


Formate Dehydrogenase Mimics as Catalysts for Carbon Dioxide Reduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
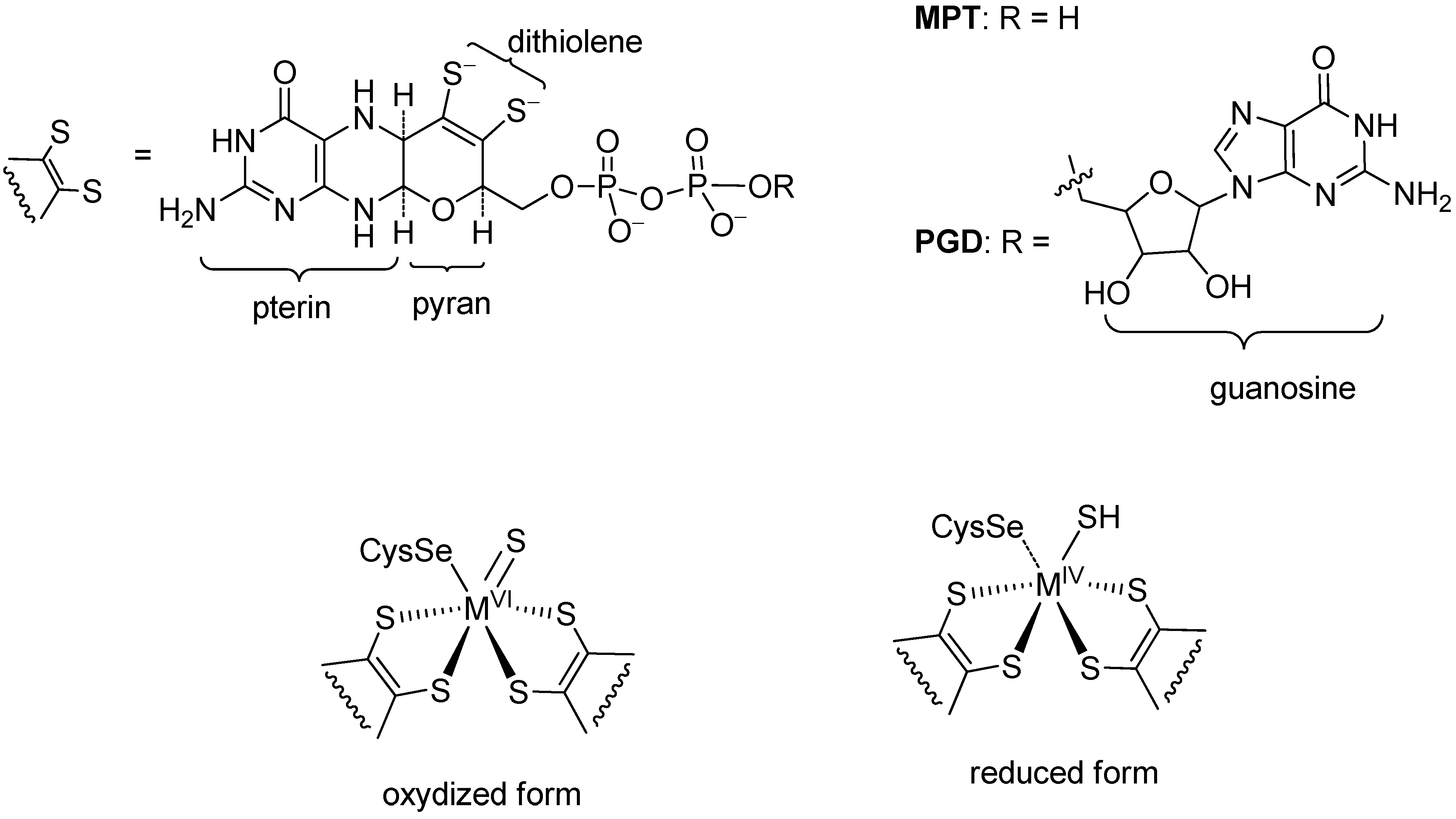
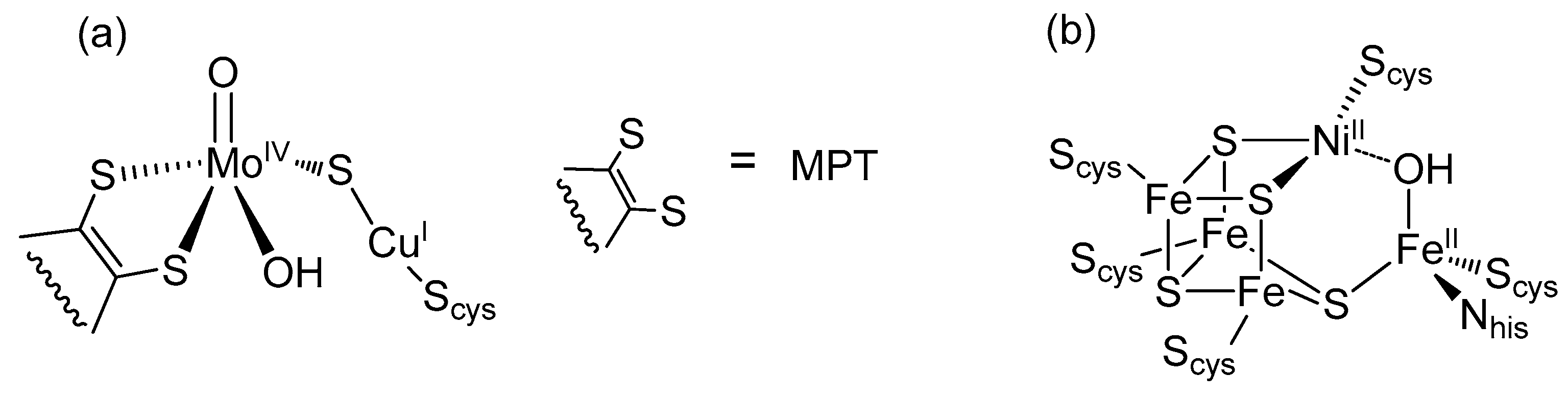
2. Formate Dehydrogenases (FDHs)
 (1)
(1)3. Structural Models of FDH
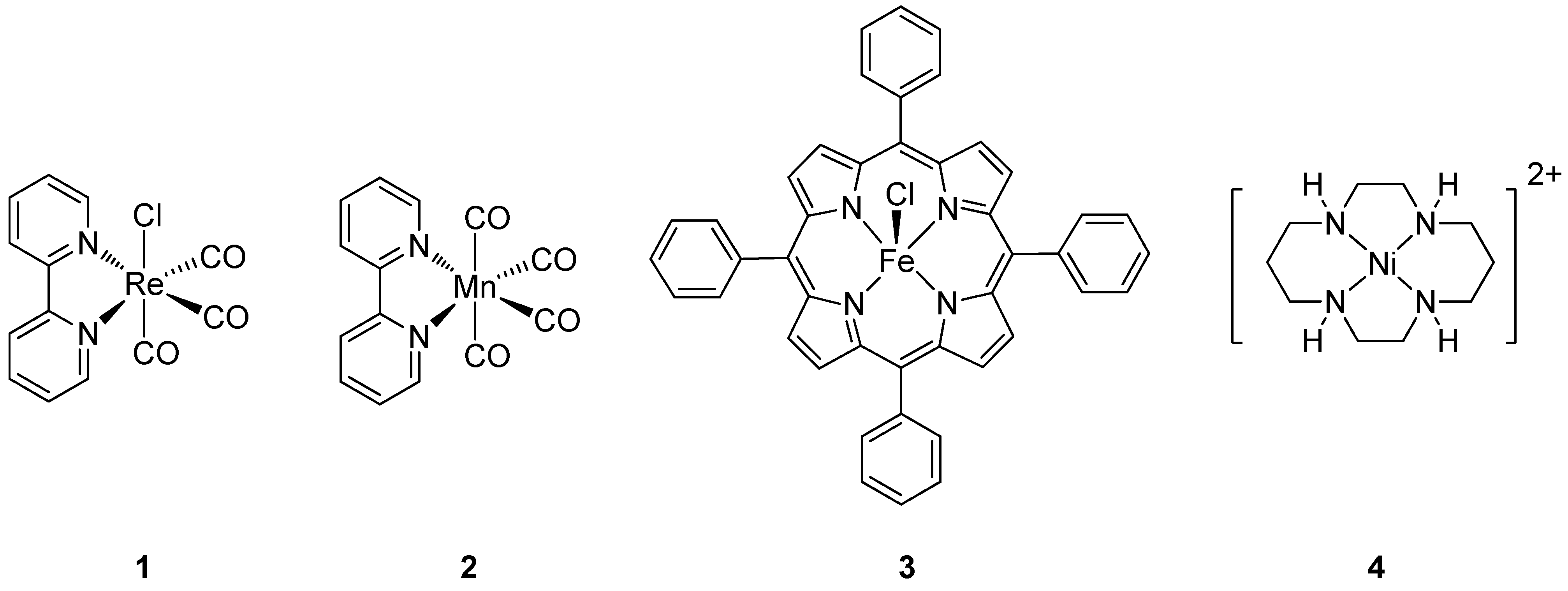
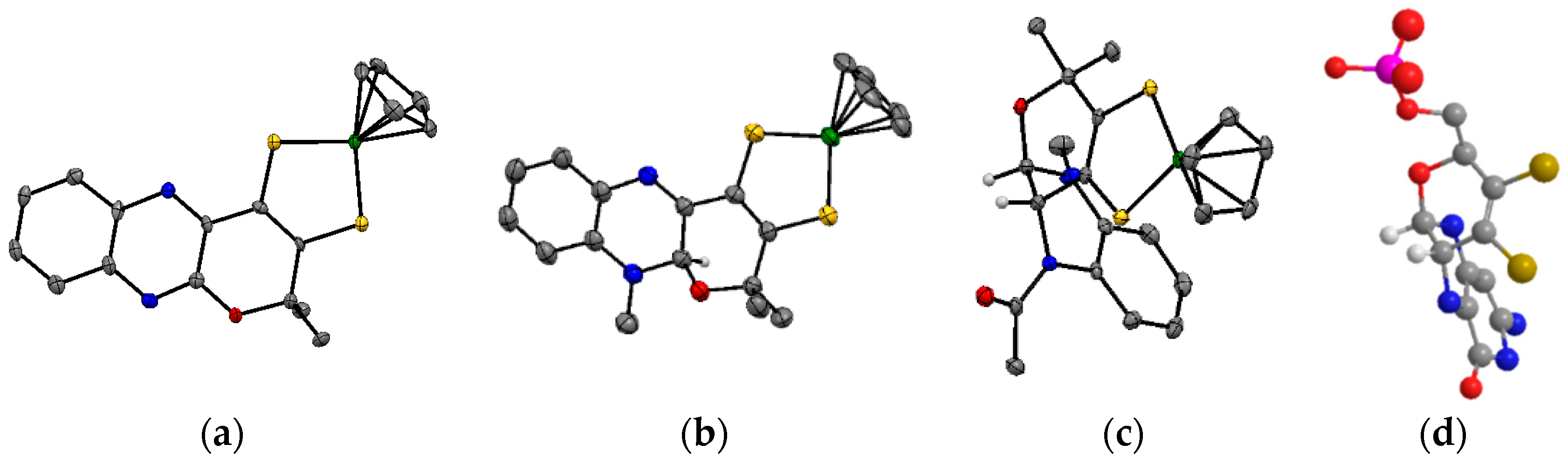
3.1. Variation of the Axial Ligands
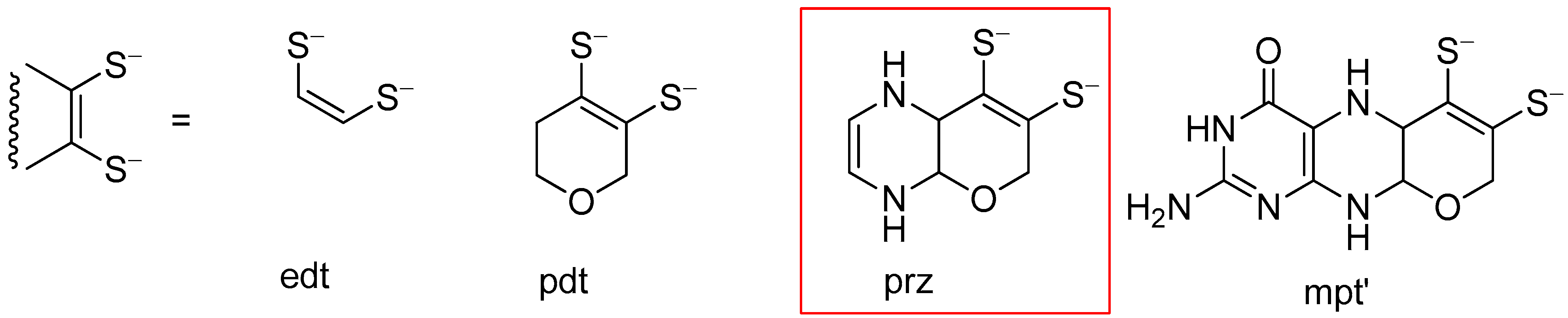
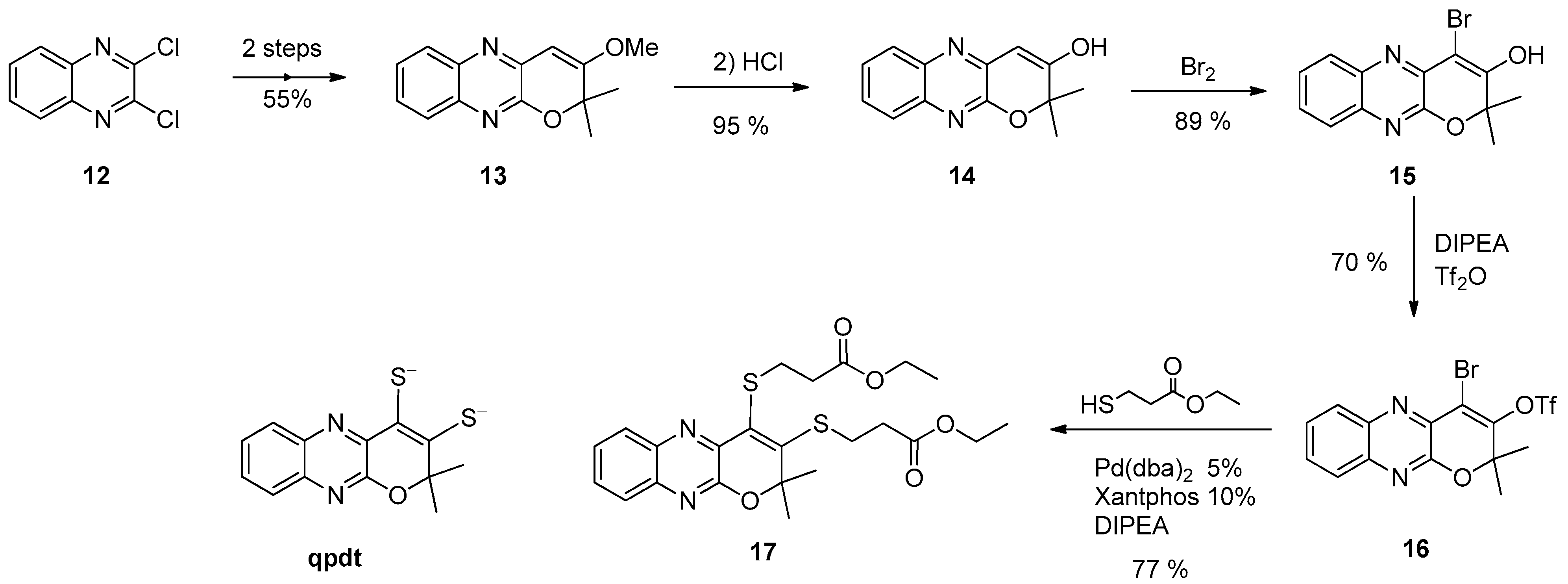
3.2. Pyrazine Containing Ligands
4. Functional Models
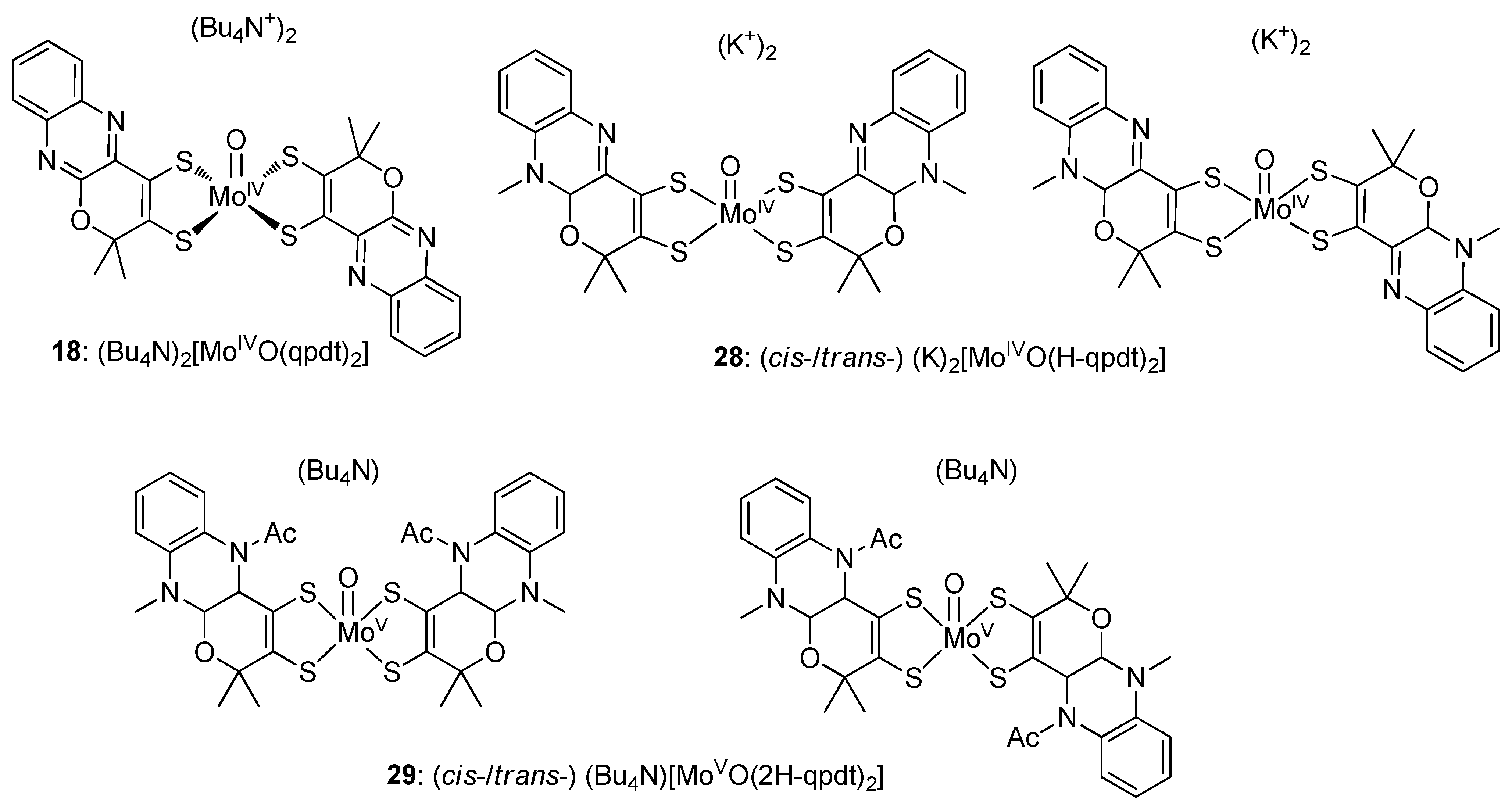
4.1. Mo/W-(Dithiolene)2 Complexes
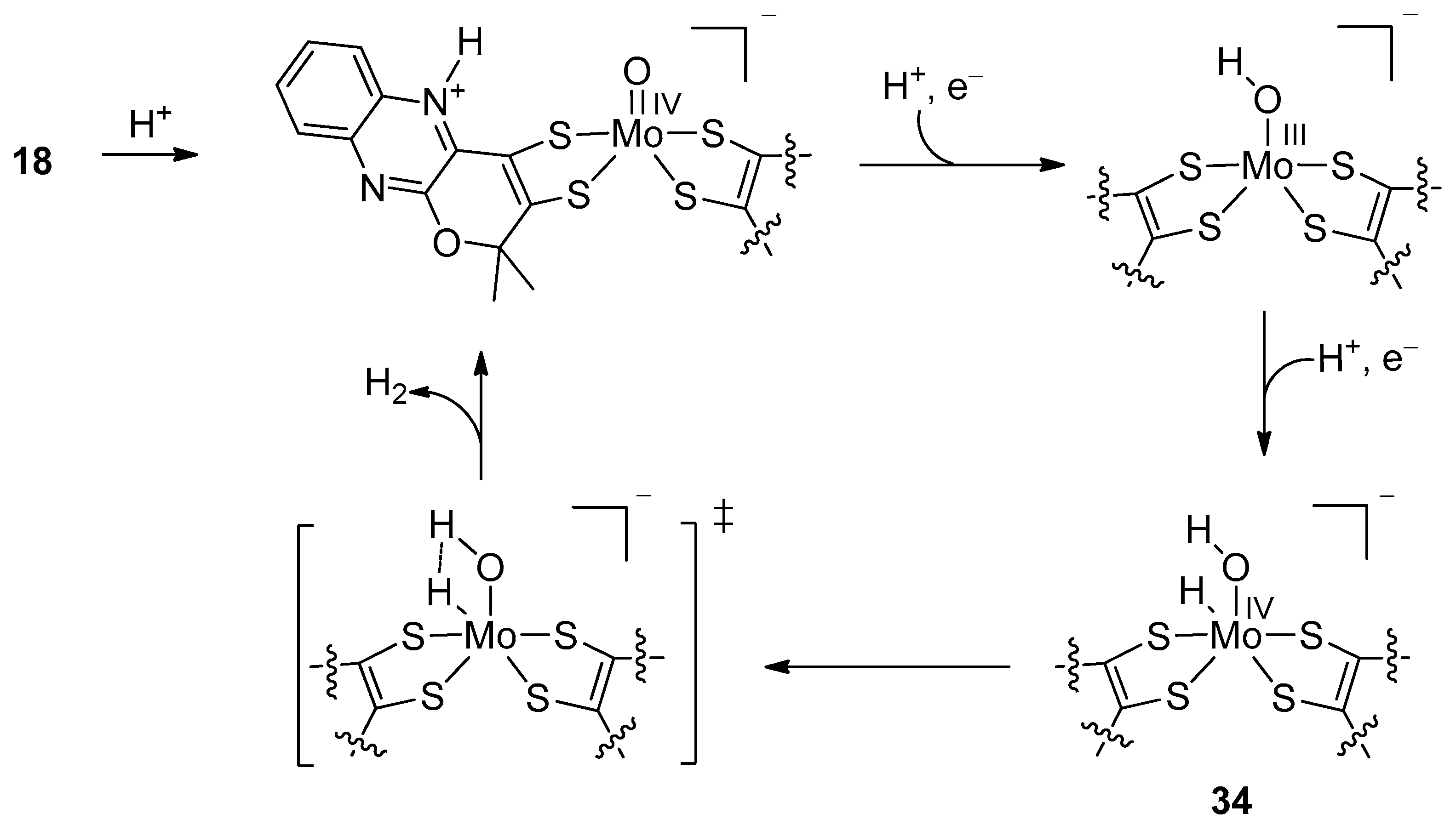
4.1.1. Equimolar Reaction with CO2
4.1.2. Catalytic Reduction of CO2
4.2. Mo/W-Cu Complexes, Models of CO-Dehydrogenases
4.3. Other Mo/W Complexes
4.4. Ni(dithiolene)2 Complexes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tan, X.Y.; Yu, C.; Ren, Y.W.; Cui, S.; Li, W.B.; Qiu, J.S. Recent advances in innovative strategies for the CO2 electroreduction reaction. Energy Environ. Sci. 2021, 14, 765–780. [Google Scholar] [CrossRef]
- Grim, R.G.; Huang, Z.; Guarnieri, M.T.; Ferrell, J.R.; Tao, L.; Schaidle, J.A. Transforming the carbon economy: Challenges and opportunities in the convergence of low-cost electricity and reductive CO2 utilization. Energy Environ. Sci. 2020, 13, 472–494. [Google Scholar] [CrossRef]
- Kinzel, N.W.; Werle, C.; Leitner, W. Transition Metal Complexes as Catalysts for the Electroconversion of CO2: An Organometallic Perspective. Angew. Chem. Int. Ed. 2021, 60, 11628–11686. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction. ACS Catal. 2017, 7, 70–88. [Google Scholar] [CrossRef]
- Romao, M.J. Molybdenum and tungsten enzymes: A crystallographic and mechanistic overview. Dalton Trans. 2009, 21, 4053–4068. [Google Scholar] [CrossRef]
