on Functionalized Silica
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Clark, J.H. Green chemistry: Challenges and opportunities. Green Chem. 1999, 1, 1–8. [Google Scholar] [CrossRef]
- Bardi, U. Peak oil: The four stages of a new idea. Energy 2009, 34, 323–326. [Google Scholar] [CrossRef]
- Lutz, C.; Lehr, U.; Wiebe, K.S. Economic effects of peak oil. Energy Policy 2012, 48, 829–834. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J.C. Green Chemistry Theory and Practice; Oxford University Press: New York, NY, USA, 1998; ISBN1 0198502346. ISBN2 9780198502340. [Google Scholar]
- Regulation (EC) No. 1907/2006 of the European Parliament and of the Council, Official Journal of the European Union 30.12.2006, L396. Available online: https://osha.europa.eu/fr/legislation/directives/regulation-ec-no-1907-2006-of-the-european-parliament-and-of-the-council (accessed on 6 August 2022).
- Sheldon, R.A.; Arends, I.; Hanefeld, U. Green Chemistry and Catalysis; Wiley-VCH: Weinheim, Germany, 2007; ISBN 978-3-527-30715-9. [Google Scholar]
- Sheldon, R.A. Green chemistry and resource efficiency: Towards a green economy. Green Chem. 2016, 18, 3180–3183. [Google Scholar] [CrossRef]
- Moraes, J.D.; Almeida, A.A.; Brito, M.R.; Marques, T.H.; Lima, T.C.; Sousa, D.P.; Nakano, E.; Mendonça, R.Z.; Freitas, R.M. Role of Catalase and Superoxide Dismutase Activities on Oxidative Stress in the Brain of a Phenylketonuria Animal Model and the Effect of Lipoic Acid. Planta Med. 2013, 79, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.K.; Dhir, A. Effect of various classes of antidepressants in behavioral paradigms of despair. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 1248–1254. [Google Scholar] [CrossRef]
- Umezu, T. Anticonflict effects of plant-derived essential oils. Pharmacol. Biochem. Behav. 1999, 64, 35–40. [Google Scholar] [CrossRef]
- De Almeida, A.A.C.; Pereira Costa, J.; de Carvalho, R.B.F.; Pergentino de Sousa, D.; Mendes de Freitas, R. Evaluation of acute toxicity of a natural compound (+)-limonene epoxide and its anxiolytic-like action. Brain Res. 2012, 1448, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Sudo, A. Development and application of novel ring-opening polymerizations to functional networked polymers. Polym. Sci. 2009, 47, 4847–4858. [Google Scholar] [CrossRef]
- Petrovic, Z.S. Polyurethanes from Vegetable Oils. Polym. Rev. 2008, 48, 109–155. [Google Scholar] [CrossRef]
- Burdock, G.A. Fenaroli’s Handbook of Flavor Ingredients, 6th ed.; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar] [CrossRef]
- Blair, M.; Tuck, K.L. A new diastereoselective entry to the (1S,4R)- and (1S,4S)-isomers of 4-isopropyl-1-methyl-2-cyclohexen-1-ol, aggregation pheromones of the ambrosia beetle Platypus quercivorus. Tetrahedron Asymmetry 2009, 20, 2149–2153. [Google Scholar] [CrossRef]
- McDonald, R.N.; Steppel, R.N.; Dorsey, J.E. m-CHLOROPERBENZOIC ACID. Org. Synth. 1970, 50, 15–18. [Google Scholar] [CrossRef]
- Rose, E.; Andrioletti, B.; Zrig, S.; Quelquejeu-Etheve, M. Enantioselective epoxidation of olefins with chiral metalloporphyrin catalysts. Chem. Soc. Rev. 2005, 34, 573–583. [Google Scholar] [CrossRef]
- Maiti, S.K.; Dinda, S.; Bhattacharyya, R. Unmatched efficiency and selectivity in the epoxidation of olefins with oxo-diperoxomolybdenum(VI) complexes as catalysts and hydrogen peroxide as terminal oxidant. Tetrahedron Lett. 2008, 49, 6205–6208. [Google Scholar] [CrossRef]
