
Discovery of Quinacrine as a Potent Topo II and Hsp90 Dual-Target Inhibitor, Repurposing for Cancer Therapy

Abstract
:1. Introduction
2. Results
2.1. The Structural Alignment on Topo IIα and Hsp90α
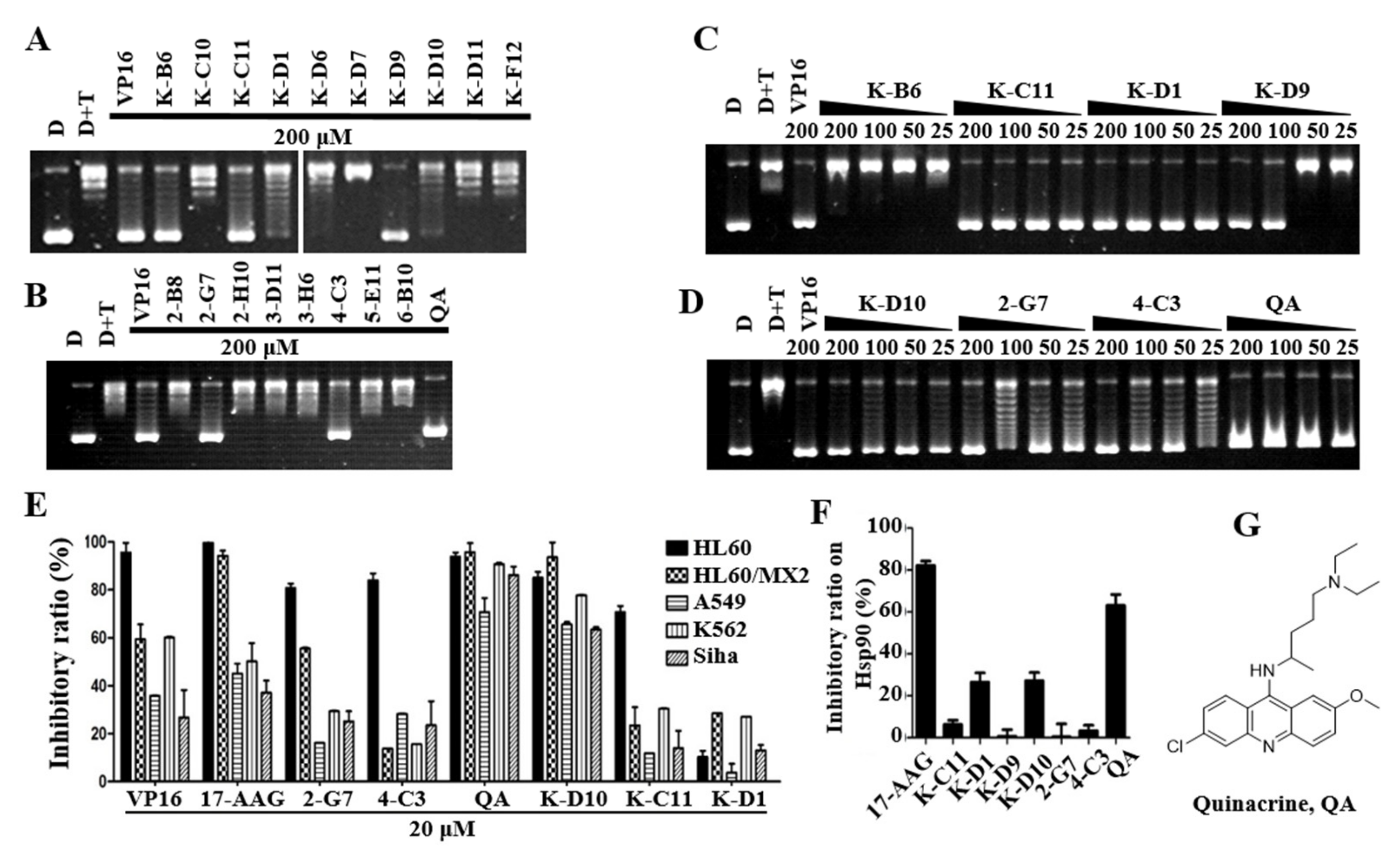
2.2. Preliminary Screening of Topo IIα Inhibitors
2.3. QA May Be a Potential Dual−Effect Inhibitor Based on Further Screening
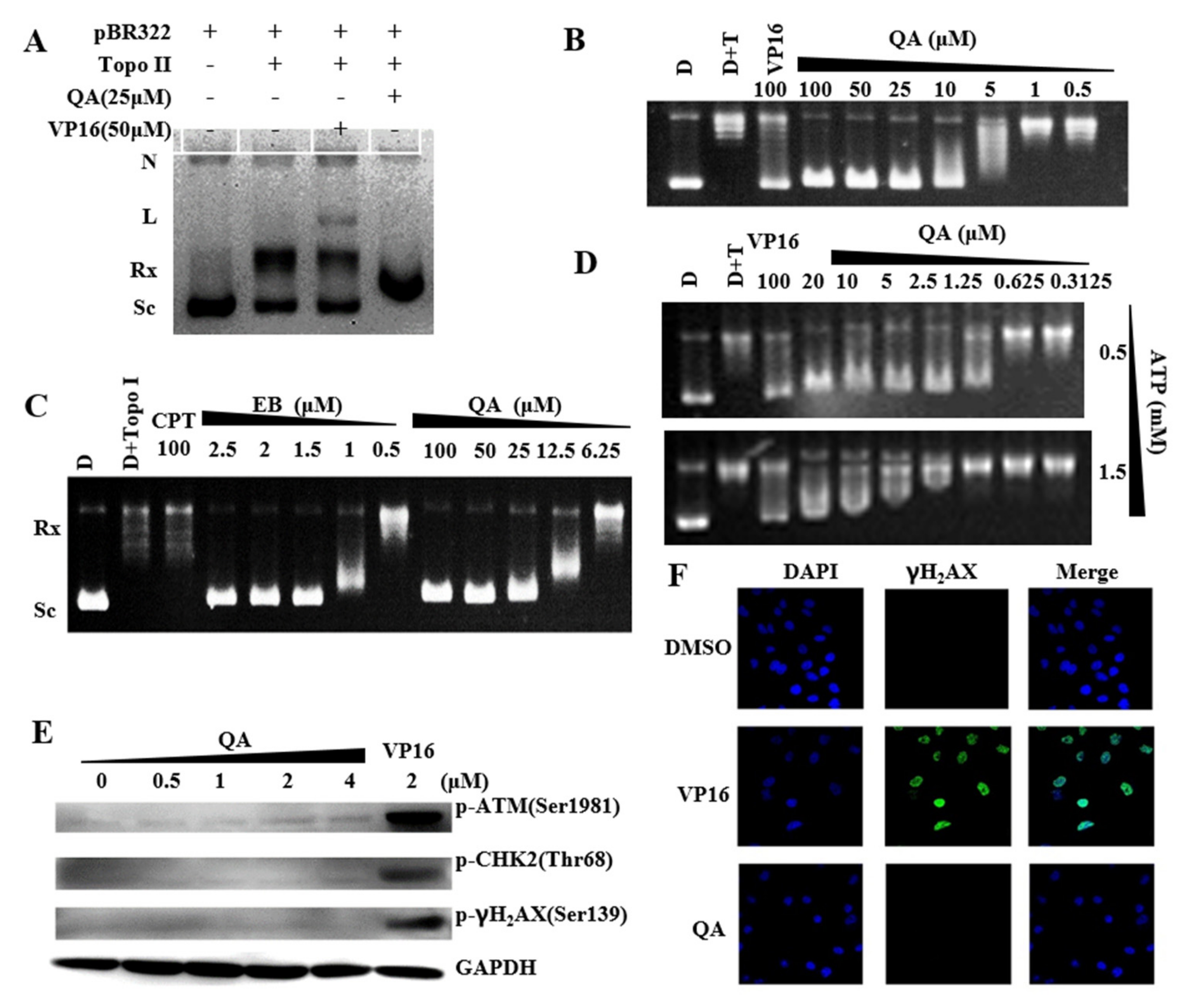
2.4. QA Is a Topo II Catalytic Inhibitor with Weak DNA Intercalation
2.5. QA Can Bind the N−Terminal ATP−Binding Site of Hsp90 and Inhibit Hsp90 Activity
2.6. Binding Model of QA on Topo II and Hsp90
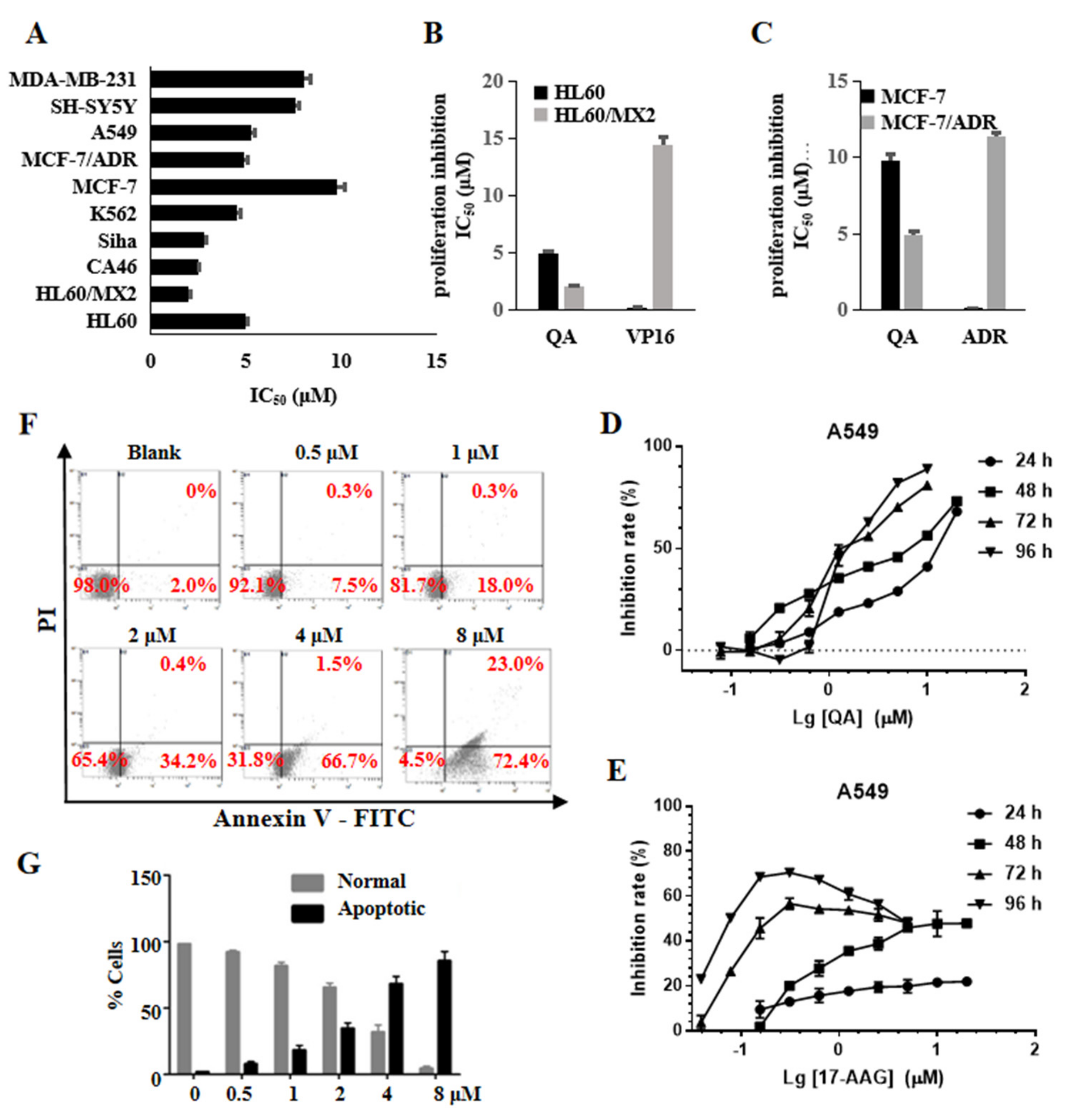
2.7. QA Has Broad Antitumor Activity and Induces Cell Apoptosis
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Cloning, Expression, and Purification
4.4. Structural Alignment of Topo IIα ATPase and Hsp90α N−ATPase
4.5. Molecular Docking Analysis
4.6. Colorimetric Determination of ATPase Activity
4.7. Cell Proliferation Assay
4.8. Topo II−Mediated DNA Relaxation Assay
4.9. Topo II−Mediated DNA Cleavage Assay
4.10. DNA−Unwinding Assay
4.11. Western Blotting
4.12. Immunofluorescence
4.13. Wound−Healing Assay
4.14. Competitive Displacement Assays Using Geldanamycin−FITC
4.15. Flow Cytometric Analysis
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barker, C.R.; McNamara, A.V.; Rackstraw, S.A.; Nelson, D.E.; White, M.R.; Watson, A.J.; Jenkins, J.R. Inhibition of Hsp90 acts synergistically with topoisomerase II poisons to increase the apoptotic killing of cells due to an increase in topoisomerase II mediated DNA damage. Nucleic Acids Res. 2006, 34, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S. The molecular perspective: DNA topoisomerases. Stem Cells 2002, 20, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Jeppsson, K.; Kanno, T.; Shirahige, K.; Sjogren, C. The maintenance of chromosome structure: Positioning and functioning of SMC complexes. Nat. Rev. Mol. Cell Biol. 2014, 15, 601–614. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, H.; Xiao, Z.; Zhang, G.; Zhang, D.; Bao, X.; Li, F.; Wu, S.; Gao, Y.; Wei, N. DNA damage and apoptosis induced by a potent orally podophyllotoxin derivative in breast cancer. Cell Commun. Signal. 2018, 16, 52. [Google Scholar] [CrossRef]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef]