- Niks, D.; Hille, R. Molybdenum- and tungsten-containing formate dehydrogenases and formylmethanofuran dehydrogenases: Structure, mechanism, and cofactor insertion. Protein Sci. 2019, 28, 111–122. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, I.; Moura, J.J.G. Molybdenum and tungsten-containing formate dehydrogenases: Aiming to inspire a catalyst for carbon dioxide utilization. Inorg. Chim. Acta 2017, 455, 350–363. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, J.J.G.; Moura, I. Molybdenum and tungsten-dependent formate dehydrogenases. J. Biol. Inorg. Chem. 2015, 20, 287–309. [Google Scholar] [CrossRef]
- Boyington, J.C.; Gladyshev, V.N.; Khangulov, S.V.; Stadtman, T.C.; Sun, P.D. Crystal structure of formate dehydrogenase H: Catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science 1997, 275, 1305–1308. [Google Scholar] [CrossRef]
- Jormakka, M.; Richardson, D.; Byrne, B.; Iwata, S. Architecture of NarGH reveals a structural classification of Mo-bisMGD enzymes. Structure 2004, 12, 95–104. [Google Scholar] [CrossRef]
- Raaijmakers, H.; Macieira, S.; Dias, J.M.; Teixeira, S.; Bursakov, S.; Huber, R.; Moura, J.J.G.; Moura, I.; Romao, M.J. Gene sequence and the 1.8 angstrom crystal structure of the tungsten-containing formate dehydrogenase from Desulfolvibrio gigas. Structure 2002, 10, 1261–1272. [Google Scholar] [CrossRef]
- Raaijmakers, H.C.A.; Romao, M.J. Formate-reduced E-coli formate dehydrogenase H: The reinterpretation of the crystal structure suggests a new reaction mechanism. J. Biol. Inorg. Chem. 2006, 11, 849–854. [Google Scholar] [CrossRef]
- Maia, L.B.; Fonseca, L.; Moura, I.; Moura, J.J.G. Reduction of Carbon Dioxide by a Molybdenum-Containing Formate Dehydrogenase: A Kinetic and Mechanistic Study. J. Am. Chem. Soc. 2016, 138, 8834–8846. [Google Scholar] [CrossRef] [PubMed]
- Enemark, J.H.; Cooney, J.J.A.; Wang, J.-J.; Holm, R.H. Synthetic Analogues and Reaction Systems Relevant to the Molybdenum and Tungsten Oxotransferases. Chem. Rev. 2004, 104, 1175–1200. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Burgmayer, S.J.N. Recent developments in the study of molybdoenzyme models. J. Biol. Inorg. Chem. 2015, 20, 373–383. [Google Scholar] [CrossRef]