- Mbeleck, R.; Ambroziak, K.; Saha, B.; Sherrington, D.C. Stability and recycling of polymer-supported Mo(VI) alkene epoxidation catalysts. React. Funct. Polym. 2007, 67, 1448–1457. [Google Scholar] [CrossRef]
- Jørgensen, K.A. Transition-Metal-Catalyzed Epoxidations. Chem. Rev. 1989, 89, 431–458. [Google Scholar] [CrossRef]
- Sherwood, J. European Restrictions on 1,2-Dichloroethane: C−H Activation Research and Development Should Be Liberated and not Limited. Angew. Chem. Int. Ed. 2018, 57, 14286–14290. [Google Scholar] [CrossRef]
- Gao, F.; Bai, R.; Ferlin, F.; Vaccaro, L.; Li, M.; Gu, Y. Replacement strategies for non-green dipolar aprotic solvents. Green Chem. 2020, 22, 6240–6257. [Google Scholar] [CrossRef]
- Welton, T. Solvents and sustainable chemistry. Proc. R. Soc. A 2015, 471, 20150502. [Google Scholar] [CrossRef] [Green Version]
- Kollar, J.; Wyckoff, N.J. Process For Preparing Glycidol. U.S. Patent 3625981A, 7 December 1971. [Google Scholar]
- Sheng, M.N.; Zajaczek, G.J. Methods of Producing Epoxides. GB1.136.923, 18 December 1968. [Google Scholar]
- Sobczak, J.M.; Glowiak, T.; Ziolkowski, J.J. The structure of binuclear molybdenum(VI) oxocomplexes with dianionic tridentate Schiff bases. Trans. Met. Chem. 1990, 15, 208–211. [Google Scholar] [CrossRef]
- Sobczak, J.M.; Ziółkowski, J.J. Molybdenum complex-catalysed epoxidation of unsaturated fatty acids by organic hydroperoxides. Appl. Catal. A Gen. 2003, 248, 261–268. [Google Scholar] [CrossRef]
- Agustin, D.; Bibal, C.; Neveux, B.; Daran, J.-C.; Poli, R. Structural Characterization and Theoretical Calculations of cis-Dioxo(N-salicylidene-2-aminophenolato)(ethanol)molybdenum(VI) Complexes MoO2(SAP)(EtOH) (SAP = N-salicylidene-2 aminophenolato). Z. Anorg. Allg. Chem. 2009, 635, 2120–2125. [Google Scholar] [CrossRef]
- Agustin, D.; Daran, J.-C.; Poli, R. Polymorph of {2-[(2-hydroxyethyl)iminiomethyl]phenolato-κO}dioxido{2-[(2-oxidoethyl) iminomethyl]phenolato-κ3O,N,O’}molybdenum(VI). Acta Cryst. 2008, 64, m101–m104. [Google Scholar] [CrossRef]
- Morlot, J.; Uyttebroeck, N.; Agustin, D.; Poli, R. Solvent-Free Epoxidation of Olefins Catalyzed by “[MoO2(SAP)]”: A New Mode of tert-Butylhydroperoxide Activation. ChemCatChem 2013, 5, 601–611. [Google Scholar] [CrossRef]
- Bartlett, P.D. Recent work on the mechanisms of peroxide reactions. Rec. Chem. Prog. 1950, 11, 47–51. [Google Scholar]
- Wang, W.; Vanderbeeken, T.; Agustin, D.; Poli, R. Tridentate ONS vs. ONO salicylideneamino(thio)phenolato [MoO2L] complexes for catalytic solvent-free epoxidation with aqueous TBHP. Catal. Commun. 2015, 63, 26–30. [Google Scholar] [CrossRef]
- Wang, W.; Guerrero, T.; Merecias, S.R.; García-Ortega, H.; Santillan, R.; Daran, J.-C.; Farfán, N.; Agustin, D.; Poli, R. Substituent effects on solvent-free epoxidation catalyzed by dioxomolybdenum(VI) complexes supported by ONO Schiff base ligands. Inorg. Chim. Acta 2015, 431, 176–183. [Google Scholar] [CrossRef]
- Wang, W.; Daran, J.-C.; Poli, R.; Agustin, D. OH-substituted tridentate ONO Schiff base ligands and related molybdenum(VI) complexes for solvent-free (ep)oxidation catalysis with TBHP as oxidant. J. Mol. Catal. A Chem. 2016, 416, 117–126. [Google Scholar] [CrossRef]