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef]
- Cowell, I.G.; Sondka, Z.; Smith, K.; Lee, K.C.; Manville, C.M.; SidorczukLesthuruge, M.; Rance, H.A.; Padget, K.; Jackson, G.H.; Adachi, N.; et al. Model for MLL translocations in therapy-related leukemia involving topoisomerase IIbeta-mediated DNA strand breaks and gene proximity. Proc. Natl. Acad. Sci. USA 2012, 109, 8989–8994. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Baglini, E.; Salerno, S.; Barresi, E.; Robello, M.; Da Settimo, F.; Taliani, S.; Marini, A.M. Multiple Topoisomerase I (TopoI), Topoisomerase II (TopoII) and Tyrosyl-DNA Phosphodiesterase (TDP) inhibitors in the development of anticancer drugs. Eur. J. Pharm. Sci. 2021, 156, 105594. [Google Scholar] [CrossRef]
- Hu, W.; Huang, X.S.; Wu, J.F.; Yang, L.; Zheng, Y.T.; Shen, Y.M.; Li, Z.Y.; Li, X. Discovery of Novel Topoisomerase II Inhibitors by Medicinal Chemistry Approaches. J. Med. Chem. 2018, 61, 8947–8980. [Google Scholar] [CrossRef]
- Young, J.C.; Moarefi, I.; Hartl, F.U. Hsp90: A specialized but essential protein-folding tool. J. Cell Biol. 2001, 154, 267–273. [Google Scholar] [CrossRef]
- Taldone, T.; Patel, H.J.; Bolaender, A.; Patel, M.R.; Chiosis, G. Protein chaperones: A composition of matter review (2008–2013). Expert Opin. Ther. Pat. 2014, 24, 501–518. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef]
- Khalil, A.A.; Kabapy, N.F.; Deraz, S.F.; Smith, C. Heat shock proteins in oncology: Diagnostic biomarkers or therapeutic targets? Biochim. Biophys. Acta 2011, 1816, 89–104. [Google Scholar] [CrossRef]
- Biamonte, M.A.; Van de Water, R.; Arndt, J.W.; Scannevin, R.H.; Perret, D.; Lee, W.C. Heat shock protein 90: Inhibitors in clinical trials. J. Med. Chem. 2010, 53, 3–17. [Google Scholar] [CrossRef]