- Patsch, S.; Correia, J.V.; Elvers, B.J.; Steuer, M.; Schulzke, C. Inspired by Nature-Functional Analogues of Molybdenum and Tungsten-Dependent Oxidoreductases. Molecules 2022, 27, 3695. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Tsukube, H. Chemical analogues relevant to molybdenum and tungsten enzyme reaction centres toward structural dynamics and reaction diversity. Chem. Soc. Rev. 2008, 37, 2609–2619. [Google Scholar] [CrossRef]
- Majumdar, A.; Sarkar, S. Bioinorganic chemistry of molybdenum and tungsten enzymes: A structural–functional modeling approach. Coord. Chem. Rev. 2011, 255, 1039–1054. [Google Scholar] [CrossRef]
- Sung, K.-M.; Holm, R.H. Oxo Transfer Reactions Mediated by Bis(dithiolene)tungsten Analogues of the Active Sites of Molybdoenzymes in the DMSO Reductase Family: Comparative Reactivity of Tungsten and Molybdenum. J. Am. Chem. Soc. 2001, 123, 1931–1943. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.-M.; Holm, R.H. Synthesis and Structures of Bis(dithiolene)−Tungsten(IV) Complexes Related to the Active Sites of Tungstoenzymes. Inorg. Chem. 2000, 39, 1275–1281. [Google Scholar] [CrossRef]
- Lim, B.S.; Holm, R.H. Bis(dithiolene)molybdenum analogues relevant to the DMSO reductase enzyme family: Synthesis, structures, and oxygen atom transfer reactions and kinetics. J. Am. Chem. Soc. 2001, 123, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.S.; Donahue, J.P.; Holm, R.H. Synthesis and Structures of Bis(dithiolene)molybdenum Complexes Related to the Active Sites of the DMSO Reductase Enzyme Family. Inorg. Chem. 2000, 39, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Goddard, C.A.; Holm, R.H. Synthesis and Reactivity Aspects of the Bis(dithiolene) Chalcogenide Series [WIVQ(S2C2R2)2]2− (Q = O, S, Se). Inorg. Chem. 1999, 38, 5389–5398. [Google Scholar] [CrossRef]
- Elvers, B.J.; Schulzke, C.; Fischer, C. Photochemical Unmasking of 1,3-Dithiol-2-ones: An Alternative Route to Heteroleptic Dithiolene Complexes from Low-Valent Molybdenum and Tungsten Precursors. Eur. J. Inorg. Chem. 2019, 2019, 2796–2805. [Google Scholar] [CrossRef]