- Cindric, M.; Pavlovic, G.; Katava, R.; Agustin, D. Towards a global greener process: From solventless synthesis of molybdenum(VI) ONO Schiff base complexes to catalyzed olefin epoxidation under organic-solvent-free conditions. New J. Chem. 2017, 41, 594–602. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Pisk, J.; Prugovecki, B.; Matkovic-Calogovic, D. Novel dioxomolybdenum(VI) and oxomolybdenum(V) complexes with pyridoxal thiosemicarbazone ligands: Synthesis and structural characterization. Inorg. Chim. Acta 2009, 362, 4059–4064. [Google Scholar] [CrossRef]
- Pisk, J.; Agustin, D.; Vrdoljak, V.; Poli, R. Epoxidation Processes by Pyridoxal Dioxomolybdenum(VI) (Pre)Catalysts Without Organic Solvent. Adv. Synth. Catal. 2011, 353, 2910–2914. [Google Scholar] [CrossRef]
- Pisk, J.; Prugovecki, B.; Matkovic-Calogovic, D.; Poli, R.; Agustin, D.; Vrdoljak, V. Charged dioxomolybdenum(VI) complexes with pyridoxal thiosemicarbazone ligands as molybdenum(V) precursors in oxygen atom transfer process and epoxidation (pre)catalysts. Polyhedron 2012, 33, 441–449. [Google Scholar] [CrossRef]
- Pisk, J.; Prugovečki, B.; Matković-Čalogović, D.; Jednačak, T.; Novak, P.; Agustin, D.; Vrdoljak, V. Pyridoxal hydrazonato molybdenum(VI) complexes: Assembly, structure and epoxidation (pre)catalyst testing under solvent-free conditions. RSC Adv. 2014, 4, 39000–39010. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Pisk, J.; Agustin, D.; Novak, P.; Parlov Vuković, J.; Matković-Čalogovic, D. Dioxomolybdenum(VI) and dioxotungsten(VI) complexes chelated with the ONO tridentate hydrazone ligand: Synthesis, structure and catalytic epoxidation activity. New J. Chem. 2014, 38, 6176–6185. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Pisk, J.; Prugovečki, B.; Agustin, D.; Novak, P.; Matković-Čalogović, D. Dioxotungsten(VI) complexes with isoniazid-related hydrazones as (pre)catalysts for olefin epoxidation: Solvent and ligand substituent effects. RSC Adv. 2016, 6, 36384–36393. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Mandarić, M.; Hrenar, T.; Đilović, I.; Pisk, J.; Pavlović, G.; Cindrić, M.; Agustin, D. Geometrically Constrained Molybdenum(VI) Metallosupramolecular Architectures: Conventional Synthesis versus Vapor and Thermally Induced Solid-State Structural Transformations. Cryst. Growth Des. 2019, 19, 3000–3011. [Google Scholar] [CrossRef]
- Pisk, J.; Agustin, D.; Vrdoljak, V. Tetranuclear molybdenum(vi) hydrazonato epoxidation (pre)catalysts: Is water always the best choice? Catal. Commun. 2020, 142, 1060272. [Google Scholar] [CrossRef]
- Cvijanović, D.; Pisk, J.; Pavlović, G.; Šišak-Jung, D.; Matković-Čalogović, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Discrete mononuclear and dinuclear compounds containing a MoO22+ core and 4-aminobenzhydrazone ligands: Synthesis, structure and organic-solvent-free epoxidation activity. New J. Chem. 2019, 43, 1791–1802. [Google Scholar] [CrossRef]
- Pisk, J.; Rubčić, M.; Kuzman, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Molybdenum(VI) complexes of hemilabile aroylhydrazone ligands as efficient catalysts for greener cyclooctene epoxidation: An experimental and theoretical approach. New J. Chem. 2019, 43, 5531–5542. [Google Scholar] [CrossRef]
- Mrkonja, S.; Topić, E.; Mandarić, M.; Agustin, D.; Pisk, J. Efficient Molybdenum Hydrazonato Epoxidation Catalysts Operating under Green Chemistry Conditions: Water vs. Decane Competition. Catalysts 2021, 11, 756. [Google Scholar] [CrossRef]
- Bafti, A.; Razum, M.; Topić, E.; Agustin, D.; Pisk, J.; Vrdoljak, V. Implication of oxidant activation on olefin epoxidation catalysed by Molybdenum catalysts with aroylhydrazonato ligands: Experimental and theoretical studies. Mol. Catal. 2021, 512, 111764. [Google Scholar] [CrossRef]
