
[1,5]-Hydride Shift Triggered N-Dealkylative Cyclization into 2-Oxo-1,2,3,4-tetrahydroquinoline-3-carboxylates via Boronate Complexes

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
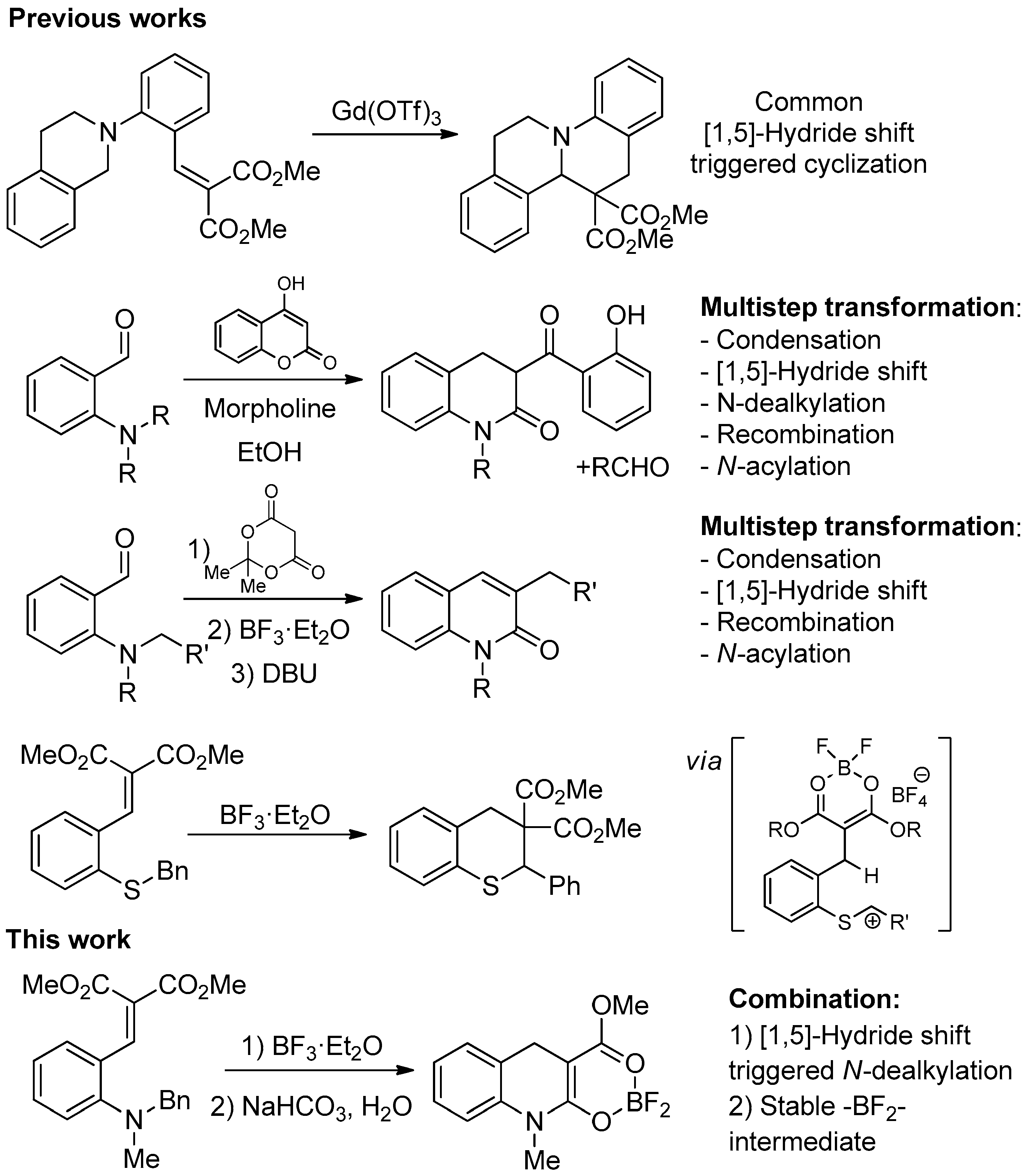
:1. Introduction
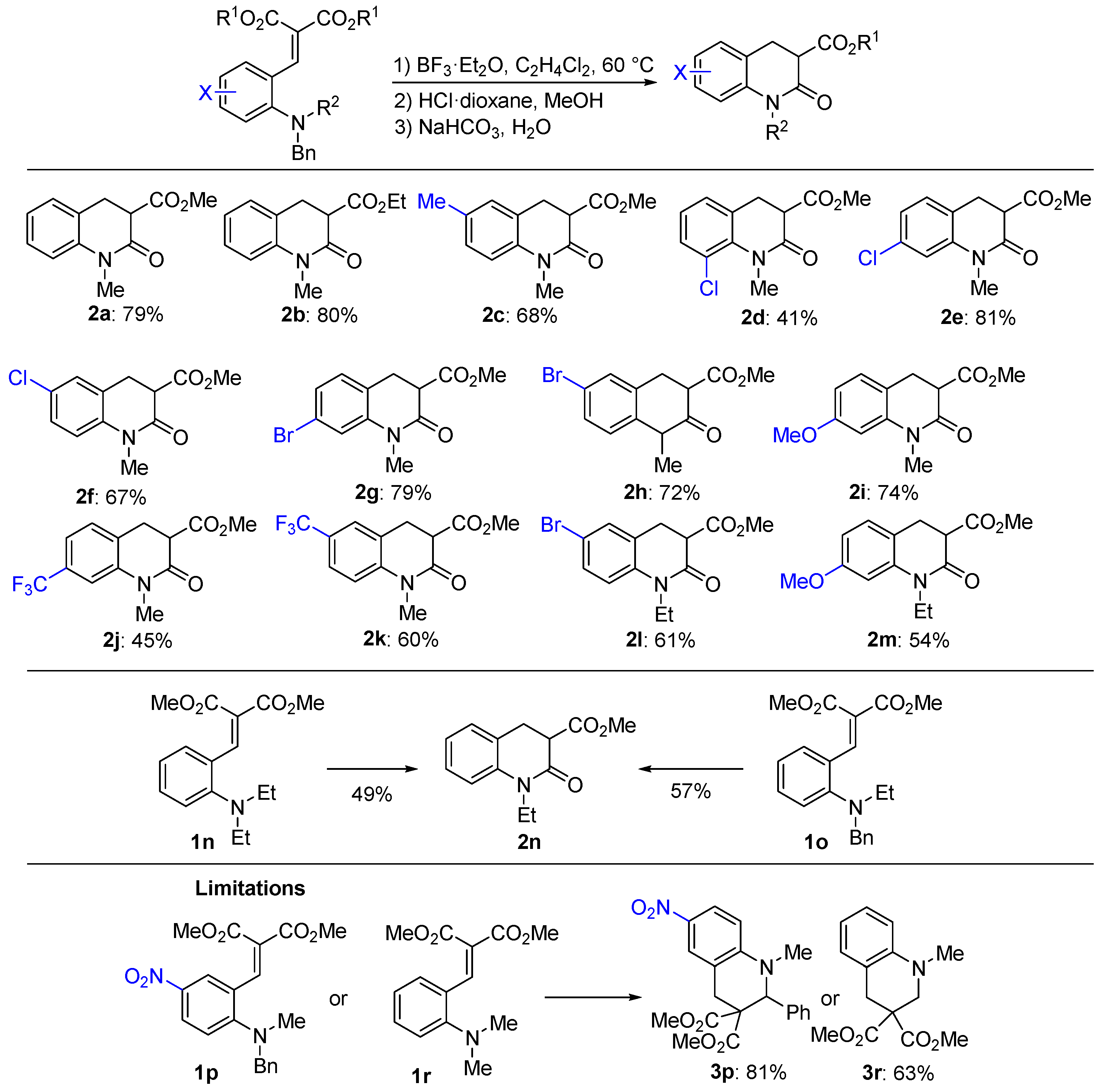
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Experimental Procedures
3.2.1. Synthesis of Methyl2-((difluoroboranyl)oxy)-1-methyl-1,4-dihydroquinoline-3-carboxylate (2a*)
3.2.2. General Procedure for Synthesis of the Compounds 2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Newhouse, T.; Baran, P.S.; Hoffmann, R.W. The economies of synthesis. Chem. Soc. Rev. 2009, 38, 3010–3021. [Google Scholar] [CrossRef]
- Burns, N.Z.; Baran, P.S.; Hoffmann, R.W. Redox Economy in Organic Synthesis. Angew. Chem. Int. Ed. 2009, 48, 2854–2867. [Google Scholar] [CrossRef]
- Seidel, D. The Azomethine Ylide Route to Amine C–H Functionalization: Redox-Versions of Classic Reactions and a Pathway to New Transformations. Acc. Chem. Res. 2015, 48, 317–328. [Google Scholar] [CrossRef]
- Fiorito, D.; Scaringi, S.; Mazet, C. Transition metal-catalyzed alkene isomerization as an enabling technology in tandem, sequential and domino processes. Chem. Soc. Rev. 2021, 50, 1391–1406. [Google Scholar] [CrossRef]
- Corma, A.; Navas, J.; Sabater, M.J. Advances in One-Pot Synthesis through Borrowing Hydrogen Catalysis. Chem. Rev. 2018, 118, 1410–1459. [Google Scholar] [CrossRef]
- Mori, K. C(sp3)–H Bond Functionalization Mediated by Hydride a Shift/Cyclization System. Bull. Chem. Soc. Jpn. 2022, 95, 296–305. [Google Scholar] [CrossRef]
- An, X.-D.; Xiao, J. Recent advances in hydride transfer-involved C(sp3)–H activation reactions. Org. Chem. Front. 2021, 8, 1364–1383. [Google Scholar] [CrossRef]
- Wang, L.; Xiao, J. Hydrogen-Atom Transfer Reactions. Top. Curr. Chem. 2016, 374, 17. [Google Scholar]