- Ryde, U.; Schulzke, C.; Starke, K. Which functional groups of the molybdopterin ligand should be considered when modeling the active sites of the molybdenum and tungsten cofactors? A density functional theory study. J. Biol. Inorg. Chem. 2009, 14, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Alphonse, F.A.; Karim, R.; Cano-Soumillac, C.; Hebray, M.; Collison, D.; Garner, C.D.; Joule, J.A. A bis(eta(5)-cyclopentadienyl)cobalt complex of a bis-dithiolene: A chemical analogue of the metal centres of the DMSO reductase family of molybdenum and tungsten enzymes, in particular ferredoxin aldehyde oxidoreductase. Tetrahedron 2005, 61, 11010–11019. [Google Scholar] [CrossRef]
- Bradshaw, B.; Collison, D.; Garner, C.D.; Joule, J.A. Stable pyrano[2,3-b]quinoxalines and pyrano[2,3-g]pteridines related to molybdopterin. Chem. Commun. 2001, 123–124. [Google Scholar] [CrossRef]
- Bradshaw, B.; Dinsmore, A.; Ajana, W.; Collison, D.; Garner, C.D.; Joule, J.A. Synthesis of the organic ligand of the molybdenum cofactor, in protected form. J. Chem. Soc. Perkin Trans. 1. 2001, 3239–3244. [Google Scholar]
- Bradshaw, B.; Dinsmore, A.; Collison, D.; Garner, C.D.; Joule, J.A. The synthesis of pyrano[2,3-b]quinoxalines related to molybdopterin. J. Chem. Soc. Perkin Trans. 1. 2001, 3232–3238. [Google Scholar]
- Bradshaw, B.; Dinsmore, A.; Garner, C.D.; Joule, J.A. Synthesis of a cobalt complex of a pyrano[2,3-b]quinoxaline-3,4-dithiolate related to molybdopterin. Chem. Commun. 1998, 417–418. [Google Scholar] [CrossRef]
- Bertero, M.G.; Rothery, R.A.; Palak, M.; Hou, C.; Lim, D.; Blasco, F.; Weiner, J.H.; Strynadka, N.C.J. Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat. Struct. Biol. 2003, 10, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Kloer, D.P.; Hagel, C.; Heider, J.; Schulz, G.E. Crystal structure of ethylbenzene dehydrogenase from Aromatoleum aromaticum. Structure 2006, 14, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Burgmayer, S.J.N. Pterin chemistry and its relationship to the molybdenum cofactor. Coord. Chem. Rev. 2011, 255, 1016–1038. [Google Scholar] [CrossRef]
- Marbella, L.; Serli-Mitasev, B.; Basu, P. Development of a Fluorescent Pb2+ Sensor. Angew. Chem. Int. Edit. 2009, 48, 3996–3998. [Google Scholar] [CrossRef] [PubMed]