- Loubidi, M.; Agustin, D.; Benharref, A.; Poli, R. Solvent-free epoxidation of himachalenes and their derivatives by TBHP using [MoO2(SAP)]2 as a catalyst. C. R. Chim. 2014, 17, 549–556. [Google Scholar] [CrossRef]
- Patrik, A.; Runeberg, P.A.; Agustin, D.C.; Eklund, P.C. Formation of Tetrahydrofurano-, Aryltetralin, and Butyrolactone Norlignans through the Epoxidation of 9-Norlignans. Molecules 2020, 25, 1160. [Google Scholar] [CrossRef]
- Balandrin, M.F.; Klocke, J.A.; Wurtele, E.S.; Bollinger, W.H. Natural plant chemicals: Sources of industrial and medicinal materials. Science 1985, 228, 1154–1160. [Google Scholar] [CrossRef]
- Satrani, B.; Aberchane, M.; Farah, A.; Chaouch, A.; Talbi, M. Composition chimique et activité antimicrobienne des huiles essentielles extraites par hydrodistillation fractionnée du bois de Cedrus atlantica Manetti. Acta Bot. Gall. 2006, 153, 97–104. [Google Scholar] [CrossRef]
- Willför, S.M.; Ahotupa, M.O.; Hemming, J.E.; Reunanen, M.H.T.; Eklund, P.C.; Sjöholm, R.E.; Eckerman, C.S.E.; Suvi, P.; Pohjamo, A.; Holmbom, B.R. Antioxidant Activity of Knotwood Extractives and Phenolic Compounds of Selected Tree Species. J. Agric. Food Chem. 2003, 51, 7600–7606. [Google Scholar] [CrossRef]
- Eklund, P.C.; Långvik, O.K.; Wärnå, J.P.; Salmi, T.O.; Willför, S.M.; Sjöholm, R.E. Chemical studies on antioxidant mechanisms and free radical scavenging properties of lignans. Org. Biomol. Chem. 2005, 3, 3336–3347. [Google Scholar] [CrossRef]
- Wang, W.; Agustin, D.; Poli, R. Influence of ligand substitution on molybdenum catalysts with tridentate Schiff base ligands for the organic solvent-free oxidation of limonene using aqueous TBHP as oxidant. Mol. Catal. 2017, 443, 52–59. [Google Scholar] [CrossRef]
- Blair, M.; Andrews, P.C.; Fraser, B.H.; Forsyth, C.M.; Junk, P.C.; Massi, M.; Tuck, K.L. Facile methods for the separation of the cis- and trans-diastereomers of limonene 1,2-oxide and convenient routes to diequatorial and diaxial 1,2-diols. Synthesis 2007, 1523–1527. [Google Scholar] [CrossRef]
- Weijers, C. Enantioselective hydrolysis of aryl, alicyclic and aliphatic epoxides by Rhodotorula glutinis. Tetrahedron Asym. 1997, 8, 639–647. [Google Scholar] [CrossRef]
- Mihalinec, J.; Pajski, M.; Guillo, P.; Mandarić, M.; Bebić, N.; Pisk, J.; Vrdoljak, V. Alcohol Oxidation Assisted by Molybdenum Hydrazonato Catalysts Employing Hydroperoxide Oxidants. Catalysts 2021, 11, 881. [Google Scholar] [CrossRef]
- Mimoun, H.; Mignard, M.; Brechot, P.; Saussine, L. Selective epoxidation of olefins by oxo[N-(2-oxidophenyl)salicylidenaminato]-vanadium(V) alkylperoxides. On the mechanism of the Halcon epoxidation process. J. Am. Chem. Soc. 1986, 108, 3711. [Google Scholar] [CrossRef]
- Rehder, D. The coordination chemistry of vanadium as related to its biological functions. Coord. Chem. Rev. 1999, 182, 297–322. [Google Scholar] [CrossRef]
- Maurya, M.R. Development of the coordination chemistry of vanadium through bis(acetylacetonato) oxovanadium(IV): Synthesis, reactivity and structural aspects. Coord. Chem. Rev. 2003, 237, 163–181. [Google Scholar] [CrossRef]
- Hartung, J. Stereoselective syntheses of functionalized cyclic ethers via (Schiff-base)vanadium(V)-catalyzed oxidations. Pure Appl. Chem. 2005, 77, 1559–1574. [Google Scholar] [CrossRef]