- Dai, J.; Chen, A.; Zhu, M.; Qi, X.; Tang, W.; Liu, M.; Li, D.; Gu, Q.; Li, J. Penicisulfuranol A, a novel C-terminal inhibitor disrupting molecular chaperone function of Hsp90 independent of ATP binding domain. Biochem. Pharmacol. 2019, 163, 404–415. [Google Scholar] [CrossRef]
- Yao, Q.; Weigel, B.; Kersey, J. Synergism between etoposide and 17-AAG in leukemia cells: Critical roles for Hsp90, FLT3, topoisomerase II, Chk1, and Rad51. Clin. Cancer Res. 2007, 13, 1591–1600. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.H.; Park, K.S.; Lee, D.H.; Alberobello, A.T.; Raffeld, M.; Pierobon, M.; Pin, E.; Petricoin Iii, E.F.; Wang, Y.; Giaccone, G. HSP-90 inhibitor ganetespib is synergistic with doxorubicin in small cell lung cancer. Oncogene 2014, 33, 4867–4876. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Basso, A.; Solit, D.; Norton, L.; Rosen, N. Modulation of Hsp90 function by ansamycins sensitizes breast cancer cells to chemotherapy-induced apoptosis in an RB- and schedule-dependent manner. See: E. A. Sausville, Combining cytotoxics and 17-allylamino, 17-demethoxygeldanamycin: Sequence and tumor biology matters, Clin. Cancer Res., 7: 2155–2158, 2001. Clin. Cancer Res. 2001, 7, 2228–2236. [Google Scholar] [PubMed]
- Skok, Z.; Zidar, N.; Kikelj, D.; Ilas, J. Dual Inhibitors of Human DNA Topoisomerase II and Other Cancer-Related Targets. J. Med. Chem. 2020, 63, 884–904. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Ma, P.F.; Lin, Y.C.; Wu, C.H.; Peng, Y.S.; Zheng, H.Y.; Lin, M.C.; Li, Y.T.; Lin, Y.W. 17-(Allylamino)-17-Demethoxygeldanamycin Enhances Etoposide-Induced Cytotoxicity via the Downregulation of Xeroderma Pigmentosum Complementation Group C Expression in Human Lung Squamous Cell Carcinoma Cells. Pharmacology 2018, 102, 91–104. [Google Scholar] [CrossRef]
- Dutta, R.; Inouye, M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 2000, 25, 24–28. [Google Scholar] [CrossRef]
- Lee, M.G.; Liu, Y.C.; Lee, Y.L.; El-Shazly, M.; Lai, K.H.; Shih, S.P.; Ke, S.C.; Hong, M.C.; Du, Y.C.; Yang, J.C.; et al. Heteronemin, a Marine Sesterterpenoid-Type Metabolite, Induces Apoptosis in Prostate LNcap Cells via Oxidative and ER Stress Combined with the Inhibition of Topoisomerase II and Hsp90. Mar. Drugs 2018, 16, 204. [Google Scholar] [CrossRef]