- Pimkov, I.V.; Serli-Mitasev, B.; Peterson, A.A.; Ratvasky, S.C.; Hammann, B.; Basu, P. Designing the Molybdopterin Core through Regioselective Coupling of Building Blocks. Chem. Eur. J. 2015, 21, 17057–17072. [Google Scholar] [CrossRef]
- Williams, B.R.; Gisewhite, D.; Kalinsky, A.; Esmail, A.; Burgmayer, S.J.N. Solvent-Dependent Pyranopterin Cyclization in Molybdenum Cofactor Model Complexes. Inorg. Chem. 2015, 54, 8214–8222. [Google Scholar] [CrossRef]
- Williams, B.R.; Fu, Y.; Yap, G.P.A.; Burgmayer, S.J.N. Structure and Reversible Pyran Formation in Molybdenum Pyranopterin Dithiolene Models of the Molybdenum Cofactor. J. Am. Chem. Soc. 2012, 134, 19584–19587. [Google Scholar] [CrossRef][Green Version]
- Gisewhite, D.R.; Nagelski, A.L.; Cummins, D.C.; Yap, G.P.A.; Burgmayer, S.J.N. Modeling Pyran Formation in the Molybdenum Cofactor: Protonation of Quinoxalyl–Dithiolene Promoting Pyran Cyclization. Inorg. Chem. 2019, 58, 5134–5144. [Google Scholar] [CrossRef]
- Porcher, J.P.; Fogeron, T.; Gomez-Mingot, M.; Chamoreau, L.M.; Li, Y.; Fontecave, M. Synthesis and Reactivity of a Bio-inspired Dithiolene Ligand and its Mo Oxo Complex. Chem. Eur. J. 2016, 22, 4447–4453. [Google Scholar] [CrossRef]
- Porcher, J.P.; Fogeron, T.; Gomez-Mingot, M.; Derat, E.; Chamoreau, L.M.; Li, Y.; Fontecave, M. A Bioinspired Molybdenum Complex as a Catalyst for the Photo- and Electroreduction of Protons. Angew. Chem. Int. Ed. 2015, 54, 14090–14093. [Google Scholar] [CrossRef]
- Ames, D.E.; Mitchell, J.C.; Takundwa, C.C. Preparation and Reactions of 2-Alkynyl-3-chloroquinoxaline. J. Chem. Res. Miniprint 1985, 1683–1696. [Google Scholar]
- Fogeron, T.; Retailleau, P.; Chamoreau, L.M.; Fontecave, M.; Li, Y. The unusual ring scission of a quinoxaline-pyran-fused dithiolene system related to molybdopterin. Dalton Trans. 2017, 46, 4161–4164. [Google Scholar] [CrossRef] [PubMed]
- Fogeron, T.; Retailleau, P.; Chamoreau, L.M.; Li, Y.; Fontecave, M. Pyranopterin Related Dithiolene Molybdenum Complexes as Homogeneous Catalysts for CO2 Photoreduction. Angew. Chem. Int. Ed. 2018, 57, 17033–17037. [Google Scholar] [CrossRef] [PubMed]