- Haibach, M.C.; Seidel, D. C-H bond functionalization through intramolecular hydride transfer. Angew. Chem. Int. Ed. 2014, 53, 5010–5036. [Google Scholar] [CrossRef]
- Peng, B.; Maulide, N. The Redox-Neutral Approach to C-H Functionalization. Chem. Eur. J. 2013, 19, 13274–13287. [Google Scholar] [CrossRef]
- Murarka, S.; Zhang, C.; Konieczynska, M.D.; Seidel, D. Lewis Acid Catalyzed Formation of Tetrahydroquinolines via an Intramolecular Redox Process. Org. Lett. 2009, 11, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, L.; Lv, J.; Cheng, J.-P.; Luo, S. Catalytic Enantioselective tert -Aminocyclization by Asymmetric Binary Acid Catalysis (ABC): Stereospecific 1,5-Hydrogen Transfer. Chem. Eur. J. 2012, 18, 8891–8895. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Liu, X.; Wang, W.; Lin, L.; Feng, X. Highly Enantioselective Synthesis of Tetrahydroquinolines via Cobalt(II)-Catalyzed Tandem 1,5-Hydride Transfer/Cyclization. Org. Lett. 2011, 13, 600–603. [Google Scholar] [CrossRef]
- Mori, K.; Kurihara, K.; Yabe, S.; Yamanaka, M.; Akiyama, T. Double C(sp3)–H Bond Functionalization Mediated by Sequential Hydride Shift/Cyclization Process: Diastereoselective Construction of Polyheterocycles. J. Am. Chem. Soc. 2014, 136, 3744–3747. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, M.; Otawa, Y.; Ido, N.; Mori, K. Highly Diastereoselective Synthesis of Medium-Sized Carbocycle-Fused Piperidines via Sequential Hydride Shift Triggered Double C(sp3)–H Bond Functionalization. Org. Lett. 2019, 21, 9334–9338. [Google Scholar] [CrossRef]
- Yokoo, K.; Sakai, D.; Mori, K. Highly Stereoselective Synthesis of Fused Tetrahydropyrans via Lewis-Acid-Promoted Double C(sp3)–H Bond Functionalization. Org. Lett. 2020, 22, 5801–5805. [Google Scholar] [CrossRef]
- Mori, K.; Isogai, R.; Kamei, Y.; Yamanaka, M.; Akiyama, T. Chiral magnesium bisphosphate-catalyzed asymmetric double C (sp3)–H bond functionalization based on sequential hydride shift/cyclization process. Am. Chem. Soc. 2018, 140, 6203–6207. [Google Scholar]