- Jun, K.Y.; Kwon, Y. Proposal of Dual Inhibitor Targeting ATPase Domains of Topoisomerase II and Heat Shock Protein 90. Biomol. Ther. 2016, 24, 453–468. [Google Scholar] [CrossRef]
- Lai, K.H.; Liu, Y.C.; Su, J.H.; El-Shazly, M.; Wu, C.F.; Du, Y.C.; Hsu, Y.M.; Yang, J.C.; Weng, M.K.; Chou, C.H.; et al. Antileukemic Scalarane Sesterterpenoids and Meroditerpenoid from Carteriospongia (Phyllospongia) sp., Induce Apoptosis via Dual Inhibitory Effects on Topoisomerase II and Hsp90. Sci. Rep. 2016, 6, 36170. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Sezer, A.; Halilovic-Alihodzic, M.; Vanwieren, A.R.; Smajkan, A.; Karic, A.; Djedovic, H.; Sutkovic, J. A review on drug repurposing in COVID-19: From antiviral drugs to herbal alternatives. J. Genet. Eng. Biotechnol. 2022, 20, 78. [Google Scholar] [CrossRef]
- Radwan, M.O.; Ciftci, H.I.; Ali, T.F.S.; Ellakwa, D.E.; Koga, R.; Tateishi, H.; Nakata, A.; Ito, A.; Yoshida, M.; Okamoto, Y.; et al. Antiproliferative S-Trityl-l-Cysteine -Derived Compounds as SIRT2 Inhibitors: Repurposing and Solubility Enhancement. Molecules 2019, 24, 3295. [Google Scholar] [CrossRef]
- Ydsten, K.A.; Hellgren, U.; Asgeirsson, H. Quinacrine Treatment of Nitroimidazole-Refractory Giardiasis. J. Infect. Dis. 2022, 225, 1773–1776. [Google Scholar] [CrossRef]
- Yan, D.; Borucki, R.; Sontheimer, R.D.; Werth, V.P. Candidate drug replacements for quinacrine in cutaneous lupus erythematosus. Lupus Sci. Med. 2020, 7, e000430. [Google Scholar] [CrossRef]
- Obermann, W.M.; Sondermann, H.; Russo, A.A.; Pavletich, N.P.; Hartl, F.U. In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J. Cell Biol. 1998, 143, 901–910. [Google Scholar] [CrossRef]
- Wu, L.; Yu, J.; Chen, R.; Liu, Y.; Lou, L.; Wu, Y.; Huang, L.; Fan, Y.; Gao, P.; Huang, M.; et al. Dual inhibition of Bcr-Abl and Hsp90 by C086 potently inhibits the proliferation of imatinib-resistant CML cells. Clin. Cancer Res. 2015, 21, 833–843. [Google Scholar] [CrossRef]