- McNamara, W.R.; Han, Z.; Alperin, P.J.; Brennessel, W.W.; Holland, P.L.; Eisenberg, R. A Cobalt–Dithiolene Complex for the Photocatalytic and Electrocatalytic Reduction of Protons. J. Am. Chem. Soc. 2011, 133, 15368–15371. [Google Scholar] [CrossRef]
- McNamara, W.R.; Han, Z.; Yin, C.-J.M.; Brennessel, W.W.; Holland, P.L.; Eisenberg, R. Cobalt-dithiolene complexes for the photocatalytic and electrocatalytic reduction of protons in aqueous solutions. Proc. Natl. Acad. Sci. USA 2012, 109, 15594–15599. [Google Scholar] [CrossRef]
- Eckenhoff, W.T.; Brennessel, W.W.; Eisenberg, R. Light-Driven Hydrogen Production from Aqueous Protons using Molybdenum Catalysts. Inorg. Chem. 2014, 53, 9860–9869. [Google Scholar] [CrossRef]
- Das, A.; Han, Z.; Brennessel, W.W.; Holland, P.L.; Eisenberg, R. Nickel Complexes for Robust Light-Driven and Electrocatalytic Hydrogen Production from Water. ACS Catal. 2015, 5, 1397–1406. [Google Scholar] [CrossRef]
- Gomez-Mingot, M.; Porcher, J.P.; Todorova, T.K.; Fogeron, T.; Mellot-Draznieks, C.; Li, Y.; Fontecave, M. Bioinspired Tungsten Dithiolene Catalysts for Hydrogen Evolution: A Combined Electrochemical, Photochemical, and Computational Study. J. Phys. Chem. B 2015, 119, 13524–13533. [Google Scholar] [CrossRef]
- Koutsouri, E.; Mitsopoulou, C.A. Photocatalytic Hydrogen Evolution by tris-dithiolene tungsten complexes. Open Chem. 2016, 14, 393–403. [Google Scholar] [CrossRef]
- Zarkadoulas, A.; Field, M.J.; Papatriantafyllopoulou, C.; Fize, J.; Artero, V.; Mitsopoulou, C.A. Experimental and Theoretical Insight into Electrocatalytic Hydrogen Evolution with Nickel Bis(aryldithiolene) Complexes as Catalysts. Inorg. Chem. 2016, 55, 432–444. [Google Scholar] [CrossRef]
- Zarkadoulas, A.; Field, M.J.; Artero, V.; Mitsopoulou, C.A. Proton-Reduction Reaction Catalyzed by Homoleptic Nickel-bis-1,2-dithiolate Complexes: Experimental and Theoretical Mechanistic Investigations. ChemCatChem 2017, 9, 2308–2317. [Google Scholar] [CrossRef]
- Sarkar, S.; Das, S.K. CO2 fixation by [WIVO(S2C2(CN)2)2]2−: Functional model for the tungsten-formate dehydrogenase of Clostridium thermoaceticum. Proc. Indian Acad. Sci. (Chem. Sci.) 1992, 104, 533–534. [Google Scholar] [CrossRef]