- Cordelle, C.; Agustin, D.; Daran, J.-C.; Poli, R. Oxo-bridged bis oxo-vanadium(V) complexes with tridentate Schiff base ligands (VOL)2O (L = SAE, SAMP, SAP): Synthesis, structure and epoxidation catalysis under solvent-free conditions. Inorg. Chim. Acta 2010, 364, 144–149. [Google Scholar] [CrossRef]
- Pisk, J.; Daran, J.-C.; Poli, R.; Agustin, D. Pyridoxal based ONS and ONO Vanadium(V) complexes: Structural analysis and catalytic application in organic solvent free epoxidation. J. Mol. Catal. A Chem. 2015, 403, 52. [Google Scholar] [CrossRef]
- Guérin, B.; Mesquita Fernandes, D.; Daran, J.-C.; Agustin, D.; Poli, R. Investigation of induction times, activity, selectivity, interface and mass transport in solvent-free epoxidation by H2O2 and TBHP: A study with organic salts of the [PMo12O40]3− anion. New J. Chem. 2013, 37, 3466–3475. [Google Scholar] [CrossRef]
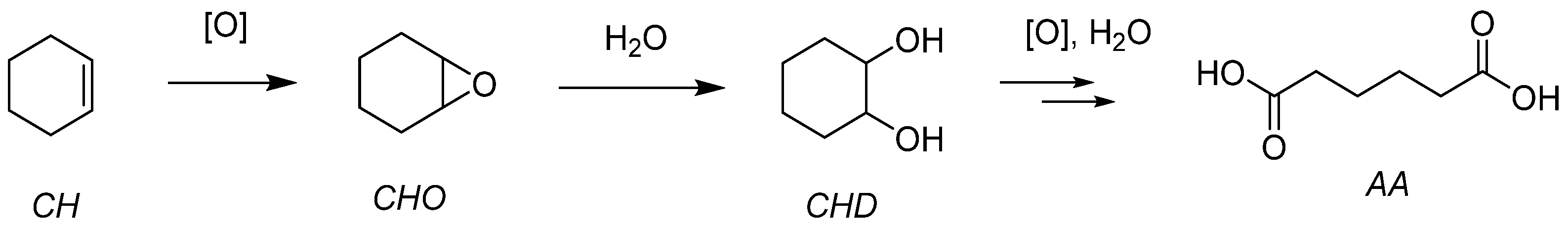
- Pisk, J.; Agustin, D.; Poli, R. Organic Salts and Merrifield Resin Supported [PM12O40]3− (M = Mo or W) as Catalysts for Adipic Acid Synthesis. Molecules 2019, 24, 783. [Google Scholar] [CrossRef]
- Wang, Y.; Gayet, F.; Guillo, P.; Agustin, D. Organic Solvent-Free Olefins and Alcohols (ep)oxidation Using Recoverable Catalysts Based on [PM12O40]3− (M = Mo or W) Ionically Grafted on Amino Functionalized Silica Nanobeads. Materials 2019, 12, 3278. [Google Scholar] [CrossRef] [Green Version]





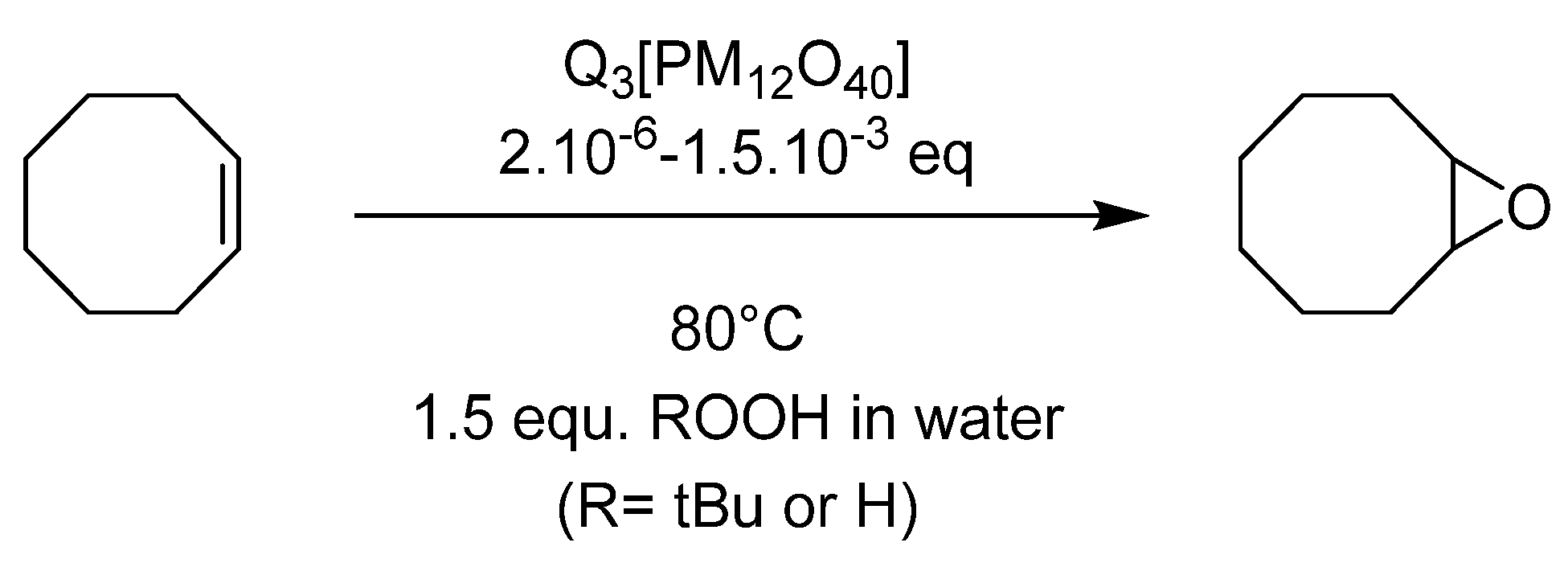


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
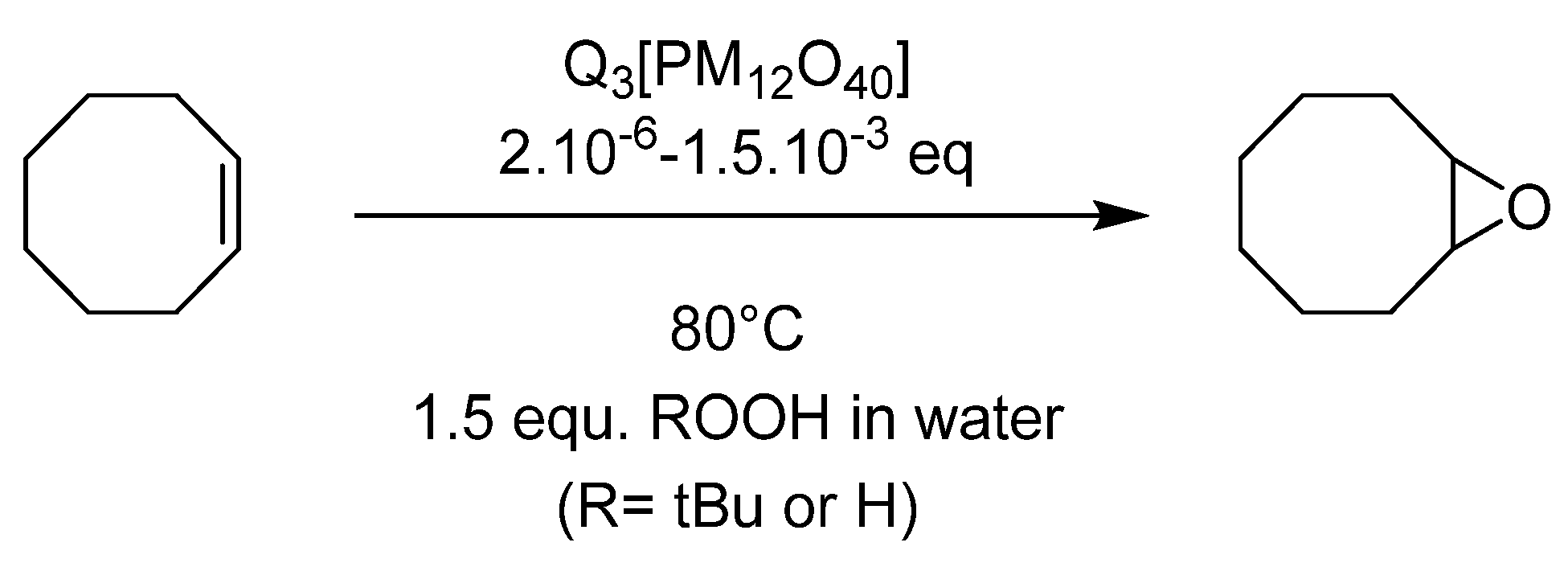{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Comparisons | Substitution | Mo (%) | COConv. (%) | COESel. (%) | TS (kcal/mol) |
|---|---|---|---|---|---|
| ONX Coordination Sphere | X = O | 1.00 | 64 | 93 | 22.5 |
| 0.60 | 66 | 90 | |||
| 0.33 | 65 | 82 | |||
| 0.24 | 63 | 79 | |||
| 0.15 | 59 | 74 | |||
| 0.10 | 50 | 73 | |||
| X = S | 1.00 | 95 | 89 | 22.3 | |
| 0.60 | 94 | 98 | |||
| 0.11 | 86 | 94 | |||
| 0.05 | 82 | 94 | |||
| 0.025 | 68 | 93 | |||
| R2 (NEt2) and/or R5 (NO2) | - | 0.25 | 71 | 94 | 22.5 |
| R5 | 86 | 96 | 21.0 | ||
| R2 | 62 | 93 | 23.8 | ||
| R5 + R2 | 73 | 91 | 22.3 | ||
| OH (R1, R2, R3) | - | 0.50 | 76 | 93 | 22.5 |
| R1 | 92 | 93 | 22.4 | ||
| R2 | 73 | 85 | 22.9 | ||
| R3 | 81 | 91 | 22.6 | ||
| OMe (R1) and/or Me (R4, R5) | - | 0.50 | 68 | 92 | 22.5 |
| R4 | 68 | 91 | n.c. | ||
| R5 | 75 | 91 | n.c. | ||
| R1 | 83 | 94 | n.c. | ||
| R1 + R4 | 74 | 94 | n.c. | ||
| R1 + R5 | 80 | 92 | n.c. |
| General Formulas | R | CO Conv. (%) | COE Sel. (%) | TOF20min | TON |
|---|---|---|---|---|---|
| MoO2L(MeOH) | Ph | 97 | 97 | 3360 | 1940 |
| [MoO2L]n | Ph | 54 | 84 | 645 | 1040 |
| Me | 71 | 92 | 960 | 1420 | |
| H | 69 | 99 | 1080 | 1380 | |
| [MoO2LH(MeOH)]Cl | Ph | 48 | 74 | 480 | 960 |
| {[MoO2LH]Cl}n | Ph | 70 | 82 | 787 | 1960 |
| Me | 78 | 86 | 1080 | 1253 | |
| H | 79 | 89 | 1680 | 1580 |
| General Formulas | X | CO Conv. (%) | COE Sel. (%) | TOF20min | TON |
|---|---|---|---|---|---|
| [MoO2L(MeOH)] | C-H | 41 | 72 | 900 | 200 |
| N | 56 | 86 | 1200 | 1080 | |
| C-OH | 67 | 75 | 900 | 1340 | |
| [MoO2L]n | C-H | 72 | 87 | 484 | 1440 |
| N | 54 | 87 | 818 | 1120 | |
| C-OH | 23 | 73 | 940 | 531 | |
| [MoO2LH]2(Mo6O19). 2 CH3CN | C-H | 72 | 58 | 44 | 99 |
| N | 72 | 53 | 44 | 97 | |
| C-OH | 76 | 55 | 72 | 105 |
| General Structures | Substitution on | Oxidant TBHP in | CO Conv. (%) | COE Sel. (%) | TOF20min | TON | |||
|---|---|---|---|---|---|---|---|---|---|