- Howes, R.; Barril, X.; Dymock, B.W.; Grant, K.; Northfield, C.J.; Robertson, A.G.; Surgenor, A.; Wayne, J.; Wright, L.; James, K.; et al. A fluorescence polarization assay for inhibitors of Hsp90. Anal. Biochem. 2006, 350, 202–213. [Google Scholar] [CrossRef]
- Carreras, C.W.; Schirmer, A.; Zhong, Z.; Santi, D.V. Filter binding assay for the geldanamycin-heat shock protein 90 interaction. Anal. Biochem. 2003, 317, 40–46. [Google Scholar] [CrossRef]
- Sadikot, T.; Swink, M.; Eskew, J.D.; Brown, D.; Zhao, H.; Kusuma, B.R.; Rajewski, R.A.; Blagg, B.S.; Matts, R.L.; Holzbeierlein, J.M.; et al. Development of a high-throughput screening cancer cell-based luciferase refolding assay for identifying Hsp90 inhibitors. Assay Drug Dev. Technol. 2013, 11, 478–488. [Google Scholar] [CrossRef]
- Patwardhan, C.A.; Alfa, E.; Lu, S.; Chadli, A. Progesterone receptor chaperone complex-based high-throughput screening assay: Identification of capsaicin as an inhibitor of the Hsp90 machine. J. Biomol. Screen. 2015, 20, 223–229. [Google Scholar] [CrossRef]
- Lindsley, J.E. Use of a real-time, coupled assay to measure the ATPase activity of DNA topoisomerase II. Methods Mol. Biol. 2001, 95, 57–64. [Google Scholar]
- Kingma, P.S.; Fortune, J.M.; Osheroff, N. Topoisomerase II-catalyzed ATP hydrolysis as monitored by thin-layer chromatography. Methods Mol. Biol. 2001, 95, 51–56. [Google Scholar]
- Geladopoulos, T.P.; Sotiroudis, T.G.; Evangelopoulos, A.E. A malachite green colorimetric assay for protein phosphatase activity. Anal. Biochem. 1991, 192, 112–116. [Google Scholar] [CrossRef]
- Rowlands, M.G.; Newbatt, Y.M.; Prodromou, C.; Pearl, L.H.; Workman, P.; Aherne, W. High-throughput screening assay for inhibitors of heat-shock protein 90 ATPase activity. Anal. Biochem. 2004, 327, 176–183. [Google Scholar] [CrossRef]
- Kimura, Y.; Kurzydlowski, K.; Tada, M.; MacLennan, D.H. Phospholamban regulates the Ca2+-ATPase through intramembrane interactions. J. Biol. Chem. 1996, 271, 21726–21731. [Google Scholar] [CrossRef]