- Seo, J.; Shearer, J.; Williard, P.G.; Kim, E. Reactivity of a biomimetic W(IV) bis-dithiolene complex with CO2 leading to formate production and structural rearrangement. Dalton Trans. 2019, 48, 17441–17444. [Google Scholar] [CrossRef] [PubMed]
- Shiekh, B.A.; Kaur, D.; Kumar, S. Bio-mimetic self-assembled computationally designed catalysts of Mo and W for hydrogenation of CO2/dehydrogenation of HCOOH inspired by the active site of formate dehydrogenase. Phys. Chem. Chem. Phys. 2019, 21, 21370–21380. [Google Scholar] [CrossRef]
- Dobbek, H.; Gremer, L.; Kiefersauer, R.; Huber, R.; Meyer, O. Catalysis at a dinuclear [CuSMo(=O)OH] cluster in a CO dehydrogenase resolved at 1.1-angstrom resolution. Proc. Natl. Acad. Sci. USA 2002, 99, 15971–15976. [Google Scholar] [CrossRef]
- Jeoung, J.H.; Dobbek, H. Carbon dioxide activation at the Ni,Fe-cluster of anaerobic carbon monoxide dehydrogenase. Science 2007, 318, 1461–1464. [Google Scholar] [CrossRef] [PubMed]
- Mouchfiq, A.; Todorova, T.K.; Dey, S.; Fontecave, M.; Mougel, V. A bioinspired molybdenum–copper molecular catalyst for CO2 electroreduction. Chem. Sci. 2020, 11, 5503–5510. [Google Scholar] [CrossRef]
- Ghosh, D.; Sinhababu, S.; Santarsiero, B.D.; Mankad, N.P. A W/Cu Synthetic Model for the Mo/Cu Cofactor of Aerobic CODH Indicates That Biochemical CO Oxidation Requires a Frustrated Lewis Acid/Base Pair. J. Am. Chem. Soc. 2020, 142, 12635–12642. [Google Scholar] [CrossRef]
- Clark, M.L.; Grice, K.A.; Moore, C.E.; Rheingold, A.L.; Kubiak, C.P. Electrocatalytic CO2 reduction by M(bpy-R)(CO)4 (M = Mo, W.; R = H, tBu) complexes. Electrochemical, spectroscopic, and computational studies and comparison with group 7 catalysts. Chem. Sci. 2014, 5, 1894. [Google Scholar] [CrossRef]
- Tory, J.; Setterfield-Price, B.; Dryfe, R.A.W.; Hartl, F. [M(CO)4(2,2′-bipyridine)] (M=Cr, Mo, W) Complexes as Efficient Catalysts for Electrochemical Reduction of CO2 at a Gold Electrode. ChemElectroChem 2015, 2, 213–217. [Google Scholar] [CrossRef]