| X | R1 | R2 | R3 | ||||||
| [MoO2L(EtOH)] | C-OH | OMe | Water (W) | 89 | 53 | 351 | 368 | ||
| OMe | 83 | 50 | 344 | 343 | |||||
| 86 | 46 | 469 | 354 | ||||||
| [WO2L(EtOH) | OMe | 17 | 13 | 190 | 70 | ||||
| OMe | 31 | 9 | 260 | 128 | |||||
| [WO2L]n | - | 16 | 15 | 169 | 68 | ||||
| [WO2L(MeOH)] | N | OMe | W | 17 | 12 | 171 | 70 | ||
| W + ACN | 51 | 2 | |||||||
| Decane (D) | 22 | 12 | |||||||
| OMe | W | 23 | 8 | 111 | 93 | ||||
| W + ACN | 63 | 2 | |||||||
| D | 25 | 19 | |||||||
| OMe | W | 22 | 6 | 168 | 90 | ||||
| W + ACN | 35 | 1 | |||||||
| D | 23 | 13 | |||||||
| [WO2L(EtOH)] | OMe | W | 20 | 3 | |||||
| W + ACN | 66 | 1 | 73 | 63 | |||||
| D | 20 | 31 | |||||||
| OMe | W | 28 | 33 | ||||||
| W + ACN | 78 | 3 | 81 | 117 | |||||
| D | 28 | 7 | |||||||
| OMe | W | 31 | 11 | ||||||
| W + ACN | 47 | 2 | 204 | 129 | |||||
| D | 31 | 19 | |||||||
| General Structures | Substitution on | Oxidant TBHP In | CO Conv. (%) | COE Sel. (%) | TOF20min | TON | |||
|---|---|---|---|---|---|---|---|---|---|
| A | B | R1 | R2 | ||||||
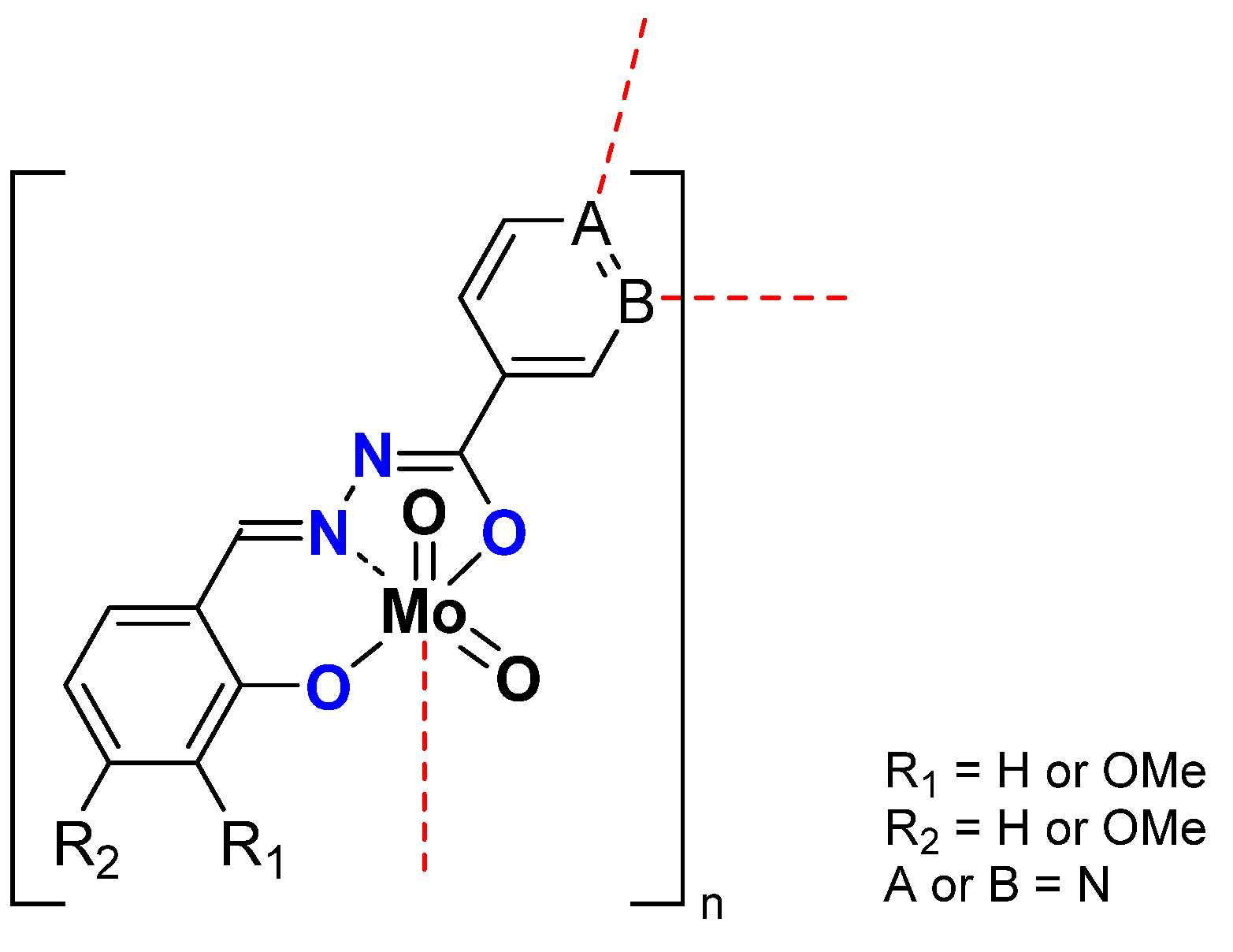
| [MoO2L]n | W | 36 | 45 | 116 | 151 | ||||
| OMe | 27 | 56 | 72 | 113 | |||||
| OMe | 49 | 67 | 119 | 192 | |||||
| [MoO2L]4 | N | OMe | 78 | 85 | 151 | 204 | |||
| [MoO2L]n | D | 66 | 78 | 225 | 273 | ||||
| OMe | 74 | 85 | 200 | 305 | |||||
| OMe | 79 | 87 | 221 | 324 | |||||
| [MoO2L]4 | OMe | 99 | 94 | 1152 | 397 | ||||
| [MoO2L]4 | N | W | 79 | 85 | 243 | 309 | |||
| OMe | 73 | 93 | 175 | 290 | |||||
| OMe | 78 | 85 | 171 | 300 | |||||
| D | >99 | 87 | 153 | 399 | |||||
| OMe | >99 | 92 | 155 | 398 | |||||
| OMe | >99 | 99 | 1152 | 397 | |||||
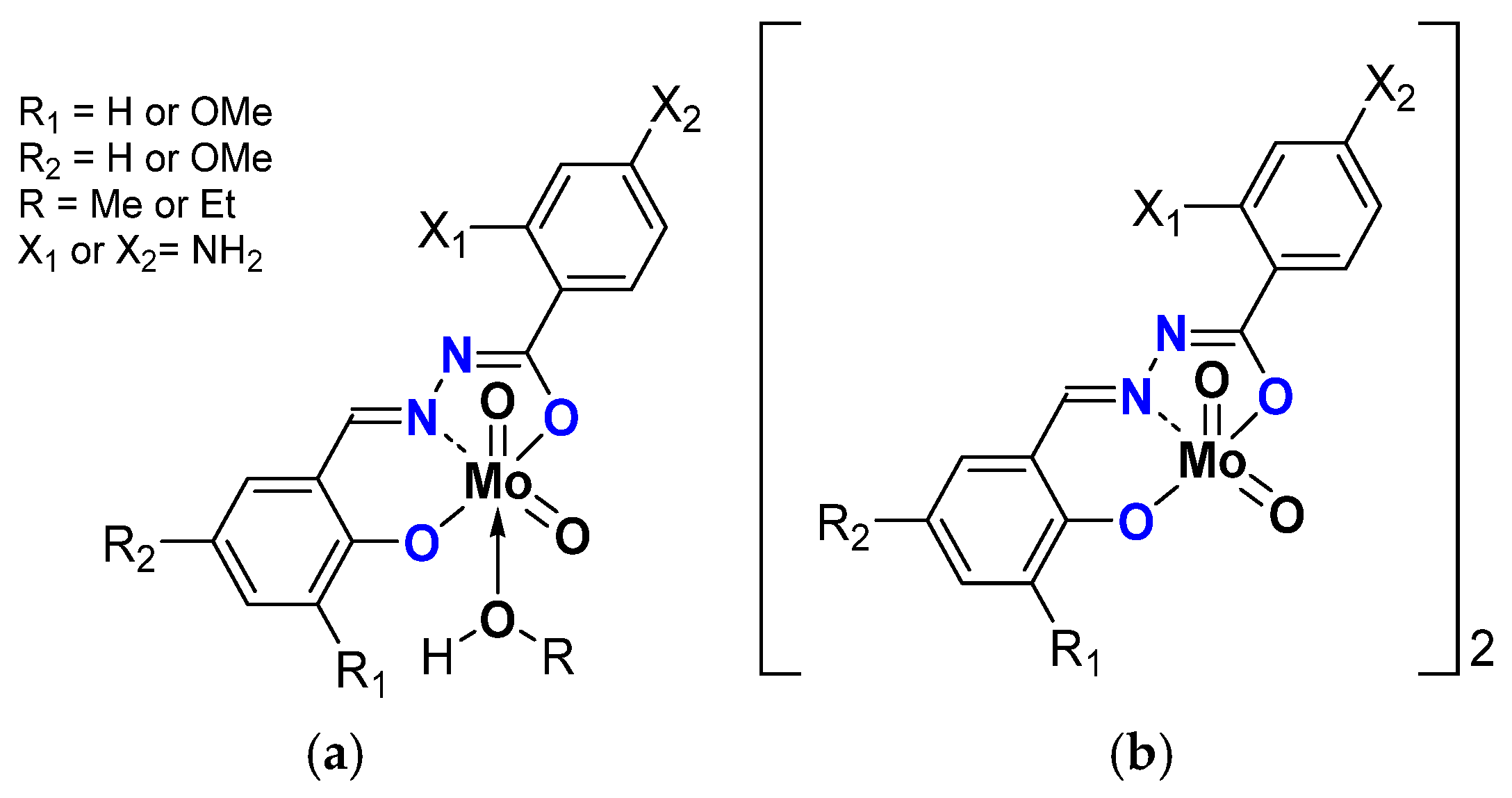
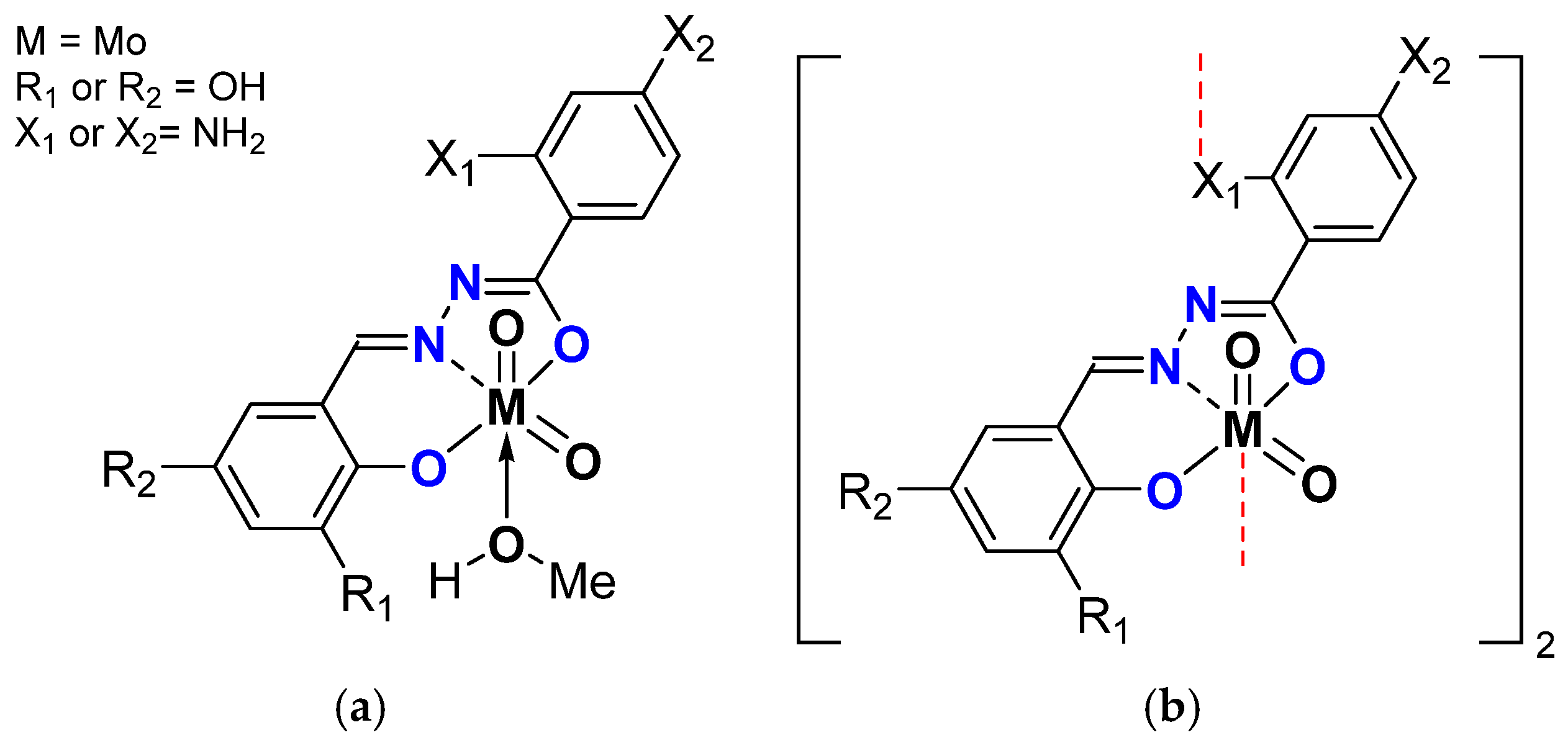
| General Structure | Mo (mol%) | Substitution on | CO Conv. (%) | COE Sel. (%) | TOF20min | TON | |||
|---|---|---|---|---|---|---|---|---|---|
| X1 | X2 | R1 | R2 | ||||||
| [MoO2L(MeOH)] | 0.25 | NH2 | 47 | 72 | 84 | 194 | |||
| OMe | 56 | 86 | 75 | 229 | |||||
| OMe | 63 | 85 | 97 | 257 | |||||
| [MoO2L(EtOH)] | 65 | 82 | 76 | 268 | |||||
| OMe | 59 | 79 | 95 | 241 | |||||
| OMe | 85 | 85 | 58 | 350 | |||||
| [MoO2L]2 | 38 | 78 | 113 | 160 | |||||
| OMe | 84 | 90 | 295 | 380 | |||||
| OMe | 89 | 86 | 298 | 386 | |||||
| [MoO2L(MeOH)] | NH2 | 94 | 93 | 702 | 390 | ||||
| [MoO2L(EtOH)] | 97 | 94 | 642 | 403 | |||||
| [MoO2L]2 | 89 | 93 | 345 | 369 | |||||
| 0.05 | 83 | 88 | 1689 | 1714 | |||||
| 0.01 | 82 | 54 | 921 | 2263 | |||||
| 0.25 | OMe | 90 | 92 | 343 | 372 | ||||
| 0.05 | 75 | 85 | 602 | 1898 | |||||
| [MoO2L(MeOH)] | 0.25 | OMe | 85 | 90 | 383 | 349 | |||
| [MoO2L]2 | 87 | 91 | 345 | 330 | |||||
| 0.05 | 69 | 93 | 954 | 1301 | |||||
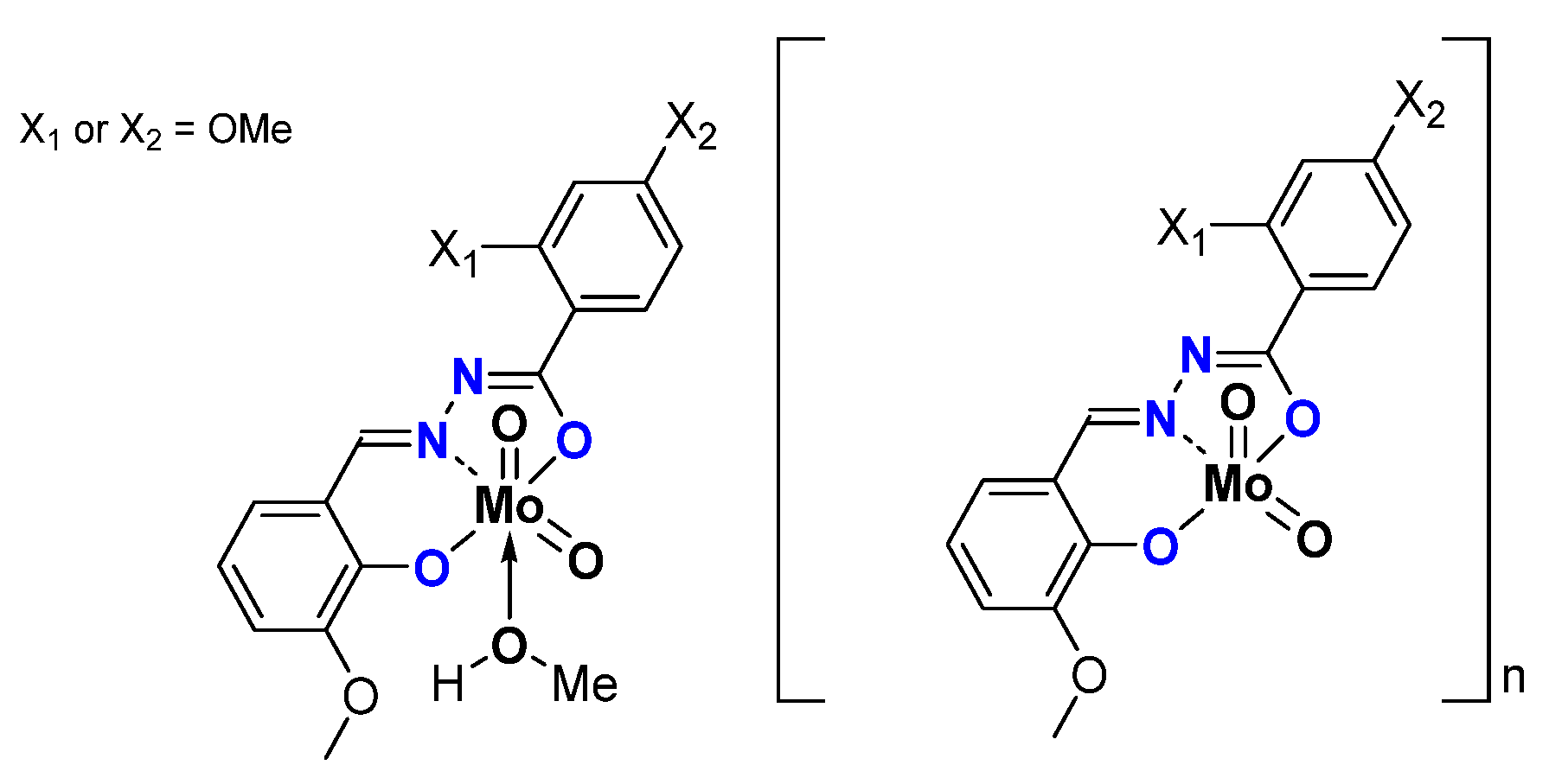
| General Structures | Substitution on | Oxidant TBHP in | CO Conv. (%) | COE Sel. (%) | TOF20min | TON | |
|---|---|---|---|---|---|---|---|
| X1 | X2 | ||||||
| [MoO2L]n | W | 90 | 88 | 290 | 361 | ||
| OMe | 92 | 90 | 274 | 368 | |||
| OMe | 96 | 93 | 391 | 386 | |||
| [MoO2L(MeOH)] | 96 | 95 | 496 | 400 | |||
| OMe | 94 | 92 | 319 | 373 | |||
| OMe | 99 | 85 | 326 | 351 | |||
| [MoO2L]n | D | 99 | 91 | 290 | 397 | ||
| OMe | 98 | 90 | 290 | 397 | |||
| OMe | 99 | 90 | 625 | 399 | |||
| [MoO2L(MeOH)] | 99 | 93 | 796 | 400 | |||
| OMe | 99 | 93 | 298 | 399 | |||
| OMe | 99 | 92 | 214 | 400 | |||
| Substitution on | |||||||||
|---|---|---|---|---|---|---|---|---|---|
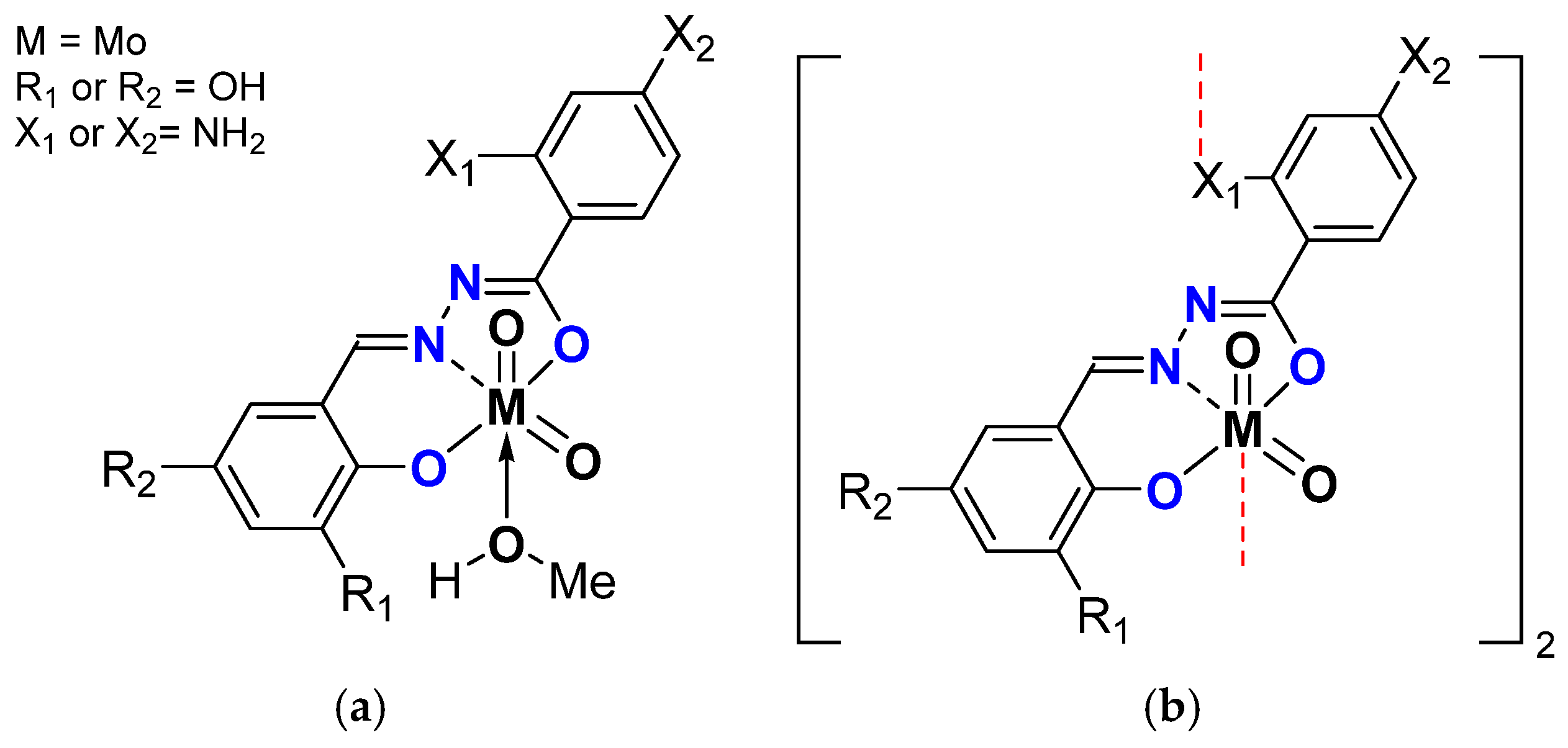