- Feng, J.; Chen, Y.; Pu, J.; Yang, X.; Zhang, C.; Zhu, S.; Zhao, Y.; Yuan, Y.; Yuan, H.; Liao, F. An improved malachite green assay of phosphate: Mechanism and application. Anal. Biochem. 2011, 409, 144–149. [Google Scholar] [CrossRef]
- Avila, C.; Kornilayev, B.A.; Blagg, B.S. Development and optimization of a useful assay for determining Hsp90′s inherent ATPase activity. Bioorganic Med. Chem. 2006, 14, 1134–1142. [Google Scholar] [CrossRef]
- Zhuo, S.T.; Li, C.Y.; Hu, M.H.; Chen, S.B.; Yao, P.F.; Huang, S.L.; Ou, T.M.; Tan, J.H.; An, L.K.; Li, D.; et al. Synthesis and biological evaluation of benzo[a]phenazine derivatives as a dual inhibitor of topoisomerase I and II. Org. Biomol. Chem. 2013, 11, 3989–4005. [Google Scholar] [CrossRef]
- Cha, S. Tight-binding inhibitors-I. Kinetic behavior. Biochem. Pharmacol. 1975, 24, 2177–2185. [Google Scholar] [CrossRef]
- Deweese, J.E.; Burgin, A.B.; Osheroff, N. Human topoisomerase IIalpha uses a two-metal-ion mechanism for DNA cleavage. Nucleic Acids Res. 2008, 36, 4883–4893. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Mimnaugh, E.G.; Xu, W.; Vos, M.; Yuan, X.; Isaacs, J.S.; Bisht, K.S.; Gius, D.; Neckers, L. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol. Cancer Ther. 2004, 3, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lin, N.U. HSP90 as a platform for the assembly of more effective cancer chemotherapy. Biochim. Biophys. Acta 2012, 1823, 756–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preet, R.; Mohapatra, P.; Mohanty, S.; Sahu, S.K.; Choudhuri, T.; Wyatt, M.D.; Kundu, C.N. Quinacrine has anticancer activity in breast cancer cells through inhibition of topoisomerase activity. Int. J. Cancer 2012, 130, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Neznanov, N.; Gorbachev, A.V.; Neznanova, L.; Komarov, A.P.; Gurova, K.V.; Gasparian, A.V.; Banerjee, A.K.; Almasan, A.; Fairchild, R.L.; Gudkov, A.V. Anti-malaria drug blocks proteotoxic stress response: Anti-cancer implications. Cell Cycle 2009, 8, 3960–3970. [Google Scholar] [CrossRef]
- Jung, D.; Khurana, A.; Roy, D.; Kalogera, E.; Bakkum-Gamez, J.; Chien, J.; Shridhar, V. Quinacrine upregulates p21/p27 independent of p53 through autophagy-mediated downregulation of p62-Skp2 axis in ovarian cancer. Sci. Rep. 2018, 8, 2487. [Google Scholar] [CrossRef]