- Yang, X.; Wang, L.; Hu, F.; Xu, L.; Li, S.; Li, S.-S. Redox-Triggered Switchable Synthesis of 3,4-Dihydroquinolin-2(1H)-one Derivatives via Hydride Transfer/N-Dealkylation/N-Acylation. Org. Lett. 2021, 23, 358–364. [Google Scholar] [CrossRef]
- Yokoo, K.; Mori, K. Expeditious Synthesis of Multisubstituted Quinolinone Derivatives Based on Ring Recombination Strategy. Org. Lett. 2020, 22, 244–248. [Google Scholar] [CrossRef]
- Zaitseva, E.; Smirnov, A.; Timashev, V.; Malyshev, W.; Zhigileva, E.; Mikhaylov, A.; Medvedev, M.; Baleeva, N.; Baranov, M.S. BF3 Mediated [1,5]-Hydride Shift Triggered Cyclization: Thioethers Join the Game. Eur. J. Org. Chem. 2022, 2022, e202200547. [Google Scholar] [CrossRef]
- Borisov, D.D.; Novikov, R.A.; Tomilov, Y.V.J. Reactions of Styrylmalonates with Aromatic Aldehydes: Detailed Synthetic and Mechanistic Studies. Org. Chem. 2021, 86, 4457–4471. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-L.; Li, X.; Qin, W.-B.; Liu, J.-J.; Huang, Y.-Y.; Wong, H.N.C.; Liu, G.-K. Air- and Light-Stable S-(Difluoromethyl)sulfonium Salts: C-Selective Electrophilic Difluoromethylation of β-Ketoesters and Malonates. Org. Lett. 2018, 20, 6925–6929. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Xu, W.; Wang, R.; Zhang, M.; Xie, C.; Zeng, X.; Wang, M. Potassium tert-Butoxide-Mediated Condensation Cascade Reaction: Transition Metal-Free Synthesis of Multisubstituted Aryl Indoles and Benzofurans. Org. Lett. 2019, 21, 3658–3662. [Google Scholar] [CrossRef] [PubMed]
- Murarka, S.; Deb, I.; Zhang, C.; Seidel, D. Catalytic enantioselective intramolecular redox reactions: Ring-fused tetrahydroquinolines. J. Am. Chem. Soc. 2009, 131, 13226–13227. [Google Scholar] [CrossRef]
- Zaitseva, E.R.; Smirnov, A.Y.; Myasnyanko, I.N.; Mineev, K.S.; Sokolov, A.I.; Volkhina, T.N.; Mikhaylov, A.A.; Baleeva, N.S.; Baranov, M.S. Imidazol-5-ones as a substrate for [1, 5]-hydride shift triggered cyclization. New J. Chem. 2021, 45, 1805–1808. [Google Scholar] [CrossRef]