| General Structure | X1 | X2 | R1 | R2 | Oxidant | COconv. (%) | COE Sel. (%) | TOFtmin | TON |
| [MoO2L]2 | NH2 | OH | TBHP in W | 94 | 83 | 370 | 378 | ||
| [MoO2L(MeOH)] | 93 | 90 | 406 | 365 | |||||
| [MoO2L]2.CH3CN | NH2 | 90 | 82 | 346 | 357 | ||||
| [MoO2L(MeOH)] | 91 | 87 | 332 | 362 | |||||
| [MoO2L]2.CH3CN | NH2 | OH | 89 | 89 | 183 | 358 | |||
| [MoO2L]2.CH3CN * | 86 | 96 | 204 | 339 | |||||
| [MoO2L(MeOH)] | 83 | 94 | 179 | 360 | |||||
| [MoO2L]2 | NH2 | 58 | 82 | 36 | 234 | ||||
| [MoO2L(MeOH)] | 50 | 85 | 42 | 270 | |||||
| [MoO2L]2 | NH2 | OH | TBHP in D | >99 | 91 | 1005 | 400 | ||
| [MoO2L(MeOH)] | >99 | 92 | 9415 | 400 | |||||
| [MoO2L]2.CH3CN | NH2 | >99 | 93 | 1197 | 400 | ||||
| [MoO2L(MeOH)] | >99 | 89 | 9556 | 400 | |||||
| [MoO2L]2.CH3CN | NH2 | OH | >99 | 90 | 8119 | 367 | |||
| [MoO2L]2.CH3CN * | >99 | 95 | 2445 | 354 | |||||
| [MoO2L]2 | NH2 | 13 | 57 | 106 | 50 | ||||
| [MoO2L]2 | NH2 | OH | H2O2 in W | 12 | 20 | 13 | 49 | ||
| [MoO2L(MeOH)] | 5 | 52 | 22 | 20 | |||||
| [MoO2L]2.CH3CN | NH2 | 10 | 26 | 7 | 41 | ||||
| [MoO2L(MeOH)] | 7 | 49 | 16 | 27 | |||||
| [MoO2L]2.CH3CN | NH2 | OH | 16 | 23 | 9 | 62 | |||
| [MoO2L]2.CH3CN * | 15 | 16 | 49 | 61 | |||||
| [MoO2L]2 | NH2 | 16 | 14 | 96 | 63 | ||||
| Substitution on | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| General Structure | R1 | R2 | A | B | Oxidant | Conv. (%) | Sel. (%) | TOFtmin | TON |
| [MoO2L]n | OH | N | H2O2 | 88 | 41 | 113 | 308 | ||
| OH | N | 87 | 42 | 294 | 339 | ||||
| OH | N | 77 | 40 | 4 | 300 | ||||
| OH | N | 85 | 44 | 34 | 329 | ||||
| [MoO2L(MeOH)] | OH | N | 87 | 41 | 23 | 360 | |||
| OH | N | 84 | 41 | 286 | 344 | ||||
| OH | N | 89 | 37 | 27 | 367 | ||||
| OH | H | 91 | 42 | 20 | 394 | ||||
| [MoO2L]n | OH | N | TBHP in water | 64 | 10 | 237 | 235 | ||
| OH | N | 66 | 10 | 29 | 270 | ||||
| [MoO2L(MeOH)] | OH | N | 62 | 11 | 29 | 270 | |||
| OH | N | 56 | 19 | 185 | 271 | ||||
| [MoO2L(MeOH)] | OH | N | TBHP in decane | 99 | 4 | 1001 | 289 | ||
| General Formula | R or X | Conv. (%) | Sel. (%) | TOF20min | TON |
|---|---|---|---|---|---|
| [V2O3L2] | R = H | 61 | 35 | 2339 | 1251 |
| [V2O3(LH)2]Cl2 | R = H | 87 | 32 | 2409 | 1804 |
| [V2O3L2] × 2 MeOH | R = Ph | 67 | 32 | 1587 | 1386 |
| [V2O3(LH)2]Cl2 × 2 MeOH | R = Ph | 74 | 36 | 1930 | 1551 |
| [VO2(LH)] × MeOH × H2O | X = C-H | 23 | 10 | 940 | 532 |
| [VO2(LH)] × MeOH × H2O | X = C-OH | 33 | 10 | 1571 | 700 |
| [VO2(LH)] | X = N | 31 | 13 | 1179 | 633 |
| General Formulas | Cat (%) | CO Conv. (%) | COE Sel. (%) | TON | TOF20min |
|---|---|---|---|---|---|
| (BP)3[PMo12O40] | 0.099 | 90.7 | 71.6 | 910 | 470 |
| 0.010 | 85.2 | 72.1 | 8200 | 2500 | |
| 0.005 | 83.5 | 75.7 | 15,000 | 5600 | |
| 0.002 | 79.4 | 75.3 | 40,000 | 11,000 | |
| 0.001 | 65.3 | 65.6 | 53,000 | 16,000 | |
| 0.0005 | 52.0 | 44.5 | 100,000 | 35,000 | |
| (CP)3[PMo12O40] | 0.149 | 77.7 | 78.0 | 520 | 140 |
| 0.080 | 77.0 | 78.6 | 960 | 180 | |
| 0.030 | 82.0 | 79.4 | 2700 | 340 | |
| 0.004 | 66.7 | 67.5 | 15,000 | 4100 | |
| 0.0005 | 51.2 | 47.1 | 100,000 | 31,000 | |
| 0.0002 | 44.4 | 53.1 | 220,000 | 66,000 |
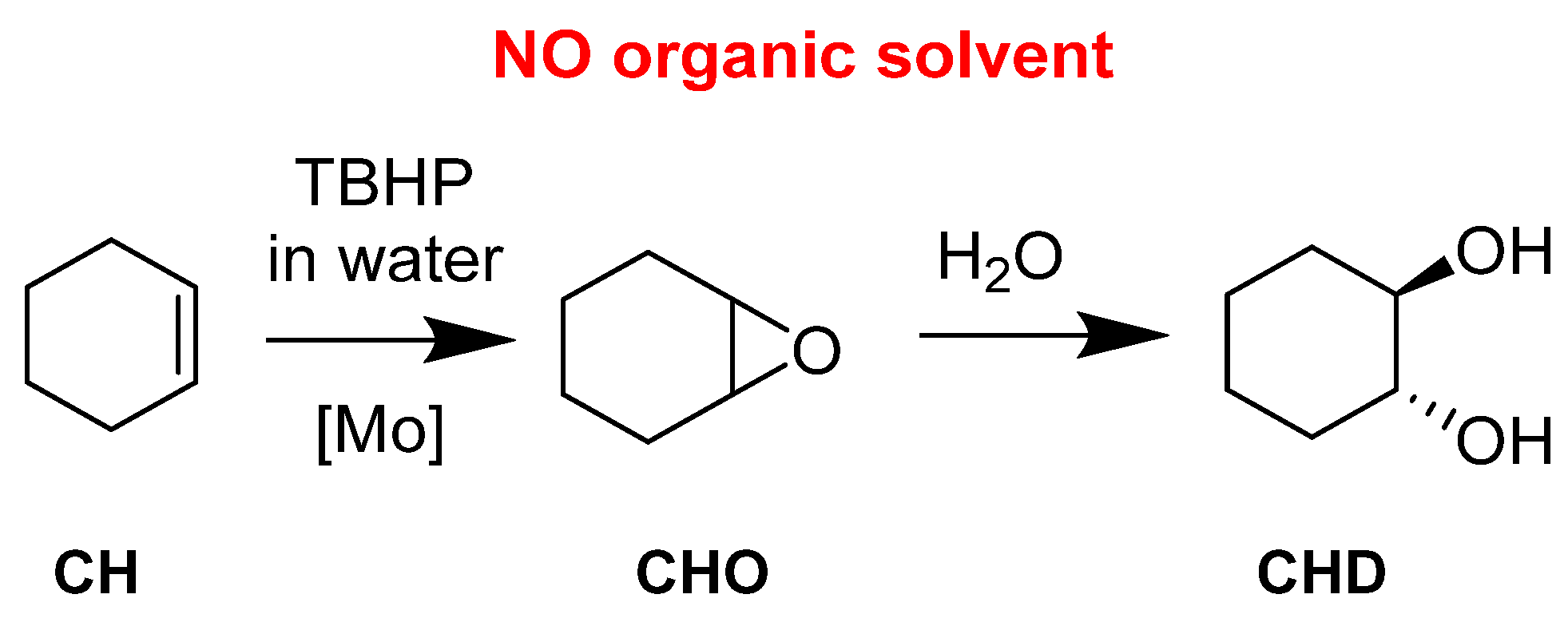
| General Structure | Substitution | Cat | H2O2 | Substrate | S Conv. | AA Yield | |
|---|---|---|---|---|---|---|---|
| M | n | (%) | (equ.) | S | (%) | (%) | |
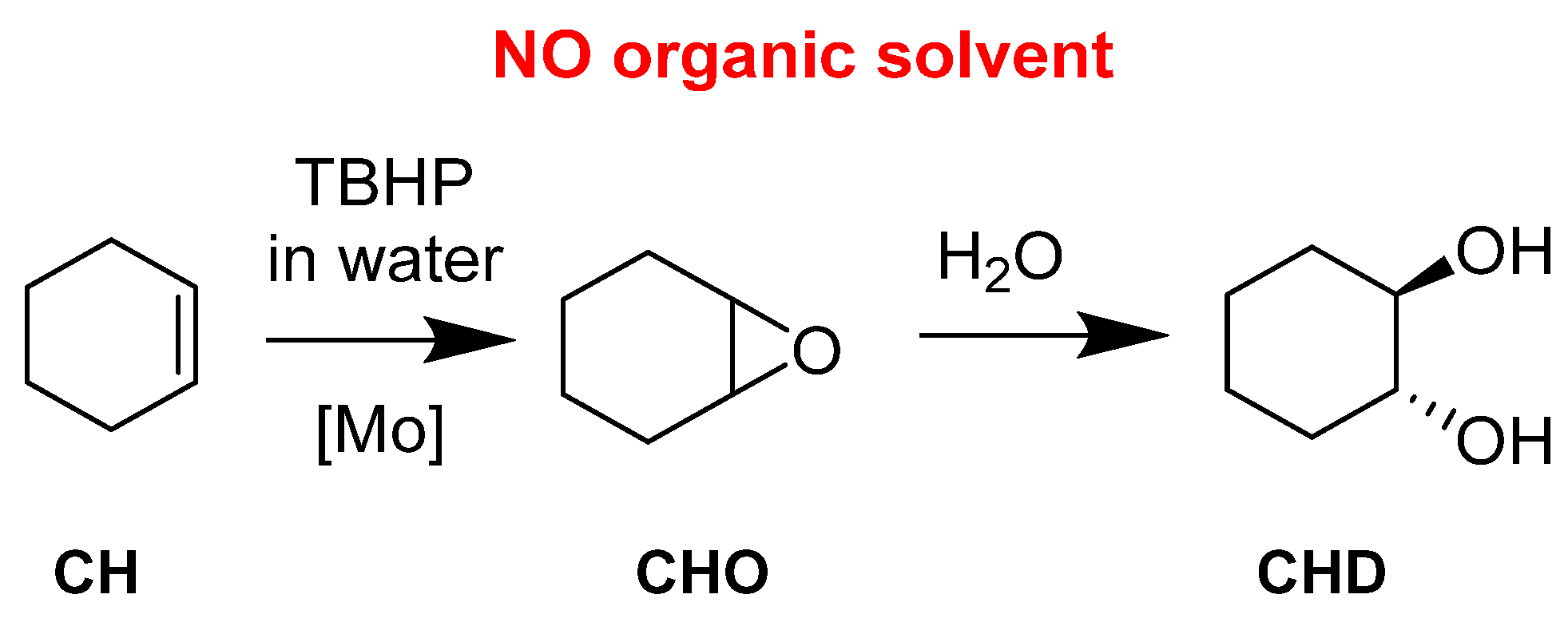
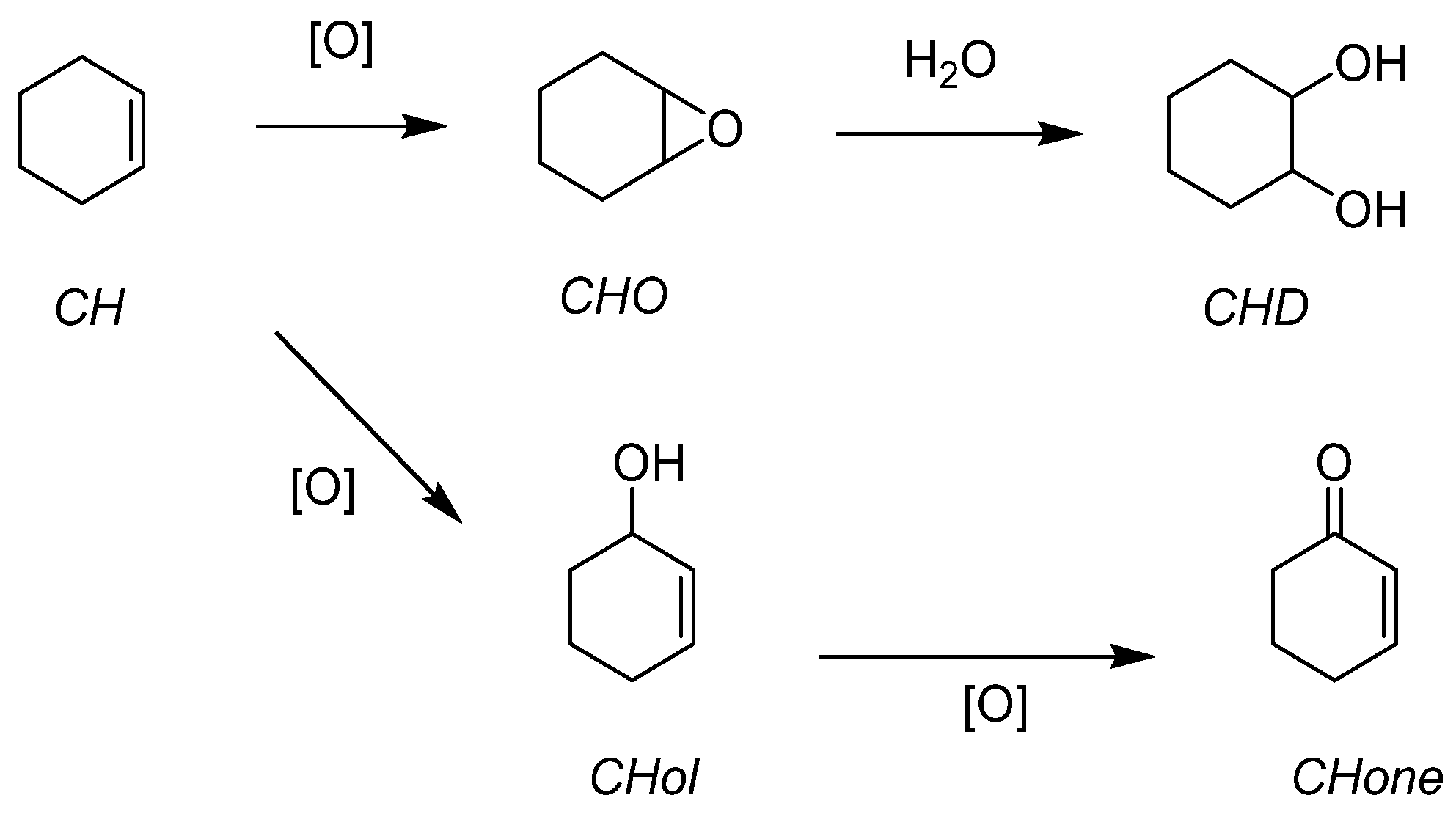
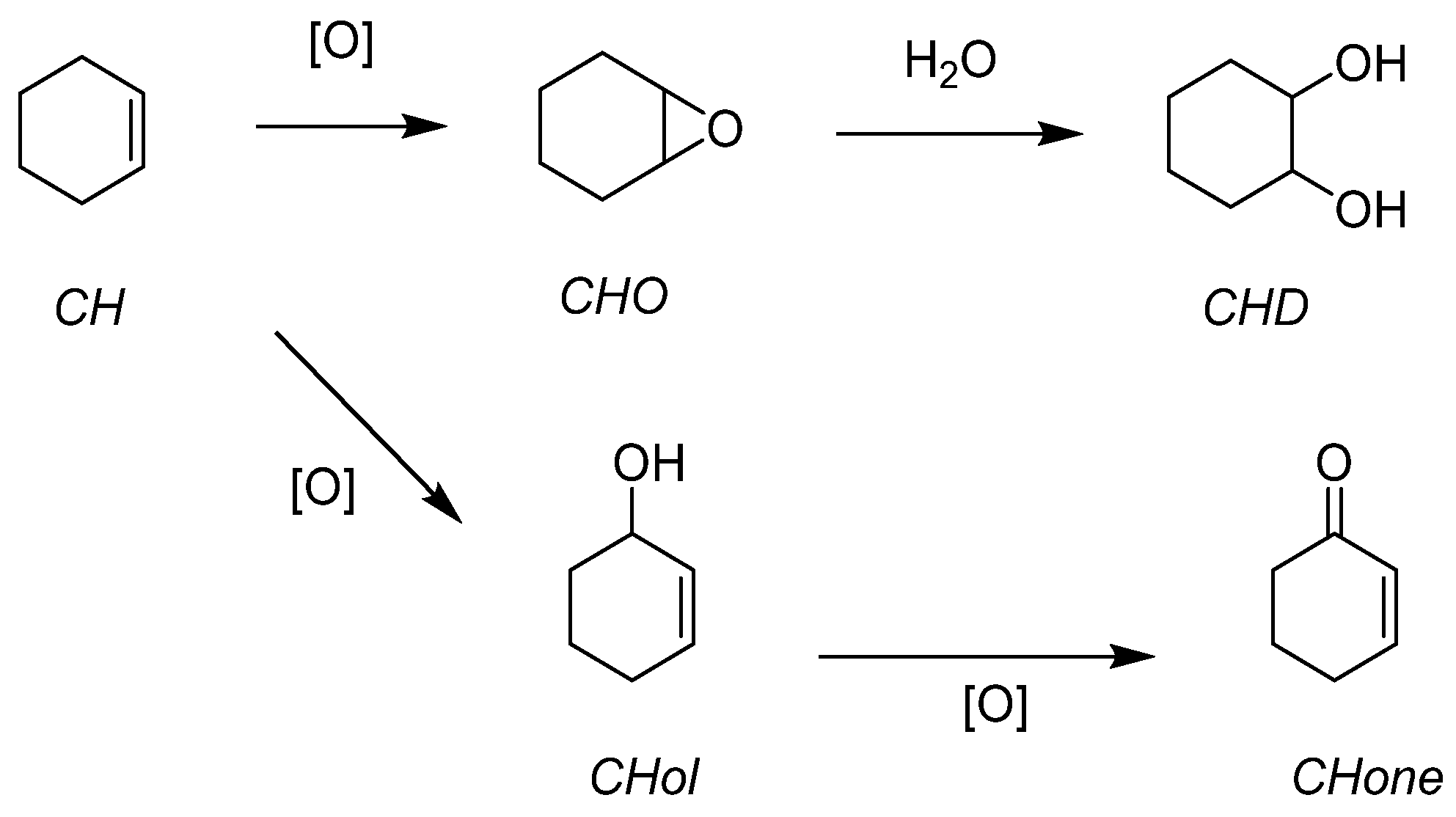
| Molecular | Mo | 4 | 0.025 | 4 | CH | 65 | 46 |