- Dermawan, J.K.; Gurova, K.; Pink, J.; Dowlati, A.; De, S.; Narla, G.; Sharma, N.; Stark, G.R. Quinacrine overcomes resistance to erlotinib by inhibiting FACT, NF-kappaB, and cell-cycle progression in non-small cell lung cancer. Mol. Cancer Ther. 2014, 13, 2203–2214. [Google Scholar] [CrossRef]
- Fontana, J.; Fulton, D.; Chen, Y.; Fairchild, T.A.; McCabe, T.J.; Fujita, N.; Tsuruo, T.; Sessa, W.C. Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ. Res. 2002, 90, 866–873. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Name | CAS | Kinase | Topo II ATPase RIR (%) * | HL60 IC50 (μM) ** |
|---|---|---|---|---|---|
| K−B6 | Staurosporine | 62996−74−1 | Pan−specific | 111.2 | <0.01 |
| K−C2 | Tyrphostin 47 | 122520−86−9 | EGFRK | 90.3 | >25 |
| K−C3 | Tyrphostin 51 | 122520−90−5 | EGFRK | 85.8 | >25 |
| K−C10 | PKC−412 | 120685−11−2 | PKC | 68.4 | 0.75 |
| K−C11 | Piceatannol | 10083−24−6 | Syk | 70.5 | 12.07 |
| K−Dl | AG−490 | 133550−35−3 | JAK−2 | 61.6 | 18.97 |
| K−D6 | Wortmannin | 19545−26−7 | PI 3−K | 66.8 | 13.32 |
| K−D7 | GF 109203X | 133052−90−1 | PKC | 64.7 | 9.51 |
| K−D8 | Hypericin | 548−04−9 | PKC | 75.8 | >25 |
| K−D9 | Ro 31−8220 | 138489−18−6 | PKC | 99.7 | 1.18 |
| K−D10 | Sphingosine | 123−78−4 | PKC | 94.3 | 11.10 |
| K−Dl 1 | H−89 | 127243−85−0 | PKA | 77.1 | 13.24 |
| K−D12 | H−8 | 84478−11−5 | PKA, PKG | 62.3 | >25 |
| K−F9 | Palmitoyl−d,L−carnitine | 6865−14−1 | PKC | 109.4 | >25 |
| K−F12 | Daidzein | 486−66−8 | CK2 | 101.2 | 3.02 |
| 2−B8 | Fluphenazine 2HC1 | 146−56−5 | 64.1% | 13.38 | |
| 2−G7 | Atovaquone | 95233−18−4 | 47.2% | 9.43 | |
| 2−H10 | Bopindolol malonate | 62658−64−4 | 41.2% | 13.23 | |
| 2−H11 | Guanfacine HCl | 29520−14−7 | 64.5% | >25 | |
| 3−C11 | Gallamine triethiodide | 65−29−2 | 56.2% | >25 | |
| 3−D11 | Flunarizine−2HCl | 30484−77−6 | 42.4% | 22.73 | |
| 3−E6 | Trifluoperazine 2HCl | 440−17−5 | 58.1% | >25 | |
| 3−H6 | Actarit | 18699−02−0 | 45.4% | 8.02 | |
| 4−C3 | Raloxifene HCl | 82640−04−8 | 47.6% | 7.20 | |