- Josa-Culleré, L.; Hirst, M.G.; Lockett, J.P.; Thompson, A.L.; Moloney, M.G. Spirocyclic tetramates by sequential knoevenagel and [1, 5]-prototropic shift. J. Org. Chem. 2019, 84, 9671–9683. [Google Scholar] [CrossRef]
- Rosevear, J.; Wilshire, J.F.K. The preparation of some 2-nitroacridines and related compounds. Aust. J. Chem. 1981, 34, 839–853. [Google Scholar] [CrossRef]
- Mori, K.; Ehara, K.; Kurihara, K.; Akiyama, T. Selective Activation of Enantiotopic C (sp3)− Hydrogen by Means of Chiral Phosphoric Acid: Asymmetric Synthesis of Tetrahydroquinoline Derivatives. J. Am. Chem. Soc. 2011, 133, 6166–6169. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaitseva, E.R.; Ivanov, D.S.; Smirnov, A.Y.; Mikhaylov, A.A.; Baleeva, N.S.; Baranov, M.S. [1,5]-Hydride Shift Triggered N-Dealkylative Cyclization into 2-Oxo-1,2,3,4-tetrahydroquinoline-3-carboxylates via Boronate Complexes. Molecules 2022, 27, 5270. https://doi.org/10.3390/molecules27165270
Zaitseva ER, Ivanov DS, Smirnov AY, Mikhaylov AA, Baleeva NS, Baranov MS. [1,5]-Hydride Shift Triggered N-Dealkylative Cyclization into 2-Oxo-1,2,3,4-tetrahydroquinoline-3-carboxylates via Boronate Complexes. Molecules. 2022; 27(16):5270. https://doi.org/10.3390/molecules27165270
Chicago/Turabian StyleZaitseva, Elvira R., Dmitrii S. Ivanov, Alexander Yu. Smirnov, Andrey A. Mikhaylov, Nadezhda S. Baleeva, and Mikhail S. Baranov. 2022. "[1,5]-Hydride Shift Triggered N-Dealkylative Cyclization into 2-Oxo-1,2,3,4-tetrahydroquinoline-3-carboxylates via Boronate Complexes" Molecules 27, no. 16: 5270. https://doi.org/10.3390/molecules27165270
APA StyleZaitseva, E. R., Ivanov, D. S., Smirnov, A. Y., Mikhaylov, A. A., Baleeva, N. S., & Baranov, M. S. (2022). [1,5]-Hydride Shift Triggered N-Dealkylative Cyclization into 2-Oxo-1,2,3,4-tetrahydroquinoline-3-carboxylates via Boronate Complexes. Molecules, 27(16), 5270. https://doi.org/10.3390/molecules27165270