- Sieh, D.; Lacy, D.C.; Peters, J.C.; Kubiak, C.P. Reduction of CO2 by Pyridine Monoimine Molybdenum Carbonyl Complexes: Cooperative Metal-Ligand Binding of CO2. Chem. Eur. J. 2015, 21, 8497–8503. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.O.; Veenstra, F.L.P.; Chippindale, A.M.; Calhorda, M.J.; Hartl, F. Group 6 Metal Complexes as Electrocatalysts of CO2 Reduction: Strong Substituent Control of the Reduction Path of [Mo(η3-allyl)(CO)2(x,x′-dimethyl-2,2′-bipyridine)(NCS)] (x = 4–6). Organometallics 2019, 38, 1372–1390. [Google Scholar] [CrossRef]
- Grice, K.A.; Saucedo, C. Electrocatalytic Reduction of CO2 by Group 6 M(CO)6 Species without "Non-Innocent" Ligands. Inorg. Chem. 2016, 55, 6240–6246. [Google Scholar] [CrossRef] [PubMed]
- Fogeron, T.; Todorova, T.K.; Porcher, J.P.; Gomez-Mingot, M.; Chamoreau, L.M.; Mellot-Draznieks, C.; Li, Y.; Fontecave, M. A Bioinspired Nickel(bis-dithiolene) Complex as a Homogeneous Catalyst for Carbon Dioxide Electroreduction. ACS Catal. 2018, 8, 2030–2038. [Google Scholar] [CrossRef]
- Fogeron, T.; Retailleau, P.; Gomez-Mingot, M.; Li, Y.; Fontecave, M. Nickel Complexes Based on Molybdopterin-like Dithiolenes: Catalysts for CO2 Electroreduction. Organometallics 2019, 38, 1344–1350. [Google Scholar] [CrossRef]























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fogeron, T.; Li, Y.; Fontecave, M. Formate Dehydrogenase Mimics as Catalysts for Carbon Dioxide Reduction. Molecules 2022, 27, 5989. https://doi.org/10.3390/molecules27185989
Fogeron T, Li Y, Fontecave M. Formate Dehydrogenase Mimics as Catalysts for Carbon Dioxide Reduction. Molecules. 2022; 27(18):5989. https://doi.org/10.3390/molecules27185989
Chicago/Turabian StyleFogeron, Thibault, Yun Li, and Marc Fontecave. 2022. "Formate Dehydrogenase Mimics as Catalysts for Carbon Dioxide Reduction" Molecules 27, no. 18: 5989. https://doi.org/10.3390/molecules27185989
APA StyleFogeron, T., Li, Y., & Fontecave, M. (2022). Formate Dehydrogenase Mimics as Catalysts for Carbon Dioxide Reduction. Molecules, 27(18), 5989. https://doi.org/10.3390/molecules27185989