| 5−E4 | 4−aminosalicylic acid | 65−49−6 | 40.2% | >25 | |
| 5−E5 | 5−aminosalicylic acid | 89−57−6 | 42.3% | >25 | |
| 5−E6 | Ampicillin trihydrate | 7177−48−2 | 42.5% | >25 | |
| 5−E11 | Atracurium besylate | 64228−81−5 | 88.9% | 22.86 | |
| 6−B10 | Doxazosin mesylate | 77883−43−3 | 47.6% | 19.73 | |
| 7−F2 | Quinacrine 2HCl 2H2O | 83−89−6 | 42.3% | 5.22 | |
| 7−H3 | Streptomycin sulfate | 3810−74−0 | 71.6% | >25 | |
| 7−H4 | Sulfadoxine | 2447−57−6 | 71.2% | >25 | |
| 7−H5 | Sulfadiazine | 68−35−9 | 71.0% | >25 | |
| 7−H6 | Sulfadimethoxine | 122−11−2 | 73.9% | >25 | |
| 7−H7 | Sulfasalazine | 599−79−1 | 70.8% | >25 | |
| 7−H8 | Tamsulosin HCl | 106463−17−6 | 71.0% | >25 | |
| 7−H9 | Telmisartan | 144701−48−4 | 79.4% | >25 | |
| 7−H10 | Tenoxicam | 59804−37−4 | 76.1% | >25 | |
| 7−H11 | Terazosin HCl | 63590−64−7 | 70.6% | >25 | |
| 8−H9 | L−thyroxine | 51−48−9 | 42.0% | >25 | |
| VP16 | 0.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, X.; Mao, T.-y.; Mai, Y.-w.; Liang, C.-c.; Huang, W.-h.; Rao, Y.; Huang, Z.-s.; Huang, S.-l. Discovery of Quinacrine as a Potent Topo II and Hsp90 Dual-Target Inhibitor, Repurposing for Cancer Therapy. Molecules 2022, 27, 5561. https://doi.org/10.3390/molecules27175561
Pan X, Mao T-y, Mai Y-w, Liang C-c, Huang W-h, Rao Y, Huang Z-s, Huang S-l. Discovery of Quinacrine as a Potent Topo II and Hsp90 Dual-Target Inhibitor, Repurposing for Cancer Therapy. Molecules. 2022; 27(17):5561. https://doi.org/10.3390/molecules27175561
Chicago/Turabian StylePan, Xin, Teng-yu Mao, Yan-wen Mai, Cheng-cheng Liang, Wei-hao Huang, Yong Rao, Zhi-shu Huang, and Shi-liang Huang. 2022. "Discovery of Quinacrine as a Potent Topo II and Hsp90 Dual-Target Inhibitor, Repurposing for Cancer Therapy" Molecules 27, no. 17: 5561. https://doi.org/10.3390/molecules27175561
APA StylePan, X., Mao, T.-y., Mai, Y.-w., Liang, C.-c., Huang, W.-h., Rao, Y., Huang, Z.-s., & Huang, S.-l. (2022). Discovery of Quinacrine as a Potent Topo II and Hsp90 Dual-Target Inhibitor, Repurposing for Cancer Therapy. Molecules, 27(17), 5561. https://doi.org/10.3390/molecules27175561