| catalysts | 2 | CHO | >99 | 36 | |||
| 2 | CHD | >99 | 74 | ||||
| W | 4 | 0.025 | 4 | CH | 75 | 61 | |
| 2 | CHO | >99 | 47 | ||||
| 2 | CHD | >99 | 72 | ||||
| Mo | 6 | 0.025 | 4 | CH | 61 | 42 | |
| 2 | CHO | >99 | 32 | ||||
| 2 | CHD | >99 | 58 | ||||
| W | 6 | 0.025 | 4 | CH | 68 | 50 | |
| 2 | CHO | >99 | 43 | ||||
| 2 | CHD | >99 | 63 | ||||
| Grafted | Mo | 4 | 0.004 | 4 | CH | 58 | 33 |
| catalysts | 2 | CHO | >99 | 28 | |||
| 2 | CHD | 71 | 46 | ||||
| W | 4 | 0.007 | 4 | CH | 61 | 43 | |
| 2 | CHO | >99 | 33 | ||||
| 2 | CHD | 94 | 60 | ||||
| Mo | 12 | 0.003 | 4 | CH | 56 | 30 | |
| 2 | CHO | >99 | 31 | ||||
| 2 | CHD | 79 | 41 | ||||
| W | 12 | 0.001 | 4 | CH | 73 | 51 | |
| 2 | CHO | >99 | 36 | ||||
| 2 | CHD | 95 | 56 | ||||
| Catalyst | Run | Cat x | Conv (%) | Sel (%) | TON |
|---|---|---|---|---|---|
| H3PW12O40 | 1 | 0.070 | 64 | 14 | 807 |
| SiO2@PW | 1 | 0.070 | 72 | 41 | 981 |
| 2 | 75 | 38 | 987 | ||
| 3 | 77 | 37 | 968 | ||
| H3PMo12O40 | 1 | 0.058 | 99 | 44 | 1712 |
| SiO2@PMo | 1 | 0.058 | 98 | 71 | 1693 |
| 2 | 96 | 72 | 1620 | ||
| 3 | 93 | 69 | 1598 |
| Catalyst | Run | Cat x | Conv | Selectivity (%) | TON | |||
|---|---|---|---|---|---|---|---|---|
| CH | CHO | CHD | CHol | CHone | ||||
| H3PW12O40 | 1 | 0.014 | 31 | <1 | 4 | 3 | 3 | 11,307 |
| SiO2@PW | 1 | 0.014 | 45 | 1 | 2 | 4 | 5 | 21,458 |
| 2 | 43 | 1 | 2 | 2 | 3 | 20,649 | ||
| 3 | 26 | 3 | 3 | 7 | 7 | 12,373 | ||
| H3PMo12O40 | 1 | 0.012 | 91 | <1 | 40 | 3 | 2 | 52,728 |
| SiO2@PMo | 1 | 0.012 | 80 | 13 | 26 | 5 | 2 | 46,732 |
| 2 | 74 | 15 | 22 | 6 | 3 | 42,487 | ||
| 3 | 60 | 26 | 20 | 5 | 2 | 36,345 | ||
| Conv | Selectivity (%) | TON | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Run | Cat x | Lim | LimO | LimD | Col | Cone | |||
| cis | trans | Ax | eq | |||||||
| H3PW12O40 | 1 | 0.070 | 67 | 0 | 0 | 5 | 3 | 1 | 4 | 1287 |
| SiO2@PW | 1 | 0.070 | 58 | 0 | 3 | 13 | 1 | 8 | 8 | 754 |
| 2 | 59 | 0 | 3 | 13 | 1 | 10 | 8 | 768 | ||
| 3 | 62 | 0 | 2 | 12 | 2 | 11 | 8 | 754 | ||
| H3PMo12O40 | 1 | 0.058 | 99 | 0 | 0 | 18 | 10 | 1 | 2 | 1859 |
| SiO2@PMo | 1 | 0.058 | 91 | 0 | 0 | 36 | 11 | 4 | 3 | 1721 |
| 2 | 86 | 0 | 0 | 32 | 8 | 6 | 7 | 1626 | ||
| 3 | 81 | 0 | <1 | 20 | 5 | 6 | 6 | 1526 | ||
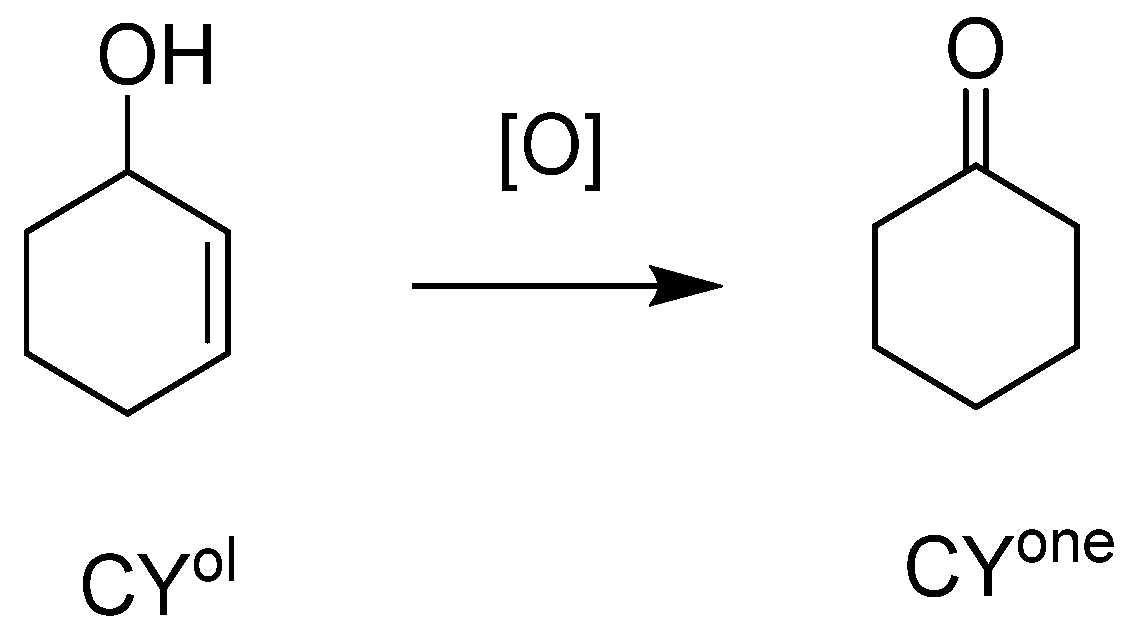
| Catalyst | Run | Cat x | Conv | Sel | TON |
|---|---|---|---|---|---|
| CYol | CYone | ||||
| H3PW12O40 | 1 | 0.070 | 44 | 34 | 525 |
| SiO2@PW | 1 | 0.070 | 11 | 51 | 137 |
| 2 | 8 | 97 | 101 | ||
| 3 | 7 | 87 | 92 | ||
| H3PMo12O40 | 1 | 0.058 | 58 | 54 | 728 |
| SiO2@PMo | 1 | 0.058 | 18 | 76 | 228 |
| 2 | 17 | 90 | 207 | ||
| 3 | 20 | 75 | 249 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisk, J.; Agustin, D. Molybdenum, Vanadium, and Tungsten-Based Catalysts for Sustainable (ep)Oxidation. Molecules 2022, 27, 6011. https://doi.org/10.3390/molecules27186011
Pisk J, Agustin D. Molybdenum, Vanadium, and Tungsten-Based Catalysts for Sustainable (ep)Oxidation. Molecules. 2022; 27(18):6011. https://doi.org/10.3390/molecules27186011
Chicago/Turabian StylePisk, Jana, and Dominique Agustin. 2022. "Molybdenum, Vanadium, and Tungsten-Based Catalysts for Sustainable (ep)Oxidation" Molecules 27, no. 18: 6011. https://doi.org/10.3390/molecules27186011
APA StylePisk, J., & Agustin, D. (2022). Molybdenum, Vanadium, and Tungsten-Based Catalysts for Sustainable (ep)Oxidation. Molecules, 27(18), 6011. https://doi.org/10.3390/molecules27186011